
Abstract
Severe fever with thrombocytopenia syndrome (SFTS) is an emerging viral infectious disease caused by a novel Bandavirus in the family Phenuiviridae. The SFTS virus (SFTSV) is transmitted to various hosts, including humans, through tick bites, leading to high fever, thrombocytopenia, and leukopenia, with a high case fatality rate (up to 30%) due to multiple organ dysfunction. Therefore, early diagnosis is crucial for effective treatment and preventing disease transmission. In this study, we aimed to develop and characterize monoclonal antibodies targeting the SFTSV nucleocapsid protein (NP). We generated recombinant NP to screen antibodies against SFTSV. Using phage display technology, we identified candidate single-chain variable fragment (scFv) sequences capable of detecting SFTSV NP. Five human IgG antibodies and six chimeric mouse antibodies exhibited strong binding ability to the recombinant NP. Furthermore, their specificity and selectivity were evaluated against NPs from different subtypes and other viral species. A sandwich enzyme–linked immunosorbent assay (ELISA) was performed to determine optimal antibody pairings for SFTSV detection. The mP01A05/hP01C09, mP01A05/hP01B10, and mP02E04/hP01A05 antibody pairs demonstrated high efficacy in diagnosing SFTSV infections. These findings provide valuable antibody resources and establish an effective platform for the diagnosis of SFTS.
Key points
• Monoclonal antibodies targeting SFTSV NP were developed using phage display technology.
• Candidate antibodies showed strong binding ability and high specificity to SFTSV NP.
• Optimized antibody pairs enabled effective SFTSV detection via sandwich ELISA.
Similar content being viewed by others
Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
Introduction
Severe fever with thrombocytopenia syndrome virus (SFTSV) is an emerging tick-borne virus that was first identified in China in 2009 (Xu et al. 2011). Since then, severe fever with thrombocytopenia syndrome (SFTS) has been observed in various Asian countries, including South Korea and Japan (Kim et al. 2018; Ongkittikul et al. 2020; Peng et al. 2020; Takahashi et al. 2014; Tran et al. 2019; Win et al. 2020; Zohaib et al. 2020). In the USA, an infection with heartland virus, which is similar to SFTSV, has been reported (McMullan et al. 2012). The term “SFTSV” is more widely mentioned in the research community to refer to the virus, despite its recent renaming as Bandavirus dabieense by the International Committee on Taxonomy of Viruses in 2022 (International Committee on Taxonomy of Viruses 2022). SFTS has some major clinical manifestations, including fever, thrombocytopenia, leukocytopenia, gastrointestinal symptoms, neurological symptoms, and bleeding tendency, with a mortality rate of up to 30% (Cao et al. 2021; Casel et al. 2021; Li et al. 2021; Zhang et al. 2013). It is transmitted by a primary host vector such as ticks and can subsequently infect various animals and humans (Luo et al. 2015). In addition, animal-to-human and human-to-human transmissions have been observed in some cases, which is probably mediated by blood or droplet contact (Jung et al. 2019; Li et al. 2024; Liu et al. 2012; Wang et al. 2014). Similar to other viruses in the family Phenuiviridae, SFTSV has a single negative-stranded RNA genome comprising three RNA segments: large (L), medium (M), and small (S) (Elliott 1990; Matsuno et al. 2013). The L segment encodes an RNA-dependent RNA polymerase, the M segment encodes glycoproteins (Gn and Gc), and the S segment encodes two proteins: a nucleocapsid protein (NP) and nonstructural (NS) protein (Sharma and Kamthania 2021; Xu et al. 2021). The NP encapsulates the three SFTSV RNA genome segments, which facilitates the packing and stabilization of the viral RNA genome to protect the virus from nucleases and the host immune system. NP is the most important target protein for viral detection because it is highly conserved among various RNA viruses. Owing to their high conservation, specific NP is used for the diagnosis of SFTSV. Multiple studies have been conducted to detect NP in SFTSV. Specifically, some groups have successfully developed antibodies capable of detecting NP for diagnosing SFTSV in laboratory settings (Lee et al. 2016, 2023a; Yu et al. 2018). Furthermore, assay systems have been developed for the detection of SFTSV-specific antibodies, such as immunoglobulin (Ig)G/IgM antibodies, utilizing recombinant NP (Jiao et al. 2012; Yu et al. 2015); however, existing resources provide limited information about antibody characterization.
In this study, we aimed to characterize and develop monoclonal antibodies targeting SFTSV NPs. Using a phage display biopanning method, scFv binders against recombinant SFTSV NPs were isolated. Two species of IgG antibodies containing the isolated sequences were produced, and their antigen-binding affinities were confirmed. These antibodies were used to assess their specificity against other viruses. Through an antibody pairing test, we identified the most effective pairs for diagnosing SFTSV. Our findings provide an important prospect to efficiently detect SFTSV for the development of SFTS diagnostics, such as rapid detection kits.
Methods and materials
Plasmid constructs
The NP gene sequences of SFTSV were obtained using strain KAJNH2 (Accession #: KU507557.1; B-3 type), which was isolated from South Korea (Park et al. 2014). DNA sequences were optimized and synthesized to enhance expression in Chinese hamster ovary cells (CHOs) (Supplemental Table S1). The full NP DNA gene, including the sequence for a C-terminal 6 × His tag, was subcloned into the pcDNA3.1 vector (Invitrogen, Carlsbad, CA, USA), a mammalian expression vector, between the AgeI-AflII sites of the plasmid. The human IgG expression vector, DNA sequences encoding the variable light (VL), or variable heavy (VH) regions, along with their cognate leader sequences, were subcloned into the pdCMV vector between the HindIII and BsiWI sites for the light chain plasmid and the EcoRI and ApaI sites for the heavy chain plasmid (Rahimizadeh et al. 2025). For the mouse IgG expression vector, DNA sequences encoding the VL and VH regions were subcloned into the pTGEX-mLC and pTGEX-mHC vector systems, respectively, according to the manufacturer’s protocol (Antibody Design Labs, San Diego, CA, USA).
Antibodies, recombinant proteins, and reagents
The antibodies used in this study for immunoblotting and enzyme-linked immunosorbent assay (ELISA) are listed in Supplemental Table S2. Protein A agarose beads were used to purify IgG antibodies (Amicogen, Jinju-si, South Korea). Ni–NTA agarose beads were used to purify the NP-6 × His protein (QIAGEN, Hilden, Germany). Recombinant NP of SFTSV (Accession #: AKI34305.1; B-2 type) and RVFV (Accession #: YP_003848707.1) were purchased from Abcam (AB256431 and AB256448, Cambridge, UK), and recombinant NP of Hantaan virus (HTNV; Accession #: AAK66740.1) was obtained from Elabscience (PKSV030173, Houston, TX, USA) for analysis of antibody cross-reactive functions (Bird et al. 2007; Yun et al. 2015). All chemicals were dissolved in appropriate reagents according to the manufacturer’s instructions.
Expression and purification of recombinant protein and full IgG antibodies
To produce antigens and antibodies, ExpiCHO cells were maintained in ExpiCHO expression medium (Thermo Fisher Scientific, Waltham, MA, USA). pcDNA3.1-NP(SFTSV)−6 × His was transiently expressed in ExpiCHO cells, following the manufacturer’s instructions. Cell harvest was performed after 8 days; subsequently, the culture was centrifuged at 5000 × g for 30 min, and the supernatant was filtered. NP proteins were purified by Ni–NTA agarose followed by anionic exchange chromatography using ÄKTA pure protein purification system (Cytiva, Marlborough, MA, USA). To produce IgG antibodies, pdCMV or pTGEX plasmids encoding the heavy chain (HC) and light chain (LC) of either human or mouse IgG were transiently expressed in ExpiCHO cells. Ten days post-transfection, the IgG proteins were purified using protein A sepharose beads.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting
Purified NP protein or IgG antibody was mixed with 5 × SDS-PAGE sample buffer and subjected to direct boiling. Proteins were separated by SDS-PAGE and stained using Coomassie Brilliant Blue R-250. Supernatant fractions from ExpiCHO cells expressing NP-6 × His or pcDNA as negative controls were separated by SDS-PAGE, and proteins were transferred to a nitrocellulose membrane. Thereafter, the membrane was incubated with an anti-His antibody at room temperature for 1 h. Bands were detected using a ChemiDoc imaging system (Invitrogen, Waltham, MA, USA).
Panning of phage-displayed scFv antibody libraries
General procedures of the phage display experiments were carried out as described in the “Phage Display: A Laboratory Manual” with some modifications (Barbas et al. 2001). Purified NP-6 × His protein was coated with immunotube (MaxiSorp, Nunc, Thermo Fisher Scientific) at 4 °C overnight. Unbound antigen was washed off with phosphate-buffered saline (PBS). After blocking the surface with 3% skim milk in phosphate-buffered saline containing 0.05% Tween 20 (PBST), the amplified scFv phage library was incubated with coated NP-6 × His in 3% skim milk at 37 °C for 1 h. After washing with PBST to remove unbound phages, the captured phages were eluted by incubation with 100 mM triethanolamine for 10 min. The eluted phages were used to infect the ER2738 Escherichia coli strain (New England Biolabs, Ipswich, MA, USA) and were amplified on an LB agar plate supplemented with carbenicillin (50 μg/mL) and 2% glucose at 30 °C overnight. Amplified ER2738 E. coli cells were used for the preparation of the next panning round. After three biopanning rounds, the output titer was measured, and single colonies in the final round were used to confirm the enzyme-linked immunosorbent assay (ELISA). Unique complementarity-determining region sequences among the high-binding clones from ELISA were determined using scFv DNA sequencing, followed by IMGT V-Quest analysis (www.imgt.org/IMGTindex/V-QUEST.php).
scFv expression and extraction
Single colonies expressing individual scFvs from binder pools of the third biopanning round were inoculated in 2 × YT medium supplemented with carbenicillin (50 μg/mL) and 2% glucose and were cultured at 37 °C for 6 h. For the induction of scFv proteins, 1 mM isopropyl β-D-1-thiogalactopyranoside was added to the cell culture medium and cultured at 30 °C overnight. After discarding the culture medium by centrifugation, the E. coli cell pellets were resuspended in 1 × TES extraction solution containing 20% w/v sucrose, 50 mM Tris, and 1 mM ethylenediaminetetraacetic acid (pH 8.0). After incubation at 4 °C for 30 min, 0.2 × TES buffer was added to the suspension. After further incubation at 4 °C for 30 min, periplasmic protein extracts containing scFv proteins were harvested from the supernatants.
Enzyme-linked immunosorbent assay
Highly bound microplates (96-well half-area plates, Corning, NY, USA) were coated with the appropriate concentration of antigens or antibodies in PBS at 4 °C overnight. After washing three times with PBST, the surface was coated with 3% skim milk in PBST (0.1% Tween 20) at 37 °C for 1 h. After blocking, the coated antigen was incubated with 30 μL of scFv extract or purified IgG in blocking buffer at 37 °C for 1 h. After washing three times with PBST, anti-HA-HRP or HRP-conjugated corresponding anti-IgG antibodies were incubated with blocking buffer at 37 °C for 1 h. After another washing, 3,3′,5,5′-tetramethylbenzidine substrate reagent (BD Biosciences, Franklin Lakes, NJ, USA) was added and incubated for 5 min, and then, the reaction was terminated using 1 N H2SO4. Individual optical densities (ODs) were measured at 450 nm using a microplate reader. For sandwich ELISA, 10 μg/mL of human IgG antibodies in the blocking buffer were coated to the microplate and washed with PBST. The mixture of 10 μg/mL mouse IgG with serial-diluted concentrations of NP or without NP in PBS was incubated. This was followed by incubation with the human IgG-coated microplate at 37 °C for 1 h. After washing three times with PBST, HRP-conjugated anti-mouse IgG antibody (Cell Signaling Technology, Danvers, MA, USA) was used to minimize the background signal with blocking buffer for sandwich ELISA detection. All ELISA experiments were independently repeated at least three times. Data are expressed as mean ± SD.
Bio-layer interferometry kinetic assay
The Octet R4 Protein Analysis System (Sartorius, Göttingen, Niedersachsen, Germany) was used for the bio-layer interferometry kinetic assay. His-tagged SFTSV NP protein (10 μg/mL) was loaded onto AR2G Biosensors. The antigen-loaded sensor was submerged in the analyte well for 240 s and subsequently dissociated in the buffer well for 600 s. An unloaded sensor was used as a reference, and its signal was subtracted in the following analysis. Data were generated using Octet acquisition software and processed using Octet analysis software. The kinetic parameters were evaluated using a 1:1 binding model, and the corresponding KD, Kon, and Koff values were calculated.
Results
Purification of recombinant NP of SFTS virus and screening of scFv binders against NP
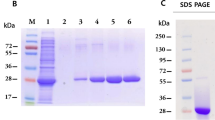
To express the recombinant NP of SFTSV, full-length NP gene sequences of SFTSV isolated in South Korea were subcloned into pcDNA3.1 mammalian expression plasmid. NP DNA sequences were also synthesized with codon-optimized sequences to achieve higher expression levels in mammalian cells (Fig. 1a). To verify the NP expression, plasmids encoding NP-6 × His were transfected into ExpiCHO cells, and the NP expression was observed by immunoblotting. NP was selectively detected in the culture medium of transiently transfected ExpiCHO cells (Fig. 1b). We then purified the recombinant protein, which consisted of the SFTSV NP with a 6 × His tag fused to its C-terminus, using Ni–NTA agarose. Purified NP had a molecular weight of 27.8 kDa (Fig. 1c). To screen for specific binders against the recombinant SFTSV NPs, biopanning was performed using a phage-displayed library containing a large synthetic human single-chain variable (scFv) fragment proteins (Yang et al. 2009). After three biopanning rounds, 20 unique scFv sequences were selected as initial binders from 600 individual clones based on primary ELISA screening, followed by sequence analysis (Fig. 1d). To select specific scFv binders for recombinant NP, ELISA was performed for dose-dependent assessment. Moreover, Gn(head)−6 × His proteins were included to distinguish between the binder and 6 × His tag. Through multiple ELISA tests, ten individual scFv clones were selected as specific binders for SFTSV NP (Fig. 1e).

Purification of SFTSV NP and screening of NP binders. a Schematic representation of the gene construct for the expression of recombinant NP isolated from the SFTSV KAJNH2 strain. b Verification of recombinant NP expression in ExpiCHO cells using immunoblotting. Each pcDNA empty vector as a negative control or pcDNA-NP-6 × His plasmid was transiently transfected to the cells; then, NP-6 × His protein in the culture medium was detected by immunoblotting using anti-His-HRP. c SDS-PAGE analysis of purified 6 × His-tagged recombinant SFTSV NP protein at approximately 28 kDa under non-reducing (NR) and reducing (R) conditions. d Schematic overview of the biopanning and selection process used to isolate NP-specific scFvs from a phage display library. e Indirect ELISA assay for selection of scFv binders. The NP protein was coated onto a 96-well ELISA plate at concentrations of 1, 10, 100, and 1000 ng/mL. Twenty clones expressing individual scFv were analyzed using indirect ELISA. Bovine serum albumin (BSA) and Gn(head)−6 × His proteins were compared to negative controls. a.u.; arbitrary units
Generation and evaluation of anti-NP antibodies
Our objective was to produce two species of IgG containing selected human scFv sequences. Initially, individual Fv DNA sequences of the VH and VL chains were subcloned into plasmids expressing either mouse or human IgG (Supplemental Table S3). Subsequently, plasmids encoding each species of full-length HC and LC were co-transfected into ExpiCHO cells. Using the protein A purification step, we successfully obtained six chimeric mouse IgG proteins (P01 A05, P01B10, P01 F08, P02E04, P04D08, and P05D07) and five fully human antibodies (P01 A05, P01B10, P01 C09, P01 F08, and P04D08) (Fig. 2a, b). We wondered whether the chimeric mouse IgG maintained its ability to bind to the NP protein when the mouse NP antibody was generated using the sequences of the human Fv. To verify the antigen-binding ability of each IgG form, a binding assay using indirect ELISA was performed. Both mouse and human NP antibodies showed strong binding against 10 μg/mL SFTSV NP (Fig. 2c, d). We further compared the binding ability of several antibodies with serially diluted NP. In Fig. 2e, P01 A05, P01B10, and P01 F08 showed a similar EC50 value in the presence of 1 μg/mL mouse and human antibodies; however, the maximum OD was slightly higher in human NP IgG compared to that in mouse IgG. Similar to the findings in Fig. 2e, serially diluted mouse and human IgGs showed increased OD values in a dose-dependent manner (Fig. 2f). To accurately compare the binding affinities of mouse and human IgG proteins, we measured the binding affinity using a bio-layer interferometry–based assay. We analyzed the binding affinities of mouse and human IgG (P01 A05) to NP. Both IgG proteins showed sufficiently high affinities, with human IgG showing a Kd value tenfold lower than that of mouse IgG (Fig. 2g). Based on these findings, we generated human monoclonal antibodies and chimeric mouse antibodies against SFTSV NP.

Generation and characterization of SFTSV NP antibodies. a, b SDS-PAGE analysis of purified mouse (a) and human (b) IgG antibodies against SFTSV NP, under reducing (R) and non-reducing (NR) conditions. c Indirect ELISA of purified mouse SFTSV IgG candidates (P01 A05, P01B10, P01 F08, P02E04, P04D08, and P05D07) to 10 μg/mL SFTSV NP. d Indirect ELISA of purified human SFTSV IgG candidates (P01 A05, P01B10, P01 C09, P01 F08, and P04D08) to the NP. e, f Comparison of binding ability between mouse and human IgG antibodies. The NP protein was coated onto a 96-well ELISA plate in a dose-dependent manner, and 10 μg/mL of each antibody (each mouse and human SFTSV IgG; P01 A05, P01B10, P01 F08, respectively) was incubated (e). Each mouse and human IgG was compared in terms of binding ability in the serial diluted concentrations (f). The error bars show the mean ± SD of the relative intensity from three independent experiments. The dose–response curve was fitted using a one-phase exponential association model in GraphPad Prism (GraphPad Software v. 5, San Diego, CA, USA). g Bio-layer interferometry sensorgram showing mouse and human IgG of P01 A05 while binding to the NP. The NP protein was immobilized on an AR2G biosensor, and the Kd value was determined by exposing the sensor to indicated concentrations of mouse and human IgG P01 A05
Assessment of the detection range of NP antibodies
An indirect ELISA was performed to assess and compare the antibody detection range against the recombinant NP of SFTSV. Initially, we utilized 1 μg/mL of each mouse and human IgG; then, we evaluated the antibody detection range by testing 10 different dilutions of NP stock solution. Most mouse antibodies have a detection limit of 1–10 ng/mL and become saturated at higher concentrations. Both P01 F08 and P02E04 exhibited lower EC50 values (88 and 78 ng/mL, respectively) and maximum OD values, indicating that they had a greater binding affinity to the NP of SFTSV than the other antibodies (Fig. 3a). Human antibodies also showed an increased binding affinity in a dose-dependent manner, with P01 C09 exhibiting the lowest detection limit and EC50 value (9.7 ng/mL; Fig. 3b). Next, we evaluated the antibody detection ranges by serially diluting mouse and human antibodies with 1 μg/mL of NP antigen coated in the well. Among the mouse IgG candidates, P02E04 showed the lowest EC50 (2.8 nM) and highest OD values (Fig. 3c). Additionally, P01 C09 human IgG exhibited a maximum OD value similar to that of P01 F08 but was determined to have the lowest EC50 (0.022 nM; Fig. 3d).

Assessment of the detection range of SFTSV NP antibodies. a, b Mouse IgG antibodies (a) and human antibodies (b) were applied to indirect ELISA at different NP concentrations (1 pg/mL to 100 μg/mL). c, d SFTSV NP (1 μg/mL) was used to assess the binding with increasing concentrations of mouse (c) and human (d) antibodies. Error bars represent SEM from experimental replicates. The error bars show the mean ± SD of the relative intensity from three independent experiments. The dose–response curve was fitted using a one-phase exponential association model in GraphPad Prism (v. 5)
Evaluating the antibody specificity and selectivity to the NP
Next, we confirmed that the selected monoclonal antibodies were specific for the diagnosis of SFTSV. First, we evaluated the broad binding ability of mouse and human NP antibodies to different NPs within the SFTSV serotype using indirect ELISA. We compared the OD values using a recombinant NP protein from SFTSV isolated from Jeju, South Korea (B-2) (Yun et al. 2015). Serotype B-3 exhibited a single amino acid variant at position V230 compared to serotype B-2, which changed from isoleucine to valine with the NP. Most mouse and human NP antibodies, with the exception of the P04D08 clones, showed similar OD values against both B-2 and B-3 serotypes. Interestingly, individual P04D08 clones in mouse and human IgG showed lower binding affinities to the B-2 NP serotype (Fig. 4a, b). In the dose-dependent ELISA, individual NP antibodies demonstrated similar binding affinities to SFTSV NPs between the B-3 and B-2 serotype viruses (Fig. 4c, d). We further investigated the specificity and selectivity of our NP antibodies against the NP of other viruses within the Bunyaviridae family. To verify cross-reactivity, we performed an indirect ELISA and compared it with SFTSV NP. We compared two different NPs within the order Bunyavirales. One of these proteins belongs to the Hantan virus family (Hantaviridae), classified in the order Bunyavirales. Additionally, we tested their binding ability to recombinant NP derived from the Rift Valley fever virus (RVFV), a member of the Phlebovirus genus within the same Phenuiviridae family (International Committee on Taxonomy of Viruses 2022). Neither the mouse nor human IgG displayed binding to the NP of either RVFV or Hantavirus. Therefore, these NP-targeting antibodies were exclusively specific and highly selective for detecting SFTSV NP (Fig. 4e, f).

Evaluation of antibody specificity and selectivity. a, b NP proteins from two different serotypes, B-3 and B-2, isolated from Kangwon and Jeju in South Korea, respectively, were coated onto an ELISA plate. The interaction of each mouse (a) or human SFTSV NP IgG (b) was determined by the detection of optical density. c, d Dose-dependent ELISA assay of SFTSV IgG candidates with NP serotypes. NP proteins from serotypes B-3 and B-2 were coated onto an ELISA plate at different concentrations. The interaction of both mouse (c) and human (d) SFTSV NP IgG antibodies was compared between the B-3 and B-2 NP types. The error bars show the mean ± SD of the relative intensity from three independent experiments. The dose–response curve was fitted using a one-phase exponential association model in GraphPad Prism (v. 5). e, f Evaluation of detection specificity in SFTSV NP IgG candidates. Each nucleoprotein derived from RVFV, HTNV, and SFTSV was coated onto a 96-well plate, and then, mouse (e) and human (f) NP IgG were incubated to investigate the interactions. The bar graph shows the mean ± SD of the relative intensity from three independent experiments
Application of sandwich ELISA for SFTSV NP detection
To determine the best pair of antibodies for detecting SFTSV NP, sandwich ELISA was performed using combinations of human IgG as the capture antibody and mouse IgG as the detector antibody. The positive/negative (P/N) values were calculated as the ratio of the absorbance obtained from the wells with NP antigen to that from wells without antigen, representing the signal-to-background ratio. It indicated that the combinations of mouse P01 A05/human P01B10, mouse P01 A05/human P01 C09, and mouse P02E04/human P01 A05 showed higher OD values with 10 μg/mL SFTSV NP compared to those in the absence of NP (Fig. 5a). Next, we further evaluated the detection ability with the SFTSV NP in the range of 1000 μg/mL to 0.001 μg/mL while using 10 μg/mL of each capture and detector antibody for sandwich ELISA. All three combinations showed similar EC50 values (3.4–5.5 μg/mL), and the highest maximum P/N value was obtained in the combination of mouse P02E04/human P01 A05 (Fig. 5b–d). Thus, SFTSV NP antibodies can be used to detect SFTSV NP and establish a detection kit based on the sandwich ELISA method.

Antibody pairing test by sandwich ELISA. a The P/N values represent a relative OD value by sandwich ELISA. The human IgG as a capture antibody was coated onto a 96-well plate, and then, mouse IgG was incubated as a detector antibody with 10 μg/mL SFTSV NP. The negative value was obtained from the indicated OD value without NP. b–d The sandwich ELISA was assessed using a different NP concentration. Three combinations showing the highest P/N value were determined to assess the P/N value in different NP concentrations. The error bars show the mean ± SD of the relative intensity from three independent experiments. The dose–response curve was fitted using a one-phase exponential association model in GraphPad Prism (v. 5)
Discussion
SFTSV is an emerging tick-borne virus that causes hemorrhagic fever, with a fatality rate in the range of 5–30% (Cao et al. 2021; Casel et al. 2021; Li et al. 2021; Zhang et al. 2013). Caring for patients is rendered difficult due to the absence of appropriate antiviral therapeutics or licensed vaccines against SFTS. Accurate and timely diagnosis plays a pivotal role in the clinical management of SFTS. Early detection enables supportive treatment to be administered before the disease progresses to more severe stages, which can significantly reduce mortality. Furthermore, due to the risk of human-to-human transmission, early identification of infected individuals is critical for infection control and public health response. Therefore, the development of effective, rapid, and reliable diagnostic tools is a key component and clinical priority in patient care (Casel et al. 2021; Kim and Park 2023; Li et al. 2021). Although various approaches, including qRT-PCR and immunoassays, have been reported for SFTSV detection, a standardized diagnostic method is still lacking. Therefore, there is an urgent need to develop a simple, rapid, and effective method for detecting SFTSV, in order to provide appropriate care to patients and prevent further viral transmission. Molecular diagnostic methods such as qRT-PCR are highly sensitive and specific, targeting the L, M, and S segments of the viral RNA genome. These methods can effectively detect SFTSV, particularly in the early phase of infection (Lu et al. 2017; Sun et al. 2012). However, they require sophisticated equipment and technical expertise, which limits their applicability in point-of-care settings. In contrast, immunological detection methods such as ELISA have also been developed and are widely used for diagnosing various viral infections, including SFTSV, due to their simplicity and rapid procedures. Several studies have demonstrated successful detection of SFTSV NP antigen during the acute phase, when viremia levels are high (Duan et al. 2020; Fukuma et al. 2016; Jiao et al. 2012; Yu et al. 2018; Zhuge et al. 2022). While conventional ELISA typically has a higher detection limit and lower sensitivity than qRT-PCR, recent advancements in ELISA-based biosensor technologies are helping to narrow this performance gap (Chang et al. 2018; Duan et al. 2020; Lin et al. 2022; Zuo et al. 2018).
In this study, we developed antibodies to detect SFTSV using a sandwich ELISA system. NP is extensively used as a target protein in many viral diagnoses because it is highly conserved and represents the most abundant protein in the virions of the Bunyaviridae family (Vapalahti et al. 1995). Mutations frequently occur in the RNA genomes of glycoproteins (Lee et al. 2023b), resulting in epitope alterations and making it difficult for antibodies to effectively detect them. Therefore, various immunoassay-based diagnostic applications preferentially target NP because of its high conservation and low variability in target proteins. Despite the advantage of targeting NP, antibodies applied in immunoassays should be highly specific to the NP serotype of the target virus, with minimal susceptibility to false negatives caused by intra-species variations. As shown in Fig. 4a, b, all candidate antibodies, with the exception of the P04D08 clone (six out of seven clones), bound to both B-2 and B-3 subtype NPs. Using ELISA, the P04D08 clone in mouse and human IgG forms was observed to have decreased values when used with B-2 subtype NP, suggesting a potential difference in the binding epitope, possibly attributable to an amino acid variant (I230). We further validated whether these antibodies could detect SFTSV NP without binding to the NP of other viruses such as RVFV and HTNV. RVFV shares taxonomic proximity with SFTSV, as both belong to the family Phenuiviridae within the order Bunyavirales, although they are classified under different genera, Phlebovirus and Bandavirus, respectively. By contrast, HTNV is more distantly related, as it belongs to the genus Orthohantavirus in the family Hantaviridae (International Committee on Taxonomy of Viruses 2022; Leventhal et al. 2021). Our data revealed that the candidate antibodies exhibited no cross-reactivity with the recombinant NP of other viruses within the order Bunyavirales (Fig. 4e, f). Despite the high degree of NP conservation among many viruses, antibody-based immunoassays have a crucial advantage in the detection of specific viral species.
In our study, we used recombinant NP of SFTSV to generate specific antibodies, as the SFTS virus is classified as a biosafety level 3 (BSL-3) pathogen. Due to the associated biosafety requirements, we chose to use recombinant protein instead of native virus for antibody screening. To preserve the native structure of the NP, we employed a mammalian expression system rather than a prokaryotic system such as E. coli. The NP gene was derived from the SFTSV strain KAJNH2 (genotype B-3), which was isolated in South Korea (Park et al. 2014), and codon optimization was performed to enhance its expression in mammalian cells. Using the resulting recombinant NP, we conducted phage display biopanning with a synthetic scFv library, instead of relying on immunization-based methods, to efficiently isolate specific scFv binders (Fig. 1). While the conventional hybridoma screening system is effective for generating high-affinity antibodies, it is time-consuming, labor-intensive, and cost-intensive, and it requires a large amount of antigen proteins for effective immunization. Phage display screening provides a reliable, labor-intensive, cost-effective, and rapid process for identifying specific binders against antigens (Lu et al. 2020; Moraes et al. 2021). Although it is common to incorporate the immunization step in the generation of an immune-based phage library to obtain high-affinity binders, non-immune and synthetic scFv libraries provide many advantages, including efficient binding affinity for candidate antibodies without the use of large amounts of antigen proteins. We did not directly compare the binding affinities of antibodies selected by biopanning versus those obtained through immunization. However, we assessed the binding affinity of one of our selected clones, P01 A05, which exhibited a moderate EC₅₀ value in the ELISA assay (Fig. 3). Both the mouse and human IgG forms of this clone demonstrated direct binding to the recombinant NP, exhibiting strong affinities ranging from several hundred picomolar to low nanomolar (Fig. 2g).
Due to the unavailability of SFTSV-positive serum samples from clinical resources, we were unable to assess the performance of our detection system using sera from confirmed SFTSV-infected patients. Therefore, further validation using well-characterized patient samples will be necessary to fully assess the clinical utility and diagnostic accuracy of the SFTSV detection kit.
In conclusion, we successfully identified scFv binders against the SFTSV NP using phage display technology. Both human and mouse antibody formats exhibited high sensitivity and binding specificity, indicating their potential applicability in the development of diagnostic tools for SFTS. Further refinement and clinical validation will be essential to assess their applicability in diagnostic settings.
Data availability
No datasets were generated or analysed during the current study.
References
Barbas CF, Burton DR, Scott JK, Silverman GJ (2001) Phage display: a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Bird BH, Khristova ML, Rollin PE, Ksiazek TG, Nichol ST (2007) Complete genome analysis of 33 ecologically and biologically diverse rift valley fever virus strains reveals widespread virus movement and low genetic diversity due to recent common ancestry. J Virol 81(6):2805–2816. https://doi.org/10.1128/JVI.02095-06
Cao J, Lu G, Wen L, Luo P, Huang Y, Liang R, Tang K, Qin Z, Chan CC, Chik KK, Du J, Yin F, Ye ZW, Chu H, Jin DY, Yuen KY, Chan JF, Yuan S (2021) Severe fever with thrombocytopenia syndrome virus (SFTSV)-host interactome screen identifies viral nucleoprotein-associated host factors as potential antiviral targets. Comput Struct Biotechnol J 19:5568–5577. https://doi.org/10.1016/j.csbj.2021.09.034
Casel MA, Park SJ, Choi YK (2021) Severe fever with thrombocytopenia syndrome virus: emerging novel phlebovirus and their control strategy. Exp Mol Med 53(5):713–722. https://doi.org/10.1038/s12276-021-00610-1
Chang YF, Wang WH, Hong YW, Yuan RY, Chen KH, Huang YW, Lu PL, Chen YH, Chen YA, Su LC, Wang SF (2018) Simple strategy for rapid and sensitive detection of avian influenza A H7N9 virus based on intensity-modulated SPR biosensor and new generated antibody. Anal Chem 90(3):1861–1869. https://doi.org/10.1021/acs.analchem.7b03934
Duan Y, Wu W, Zhao Q, Liu S, Liu H, Huang M, Wang T, Liang M, Wang Z (2020) Enzyme-antibody-modified gold nanoparticle probes for the ultrasensitive detection of nucleocapsid protein in SFTSV. Int J Environ Res Public Health 17(12):4427. https://doi.org/10.3390/ijerph17124427
Elliott RM (1990) Molecular biology of the Bunyaviridae. J Gen Virol 71(Pt 3):501–522. https://doi.org/10.1099/0022-1317-71-3-501
Fukuma A, Fukushi S, Yoshikawa T, Tani H, Taniguchi S, Kurosu T, Egawa K, Suda Y, Singh H, Nomachi T, Gokuden M, Ando K, Kida K, Kan M, Kato N, Yoshikawa A, Kitamoto H, Sato Y, Suzuki T, Hasegawa H, Morikawa S, Shimojima M, Saijo M (2016) Severe fever with thrombocytopenia syndrome virus antigen detection using monoclonal antibodies to the nucleocapsid protein. PLoS Negl Trop Dis 10(4):e0004595. https://doi.org/10.1371/journal.pntd.0004595
International Committee on Taxonomy of Viruses (2022) History of the taxon. publisher. https://ictv.global/taxonomy/taxondetails?taxnode_id=202200166&taxon_name=Bandavirus%20dabieense. Accessed 01 Sep 2024
Jiao Y, Zeng X, Guo X, Qi X, Zhang X, Shi Z, Zhou M, Bao C, Zhang W, Xu Y, Wang H (2012) Preparation and evaluation of recombinant severe fever with thrombocytopenia syndrome virus nucleocapsid protein for detection of total antibodies in human and animal sera by double-antigen sandwich enzyme-linked immunosorbent assay. J Clin Microbiol 50(2):372–377. https://doi.org/10.1128/JCM.01319-11
Jung IY, Choi W, Kim J, Wang E, Park SW, Lee WJ, Choi JY, Kim HY, Uh Y, Kim YK (2019) Nosocomial person-to-person transmission of severe fever with thrombocytopenia syndrome. Clin Microbiol Infect 25(5):633 e1-633 e4. https://doi.org/10.1016/j.cmi.2019.01.006
Kim EH, Park SJ (2023) Emerging tick-borne Dabie bandavirus: virology, epidemiology, and prevention. Microorganisms 11(9):2309. https://doi.org/10.3390/microorganisms11092309
Kim YR, Yun Y, Bae SG, Park D, Kim S, Lee JM, Cho NH, Kim YS, Lee KH (2018) Severe fever with thrombocytopenia syndrome virus infection, South Korea, 2010. Emerg Infect Dis 24(11):2103–2105. https://doi.org/10.3201/eid2411.170756
Lee H, Kim EJ, Song JY, Choi JS, Lee JY, Cho IS, Shin YK (2016) Development and evaluation of a competitive enzyme-linked immunosorbent assay using a monoclonal antibody for diagnosis of severe fever with thrombocytopenia syndrome virus in bovine sera. J Vet Sci 17(3):307–314. https://doi.org/10.4142/jvs.2016.17.3.307
Lee K, Choi MJ, Cho MH, Choi DO, Bhoo SH (2023) Antibody production and characterization of the nucleoprotein of sever fever with thrombocytopenia syndrome virus (SFTSV) for effective diagnosis of SFTSV. Virol J 20(1):206. https://doi.org/10.1186/s12985-023-02173-1
Lee K, Seok JH, Kim H, Park S, Lee S, Bae JY, Jeon K, Kang JG, Yoo JR, Heo ST, Cho NH, Lee KH, Kim K, Park MS, Kim JI (2023) Genome-informed investigation of the molecular evolution and genetic reassortment of severe fever with thrombocytopenia syndrome virus. PLoS Negl Trop Dis 17(9):e0011630. https://doi.org/10.1371/journal.pntd.0011630
Leventhal SS, Wilson D, Feldmann H, Hawman DW (2021) A look into Bunyavirales genomes: functions of non-structural (NS) proteins. Viruses 13(2):314. https://doi.org/10.3390/v13020314
Li J, Li S, Yang L, Cao P, Lu J (2021) Severe fever with thrombocytopenia syndrome virus: a highly lethal bunyavirus. Crit Rev Microbiol 47(1):112–125. https://doi.org/10.1080/1040841X.2020.1847037
Li J, Wang C, Li X, Zhang G, Sun S, Wang Z, Zhao J, Xiu L, Jiang N, Zhang H, Yang Z, Zhang J (2024) Direct transmission of severe fever with thrombocytopenia syndrome virus from farm-raised fur animals to workers in Weihai. China Virol J 21(1):113. https://doi.org/10.1186/s12985-024-02387-x
Lin CY, Wang WH, Li MC, Lin YT, Yang ZS, Urbina AN, Assavalapsakul W, Thitithanyanont A, Chen KR, Kuo CC, Lin YX, Hsiao HH, Lin KD, Lin SY, Chen YH, Yu ML, Su LC, Wang SF (2022) Boosting the detection performance of severe acute respiratory syndrome coronavirus 2 test through a sensitive optical biosensor with new superior antibody. Bioeng Transl Med 8(5):e10410. https://doi.org/10.1002/btm2.10410
Liu Y, Li Q, Hu W, Wu J, Wang Y, Mei L, Walker DH, Ren J, Wang Y, Yu XJ (2012) Person-to-person transmission of severe fever with thrombocytopenia syndrome virus. Vector Borne Zoonotic Dis 12(2):156–160. https://doi.org/10.1089/vbz.2011.0758
Lu X, Wang L, Bai D, Li Y (2017) Establishment of national reference for bunyavirus nucleic acid detection kits for diagnosis of SFTS virus. Virol J 14(1):32. https://doi.org/10.1186/s12985-017-0682-z
Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, Wu HC (2020) Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci 27(1):1. https://doi.org/10.1186/s12929-019-0592-z
Luo LM, Zhao L, Wen HL, Zhang ZT, Liu JW, Fang LZ, Xue ZF, Ma DQ, Zhang XS, Ding SJ, Lei XY, Yu XJ (2015) Haemaphysalis longicornis ticks as reservoir and vector of severe fever with thrombocytopenia syndrome virus in China. Emerg Infect Dis 21(10):1770–1776. https://doi.org/10.3201/eid2110.150126
Matsuno K, Weisend C, Travassos da Rosa AP, Anzick SL, Dahlstrom E, Porcella SF, Dorward DW, Yu XJ, Tesh RB, Ebihara H (2013) Characterization of the Bhanja serogroup viruses (Bunyaviridae): a novel species of the genus Phlebovirus and its relationship with other emerging tick-borne phleboviruses. J Virol 87(7):3719–3728. https://doi.org/10.1128/JVI.02845-12
McMullan LK, Folk SM, Kelly AJ, MacNeil A, Goldsmith CS, Metcalfe MG, Batten BC, Albarino CG, Zaki SR, Rollin PE, Nicholson WL, Nichol ST (2012) A new phlebovirus associated with severe febrile illness in Missouri. N Engl J Med 367(9):834–841. https://doi.org/10.1056/NEJMoa1203378
Moraes JZ, Hamaguchi B, Braggion C, Speciale ER, Cesar FBV, Soares G, Osaki JH, Pereira TM, Aguiar RB (2021) Hybridoma technology: is it still useful? Curr Res Immunol 2:32–40. https://doi.org/10.1016/j.crimmu.2021.03.002
Ongkittikul MDS, Watanawong MDR, Rompho RNP (2020) Severe fever with thrombocytopenia syndrome virus: the first case report in Thailand. Bankk Med J 16(2):204. https://doi.org/10.31524/bkkmedj.2020.22.001
Park SW, Han MG, Yun SM, Park C, Lee WJ, Ryou J (2014) Severe fever with thrombocytopenia syndrome virus, South Korea, 2013. Emerg Infect Dis 20(11):1880–1882. https://doi.org/10.3201/eid2011.140888
Peng SH, Yang SL, Tang SE, Wang TC, Hsu TC, Su CL, Chen MY, Shimojima M, Yoshikawa T, Shu PY (2020) Human case of severe fever with thrombocytopenia syndrome virus infection, Taiwan, 2019. Emerg Infect Dis 26(7):1612–1614. https://doi.org/10.3201/eid2607.200104
Rahimizadeh P, Kim S, Yoon BJ, Jeong Y, Lim S, Jeon H, Lim HJ, Park SH, Park SI, Kong DH, Park JR, Song YB (2025) Novel CXCR2 antibodies exhibit enhanced anti-tumor activity in pancreatic cancer. Biomed Pharmacother 185:117966. https://doi.org/10.1016/j.biopha.2025.117966
Sharma D, Kamthania M (2021) A new emerging pandemic of severe fever with thrombocytopenia syndrome (SFTS). Virusdisease 32(2):220–227. https://doi.org/10.1007/s13337-021-00656-9
Sun Y, Liang M, Qu J, Jin C, Zhang Q, Li J, Jiang X, Wang Q, Lu J, Gu W, Zhang S, Li C, Wang X, Zhan F, Yao W, Bi Z, Wang S, Li D (2012) Early diagnosis of novel SFTS bunyavirus infection by quantitative real-time RT-PCR assay. J Clin Virol 53(1):48–53. https://doi.org/10.1016/j.jcv.2011.09.031
Takahashi T, Maeda K, Suzuki T, Ishido A, Shigeoka T, Tominaga T, Kamei T, Honda M, Ninomiya D, Sakai T, Senba T, Kaneyuki S, Sakaguchi S, Satoh A, Hosokawa T, Kawabe Y, Kurihara S, Izumikawa K, Kohno S, Azuma T, Suemori K, Yasukawa M, Mizutani T, Omatsu T, Katayama Y, Miyahara M, Ijuin M, Doi K, Okuda M, Umeki K, Saito T, Fukushima K, Nakajima K, Yoshikawa T, Tani H, Fukushi S, Fukuma A, Ogata M, Shimojima M, Nakajima N, Nagata N, Katano H, Fukumoto H, Sato Y, Hasegawa H, Yamagishi T, Oishi K, Kurane I, Morikawa S, Saijo M (2014) The first identification and retrospective study of severe fever with thrombocytopenia syndrome in Japan. J Infect Dis 209(6):816–827. https://doi.org/10.1093/infdis/jit603
Tran XC, Yun Y, Van An L, Kim SH, Thao NTP, Man PKC, Yoo JR, Heo ST, Cho NH, Lee KH (2019) Endemic severe fever with thrombocytopenia syndrome, Vietnam. Emerg Infect Dis 25(5):1029–1031. https://doi.org/10.3201/eid2505.181463
Vapalahti O, Kallio-Kokko H, Narvanen A, Julkunen I, Lundkvist A, Plyusnin A, Lehvaslaiho H, Brummer-Korvenkontio M, Vaheri A, Lankinen H (1995) Human B-cell epitopes of puumala virus nucleocapsid protein, the major antigen in early serological response. J Med Virol 46(4):293–303. https://doi.org/10.1002/jmv.1890460402
Wang Y, Deng B, Zhang J, Cui W, Yao W, Liu P (2014) Person-to-person asymptomatic infection of severe fever with thrombocytopenia syndrome virus through blood contact. Intern Med 53(8):903–906. https://doi.org/10.2169/internalmedicine.53.1164
Win AM, Nguyen YTH, Kim Y, Ha NY, Kang JG, Kim H, San B, Kyaw O, Htike WW, Choi DO, Lee KH, Cho NH (2020) Genotypic heterogeneity of Orientia tsutsugamushi in scrub typhus patients and thrombocytopenia syndrome co-infection, Myanmar. Emerg Infect Dis 26(8):1878–1881. https://doi.org/10.3201/eid2608.200135
Xu B, Liu L, Huang X, Ma H, Zhang Y, Du Y, Wang P, Tang X, Wang H, Kang K, Zhang S, Zhao G, Wu W, Yang Y, Chen H, Mu F, Chen W (2011) Metagenomic analysis of fever, thrombocytopenia and leukopenia syndrome (FTLS) in Henan Province, China: discovery of a new bunyavirus. PLoS Pathog 7(11):e1002369. https://doi.org/10.1371/journal.ppat.1002369
Xu M, Wang B, Deng F, Wang H, Wang M, Hu Z, Liu J (2021) Establishment of a reverse genetic system of severe fever with thrombocytopenia syndrome virus based on a C4 strain. Virol Sin 36(5):958–967. https://doi.org/10.1007/s12250-021-00359-x
Yang HY, Kang KJ, Chung JE, Shim H (2009) Construction of a large synthetic human scFv library with six diversified CDRs and high functional diversity. Mol Cells 27(2):225–235. https://doi.org/10.1007/s10059-009-0028-9
Yu F, Du Y, Huang X, Ma H, Xu B, Adungo F, Hayasaka D, Buerano CC, Morita K (2015) Application of recombinant severe fever with thrombocytopenia syndrome virus nucleocapsid protein for the detection of SFTSV-specific human IgG and IgM antibodies by indirect ELISA. Virol J 12:117. https://doi.org/10.1186/s12985-015-0350-0
Yu MA, Jeong HW, Park SJ, Kim YI, Kwon HI, Kim EH, Si YJ, Yu KM, Robles NJ, Han HJ, Choi YK (2018) Evaluation of two different enzyme-linked immunosorbent assay for severe fever with thrombocytopenia syndrome virus diagnosis. Clin Exp Vaccine Res 7(1):82–86. https://doi.org/10.7774/cevr.2018.7.1.82
Yun Y, Heo ST, Kim G, Hewson R, Kim H, Park D, Cho NH, Oh WS, Ryu SY, Kwon KT, Medlock JM, Lee KH (2015) Phylogenetic analysis of severe fever with thrombocytopenia syndrome virus in South Korea and migratory bird routes between china, South Korea, and Japan. Am J Trop Med Hyg 93(3):468–474. https://doi.org/10.4269/ajtmh.15-0047
Zhang X, Liu Y, Zhao L, Li B, Yu H, Wen H, Yu XJ (2013) An emerging hemorrhagic fever in China caused by a novel bunyavirus SFTSV. Sci China Life Sci 56(8):697–700. https://doi.org/10.1007/s11427-013-4518-9
Zhuge Y, Ding C, Gong X, Hu D, Zhu J, Wang C (2022) Development and evaluation of two double-antibody sandwich ELISAs for detecting severe fever with thrombocytopenia syndrome virus infection. Jpn J Infect Dis 75(1):49–55. https://doi.org/10.7883/yoken.JJID.2020.1109
Zohaib A, Zhang J, Saqib M, Athar MA, Hussain MH, Chen J, Sial AU, Tayyab MH, Batool M, Khan S, Luo Y, Waruhiu C, Taj Z, Hayder Z, Ahmed R, Siddique AB, Yang X, Qureshi MA, Ujjan IU, Lail A, Khan I, Sajjad Ur R, Zhang T, Deng F, Shi Z, Shen S (2020) Serologic evidence of severe fever with thrombocytopenia syndrome virus and related viruses in Pakistan. Emerg Infect Dis 26(7):1513–1516. https://doi.org/10.3201/eid2607.190611
Zuo JY, Jiao YJ, Zhu J, Ding SN (2018) Rapid detection of severe fever with thrombocytopenia syndrome virus via colloidal gold immunochromatography assay. ACS Omega 3(11):15399–15406. https://doi.org/10.1021/acsomega.8b02366
Acknowledgements
We thank to the colleagues in the Scripps Korea Antibody Institute for their support of this study.
Funding
This study was supported by a Research Grantresearch grant from the Scripps Korea Antibody Institute (SKAI-22 TS41), a Korea Institute of Science and Technology Institutional Program (KIST-2E33310) to YBS, and the National Research Foundation (NRF) from the Korean government (MSIT) to SGL (RS-2024-00400771).
Author information
Authors and Affiliations
Contributions
Conception and design, EMP, PR, SGL, YBS.; acquisition of data, SHK, PR, SYL, HYJ, HJL; analysis and interpretation of data, EMP, SYL, PR, SGL, YBS; drafting or revising the manuscript, EMP, PR, YBS.; project administration and supervision, YBS.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
ESM 1
(PDF 82.8 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Park, EM., Kim, S., Lim, S. et al. Development of antibodies against severe fever with thrombocytopenia syndrome virus nucleoprotein for diagnosis. Appl Microbiol Biotechnol 109, 139 (2025). https://doi.org/10.1007/s00253-025-13530-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00253-025-13530-1