
Abstract
Sideroblastic anemias (SAs) represent a heterogeneous group of rare hematological disorders characterized by iron accumulation in mitochondria of erythroblasts with ineffective erythropoiesis. SAs are categorized into acquired and congenital forms. Acquired, secondary, and clonal, SA is rare in pediatric populations. Congenital SA (CSA) is classified into syndromic and non-syndromic forms. Herein, we describe three cases of pediatric patients with SA. The diagnosis of SA was based on the presence of type 3 sideroblasts on BM aspirate smear (greater than 15%) and genetic tests. In the first case, the diagnosis of myelodysplastic syndrome with ring sideroblasts (MDS-RS) with somatic SF3B1 mutation was made at the age of 11 years. A whole exome sequencing did not reveal any germinal predisposition for MDS. A wait-and-see strategy was adopted. After one year- of follow-up, no blood transfusion was needed and no further cytopenia occurred. The two other children had presented anemia at an early age and were diagnosed with CSA. The first case was a girl with SCL25A38 gene mutation. For the second one, the diagnosis of aminolevulinic acid synthase 2 deficiency was considered the most plausible given the family history and the favourable response to pyridoxine. Iron overload occurred in both patients with CSA, requiring chelation therapy. In conclusion, Perls' stain remains a valuable tool for guiding the diagnosis of unexplained anemia in pediatric patients. Genetic testing is crucial for the characterization of congenital sideroblastic anemias. The incidence of myeloid neoplasms with ring sideroblasts is exceptional in children, and the long-term prognosis remains undefined.
Similar content being viewed by others


Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
Background
Sideroblastic anemias (SAs) constitute a heterogeneous group of rare hematological disorders [1]. They result from a defect in incorporation of iron into protoporphyrin IX for heme synthesis during erythropoiesis [2]. The key feature for the diagnosis of SAs is the presence of ring sideroblasts on a bone marrow (BM) aspirate smear. It corresponds to erythroblasts with blue stained granules of iron clustered in perinuclear [3]. SAs are categorized into acquired and congenital forms. Epidemiological data on these disorders are not available, sideroblastic anemia is considered as a rare disease [1]. Hereditary forms are categorized into syndromic and non-syndromic forms. Among non-syndromic forms, X linked sideroblastic anemia (XLSA) in relation with mutation in the 5-aminolevulinic acid synthase gene (ALAS2), is the most common subcategory [1]. As for acquired SA, it is either secondary to non-clonal conditions (exposure to drugs, zinc overdose, lead toxicity, copper and pyridoxine deficiency) or clonal myeloid disorders including myelodysplastic syndrome with ring sideroblasts (MDS-RS) and MDS/MPN with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T), as per the revised fourth edition of the World Health Organization (WHO) classification of myeloid neoplasms [4]. Approximately 75 to 82% of MDS-RS patients harbor mutation in the splicing factor SF3B1. Thus, an entity of MDS with low blasts and SF3B1 mutation has been included in WHO fifth edition [5]. Acquired clonal SA is mostly encountered in adulthood. It’s uncommon in childhood and unlike adults MDS is frequently revealed by thrombocytopenia and neutropenia [6]. We report three cases of pediatric patients with SAs from different Tunisian families. In view of the rarity of this condition in children, we aimed, through this paper to describe the diagnostic approach to sideroblastic anemia and the challenges in the management of these patients.
Methods
Data were collected from the files of children followed for sideroblastic anemia at the department of Pediatric immunohematology and stem cell transplantation of the national center of bone marrow transplantation in Tunisia. The diagnosis of sideroblastic anemia was based on the presence of type 3 sideroblasts on BM aspirate smear (greater than 15%) or on genetic tests. The diagnostic of myelodysplastic syndrome was established based on the combination of the following criteria: clinical (cytopenia), morphological (dysplasia involving 10% or more of the cells, blasts < 20%), histological (erythroid hyperplasia or BM fibrosis), cytogenetic and molecular (specific genetic and chromosomal mutations or deletions).
Results
Case 1
A 11-year-old child born from a third-degree consanguineous parent with no past medical history. He presented with chronic pallor. Physical examination found an enlargement of spleen. The red blood cell count revealed normochromic normocytic non-regenerative anemia: hemoglobin (Hb) = 8.9 g/dl, mean cell volume (MCV) = 82 fl, mean corpuscular hemoglobin MCH = 27 pg, reticulocyte count = 60,000/mm3. Neutrophil and platelet counts were within normal range. Peripheral blood smear showed pronounced anisocytosis, anisochromia, and poikilocytosis. There were no signs of biological hemolysis. The lactate dehydrogenase level was 352 IU/L, and the total and direct bilirubin levels were 22 µmol/L and 9 µmol/L, respectively. Serum Ferritin level was 87 ug/l. Bone marrow aspirate revealed erythroid hyperplasia, dyserythropoiesis (erythroblasts with laminated cytoplasm and others with frayed cytoplasm), few neutrophils with hyposegmented nuclei, and absence of blasts. Ring sideroblasts (Type 3) constituted 45% of nucleated erythroblasts. No chromosomal aberrations were detected on karyotype. The bone marrow biopsy revealed erythroid and megakaryocytic hyperplasia with morphological features of erythroid and megakaryocytic dysplasia. Additionally, no evidence of myelofibrosis or excess of blasts was observed. MDS new generation sequencing (NGS) panel including the analysis of 41 genes detected a somatic mutation of Spliceosome Factor 3B subunit 1 (SF3B1). The diagnosis of myelodysplastic syndrome with ring sideroblasts (MDS-RS) and SF3B1 somatic mutation was retained. Whole exome sequencing performed on DNA extracted from cheek cells did not reveal any germinal predisposition for MDS. The patient didn’t have a matched family donor. Wait and see strategy was adopted. After a follow up of one year he didn’t require blood transfusions and he maintained normal neutrophil and platelet counts.
Case 2
A 7-year-old girl was evaluated for chronic hypochromic microcytic anemia. Examination showed pallor and enlargement of spleen, with no other finding particularly no bone abnormalities. Complete blood count showed hypochromic microcytic anemia Hb = 7.6 g/dl, MCV = 59.5 fl, CMH = 15.1 pg reticulocyte = 33,400/mm3. Serum ferritin level was elevated to 169 ng/ml. No evidence of hemolysis was noted. Hemoglobin electrophoresis was normal. Assessment of iron status indicated elevated serum iron (40 µmol/l) and transferrin saturation (86.9%), decreased transferrin concentration 1.77 g/l, while TIBC was at the lower limit of normal range (46 µmol/l). The bone marrow smear showed 28% erythroblasts, dyserythropoiesis and no ring sideroblasts on Perls’ stain. Normal concentration of serum ceruloplasmin and serum copper ruled out aceruloplasminemia. At this point, inherited iron metabolism defects were considered. Genetic analysis of TMPRSS6 and SLC11A2 genes excluded the diagnosis of Iron-refractory iron deficiency anemia (IRIDA) and DMT1 deficiency. The whole exome sequencing showed a homozygous missense variant mutation in SLC25A38 gene: c.587 T > C(p.Leu196Pro). A review of bone marrow cytological stain revealed the presence of ring sideroblasts.
She received regular blood transfusions for 5 years since the age of seven years. Liver MRI T2* showed an iron overload of 13.69 mg/gr dry weight before any transfusion. Serum ferritin and LIC increased under transfusion therapy (Table 1). Chelation therapy was initiated at the age of 9 years with Deferasirox on monotherapy, then a combination of Deferiprone and Deferoxamine was prescribed. Since the age of 12-year-old, regular transfusion regimen was withdrawal, hemoglobin levels stabilized between 7,5–9 g/dl. She developed osteopenia (Z-score −1.1 DS) that was managed by vitamin D and calcium supplementation. She had an HLA matched sibling donor, but since she had been independent of transfusions for the past three years, the hematopoietic stem cell transplant was not re-discussed. At last follow up she had 17 years of age, transfusion free since 3 years, with a mean Hb level at 7.5 g/dl.
Case 3
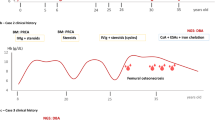
A 13-year-old boy, referred to our department for non-transfusion-dependent chronic hypochromic microcytic anemia. There is no parental consanguinity. Anemia was first diagnosed at 6 months of age. Irregular iron supplementation was prescribed until the age of 4 years but without improvement. Investigations undertaken have included hemoglobin electrophoresis (HbA 97.9% HbA2 2.1%), high serum ferritin level (321 ng/ml). No evidence of hemolysis in laboratory assessments. Aside for pallor, examination was normal particularly neurological exam. Complete blood count showed persistent hypochromic microcytic non-regenerative anemia: hemoglobin (Hb) = 7.9 g/dl, mean cell volume (MCV) = 54.9 fl, MCH = 15.5 pg, reticulocyte count 28,000/mm3. Absolute neutrophil count and platelets were within normal range. Biochemical assessment of iron status indicated serum iron of 45.47 µmol/l (Normal value: (NV 9–28), total iron binding capacity (TIBC) of 61.58 µmol/l (NV 45–65), transferrin saturation (TS) of 73.84% (NV 25–50) and serum ferritin = 535 ng/ml. Blood smear aspirate revealed erythroid hyperplasia (61%), marked dyserythropoiesis (karyorrhexis, erythroblasts are small having a ragged cytoplasm, nuclear fragmentation) (Fig. 3). Granulocytic and megakaryocytic lineages showed no anomaly. Perls’ stain of BM identified 53% of ring sideroblasts and increased numbers of siderocytes. No karyotype abnormality was observed. Blood levels of ceruloplasmin, copper and lead were normal, excluding copper deficiency and lead poisoning. During the follow-up period, the patient's hemoglobin level was maintained around 7 g/dl until the age of 15 years, with no impact on growth. Subsequently, the hemoglobin level declined to 5.5 g/dl. The addition of pyridoxine at the dose of 100 mg/d resulted in an increase of hemoglobin levels (Fig. 1).

Follow-up of hemoglobin levels in patient 3 of a boy with sideroblastic anemia, over 9 years. Hatched line represents pyridoxine interruption due drug unavailability
The diagnosis of X-linked sideroblastic anaemia caused by ALAS2 deficiency was considered the most plausible. This was corroborated by the mother's assessment revealing a moderate microcytic anemia (Hb = 11 g/dL, MCV = 73.5 fL, MCH = 23 pg, and red cell distribution width (RDW) = 26%), a marked anisocytosis (presence of normocytes and microcytes in blood smear test) (Fig. 2) and a normal serum ferritin level (36 ng/ml).

Blood smear of the mother of patient 3: anisocytosis, normocytes, hypochromic microcytes, poikilocytosis
At the last follow up, the patient was 21 years old and still on pyridoxine. He was non transfusion dependent with a stable Hb level of 9,8–10 g/dl.
Liver MRI T2* revealed moderate iron overload (Liver iron concentration = 9 mg/g dry weight) at the age of 16 after four blood transfusions. Thus, iron chelation therapy was initiated and subsequently adjusted based on availability and tolerance of iron chelators (Fig. 3).

Kinetics of serum ferritin level in patient 3 with non-transfusion-dependent sideroblastic anemia
Clinical and biological characteristics of the three patients at diagnosis are summarized in Table 2.
Discussion
Three patients with sideroblastic anemias were enrolled in this single-centre study conducted in the pediatric hematology department. Two patients were diagnosed with congenital non syndromic sideroblastic anemia (CSA). The third patient was diagnosed with myelodysplastic syndrome (MDS) at the age of 11 years. Sideroblastic anemias constitute a rare and heterogeneous group of disorders characterized by iron accumulation in mitochondria of erythroblasts with ineffective erythropoiesis [1]. SAs can be congenital or acquired. Congenital sideroblastic anemias caused by defects in heme biosynthesis, iron-sulfur cluster (ISC) biogenesis, mitochondrial protein synthesis or dysfunction [7]. Acquired SA is associated with clonal hematopoiesis (MDS, MPS) or various reversible conditions including drugs or alcohol exposure, vitamin B6 or copper deficiency, Zinc overdose and heavy metal poisoning [1].
For clonal sideroblastic anemia, the 2016 revision to the WHO of myeloid neoplasms distinguished three entities of myeloid neoplasia with sideroblastic anaemia: MDS-RS with single lineage dysplasia, MDS-RS with multilineage dysplasia and MDS/MPN with ring sideroblasts and thrombocytosis (MDS/MPN-R) [4]. Somatic mutation of Splicing Factor 3B Subunit 1A (SF3B1) is strongly associated with MDS-RS, thus, it was used to distinguish clonal SA from other causes of adults SAs. In the fifth edition of WHO MDS classification, MDS with SF3B1 mutations was identified as a distinct subtype [8]. Our patient was diagnosed with MDS-RS-MDL with SF3B1 somatic mutation at the age of 11.8 years without any germinal predisposition. Unlike to its prevalence in adults, sideroblastic anemia represents an extremely rare manifestation of MDS in paediatric patients. In addition, common genes somatic mutations involved in adult MDS have not been identified in pediatric patients with MDS specially SF3B1 [9]. In our knowledge, to date, only one case of MDS-RS with SF3B1 mutation has been reported in pediatric population: a 17-year-old patient with Fanconi anamia [10]. For our patient a wait and see approach was adopted due to the absence of severe cytopenia, and the absence of blasts and cytogenetic abnormalities. The impact of SF3B1 mutation in MDS patients was assessed in adults’ population. No independent effect was found on overall survival and Leukemia free survival [11, 12].
We have identified only two cases of congenital sideroblastic anemia in our pediatric hematology department, a reference centre for pediatric hematology in Tunisia. This low incidence underscores the rarity of the condition and may also suggest a possible underdiagnosis. In pediatric cases of microcytic anaemia where iron deficiency and hemoglobinopathies have been excluded, monogenic microcytic anemias, including iron metabolism defects and sideroblastic anemias, should be considered as potential underlying causes. First-line evaluation including serum iron, transferrin, ferritin, ratio of serum iron/total iron-binding capacity, and eventually serum sTfR and hepcidin level, guided diagnosis establishment [13]. Bone marrow smears with PERLS’ stain is required to confirm the diagnosis of sideroblastic anemia, although it is not mandatory in the presence of genetic testing [13]. It remains a valuable diagnostic tool in settings where genetic and other diagnostic tests may be inaccessible or delayed.
Recent advancements in understanding of iron metabolism, coupled with the application of next-generation sequencing technologies, have significantly improved the characterization of congenital sideroblastic anemias [7, 14]. These anemias are classified according to their mode of inheritance into X-linked, autosomal, or mitochondrial mutations. Furthermore, they are categorized as either syndromic (XLSA/A, MLASA) or non-syndromic (ALAS2, SLC25A38, HSPA9, HSCB, GLRX5) based on the presence or absence of associated extra-hematological features [7, 14, 15].
Non-syndromic X-linked SA due to ALAS 2 deficiency is the most common form of congenital SAs (CSA), account for 40% of cases of CSA, around two hundred cases have been reported [1, 16]. About two-thirds of cases occur in males during early childhood or adolescence, while one-third appear in young to middle-aged females [1]. ALA synthase catalyses the first step of heme synthesis in mitochondria leading to the production of D-aminolevulinic acid (ALA). This reaction requires glycine and succinyl-coenzyme A as substrates and pyridoxal phosphate as cofactor [7, 17, 17, 18]. Its defect results in heme deficiency leading to ineffective erythropoiesis and ultimately causes microcytic hypochromic sideroblastic anemia and iron overload. ALAS2 synthase deficiency was considered the most plausible diagnosis for our third patient given the non-syndromic nature of sideroblastic anaemia and the positive response to pyridoxine treatment. About two-thirds of patient with ALAS2 deficiency responded to pyridoxine supplementation [7, 18]. The presence of dimorphic red blood cell populations in the blood smear of mother’s patient is also indicative of ALAS2 deficiency [7].
The second patient in our series was diagnosed with congenital sideroblastic anaemia due to SLC25A38 gene mutation. She was diagnosed with Chronic microcytic hypochromic anaemia at the age of 7 years. Blood transfusion therapy was used for 5 years, subsequently her Hb level stabilized at 8.5–9 g/dl. SLC25A38 gene mutation is the second cause of CSA with autosomal recessive transmission. SLC25A38 is a mitochondrial glycine transporter, its absence has been impeded aminolevulinate (ALA) synthesis despite normal ALAS2 enzymatic activity [19]. Around a hundred cases were reported: unlike to ALAS deficiency patients presented early in the life at birth or in infant with non-syndromic hypochromic microcytic anaemia, that usually require chronic transfusion [15, 19]. Heeney et al. reported that of the 28 patients in whom data are available, 25 patients received their first transfusion in the first year of life and all but one was maintained on chronic regular transfusions [19]. In all 20 patients for whom oral pyridoxine was prescribed, no improvement of the Hb level was observed [19]. Like ALAS2 deficiency iron overload represent a significant morbidity in SLC25A38 deficiency [19, 20]. Le Rouzic et al. reported a cohort of thirteen patients with ALAS2 and SLC25A38 deficiencies, among whom five had undergone chronic transfusion therapy and nine had been diagnosed with secondary hemochromatosis [20]. Among inherited sideroblastic anemias systemic iron overload occurs mainly when the inherited defect is restricted to the erythroid cell line (XLSA, GLRX5, and SLC25A38 mutations), as a consequence of inefficient erythropoiesis and increased iron absorption[18].
Hemochromatosis must be managed to prevent organ damage, as iron overload can aggravate anemia. Iron chelators such as deferoxamine, deferasirox, and deferiprone may be used alone or in combination. If iron chelator drugs are not available, phlebotomy is considered in ALAS 2 deficiency with good response to pyridoxine [19,20,21].
Finally, defects in mitochondrial protein translation or in respiratory chain complex proteins are associated with syndromic sideroblastic anemias. In these conditions, extra-hematological features such as neuromuscular disorders, exocrine pancreatic insufficiency, lactic acidosis, and immunodeficiency are often at the forefront, systemic iron overload is usually absent in these cases [7, 17].
Conclusions
Acquired sideroblastic anemias, either clonal or non-clonal disorders, are extremely rare in children. The SF3B1 gene mutation is strongly associated to myeloid neoplasms with ring sideroblasts. While MDS with SF3B1 mutations has a favourable prognosis in adults, the long-term prognosis for childhood MDS with SF3B1 mutations is still unknown.
ALAS2 deficiency and SLC25A38 gene mutations are the most common causes of congenital sideroblastic anemias. Pyridoxine improves hemoglobin levels in approximately two-thirds of ALAS2 deficiency patients. Genetic testing should be implemented to investigate anemia with no evident causes. Iron overload should be screened for, even in non-transfusion-dependent patients with sideroblastic anemias. Hematopoietic stem cell transplantation is to be considered in non-syndromic transfusion-dependent sideroblastic anemias.
Data availability
No datasets were generated or analysed during the current study.
References
Abu-Zeinah G, DeSancho MT (2020) Understanding Sideroblastic Anemia: An overview of genetics, epidemiology, pathophysiology and current therapeutic options. J Blood Med 11:305–318. https://doi.org/10.2147/JBM.S232644
Ashorobi D, Chhabra A (2024) Sideroblastic anemia. StatPearls Publishing, StatPearls, Treasure Island (FL)
Bottomley SS, Fleming MD (2014) Sideroblastic anemia: diagnosis and management. Hematol Oncol Clin North Am 28:653–70), v. https://doi.org/10.1016/j.hoc.2014.04.008
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM et al (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127:2391–2405. https://doi.org/10.1182/blood-2016-03-643544
Hasserjian RP, Germing U, Malcovati L (2023) Diagnosis and classification of myelodysplastic syndromes. Blood 142:2247–2257. https://doi.org/10.1182/blood.2023020078
Kardos G, Baumann I, Passmore SJ, Locatelli F, Hasle H, Schultz KR et al (2003) Refractory anemia in childhood: a retrospective analysis of 67 patients with particular reference to monosomy 7. Blood 102:1997–2003. https://doi.org/10.1182/blood-2002-11-3444
Ducamp S, Fleming MD (2019) The molecular genetics of sideroblastic anemia. Blood 133:59–69. https://doi.org/10.1182/blood-2018-08-815951
Rudelius M, Weinberg OK, Niemeyer CM, Shimamura A, Calvo KR (2023) The international consensus classification (ICC) of hematologic neoplasms with germline predisposition, pediatric myelodysplastic syndrome, and juvenile myelomonocytic leukemia. Virchows Arch Int J Pathol 482:113–130. https://doi.org/10.1007/s00428-022-03447-9
Chatterjee T, Choudhry VP (2013) Childhood myelodysplastic syndrome. Indian J Pediatr 80:764–771. https://doi.org/10.1007/s12098-013-1130-8
Boles B, Shiel M, Gardner J-A, Conant JL (2023) Pediatric myelodysplastic syndrome with SF3B1 mutation. J Assoc Genet Technol 49:69–72
Farrukh F, Abdelmagid M, Mangaonkar A, Patnaik M, Al-Kali A, Elliott MA et al (2020) Prognostic impact of SF3B1 mutation and multilineage dysplasia in myelodysplastic syndromes with ring sideroblasts: a Mayo Clinic study of 170 informative cases. Haematologica 2525–32. https://doi.org/10.3324/haematol.2023.284719
Jafari PA, Sadeghian MH, Miri HH, Sadeghi R, Bagheri R, Lavasani S et al (2020) Prognostic significance of SF3B1 mutations in patients with myelodysplastic syndromes: a meta-analysis. Crit Rev Oncol Hematol 145. https://doi.org/10.1016/j.critrevonc.2019.102832
Cappellini MD, Russo R, Andolfo I, Iolascon A (2020) Inherited microcytic anemias. Hematol Am Soc Hematol Educ Program 2020:465–470. https://doi.org/10.1182/hematology.2020000158
Fujiwara T, Harigae H (2019) Molecular pathophysiology and genetic mutations in congenital sideroblastic anemia. Free Radic Biol Med 133:179–185. https://doi.org/10.1016/j.freeradbiomed.2018.08.008
Fouquet C, Le Rouzic M-A, Leblanc T, Fouyssac F, Leverger G, Hessissen L et al (2019) Genotype/phenotype correlations of childhood-onset congenital sideroblastic anaemia in a European cohort. Br J Haematol 187:530–542. https://doi.org/10.1111/bjh.16100
Bergmann AK, Campagna DR, McLoughlin EM, Agarwal S, Fleming MD, Bottomley SS et al (2010) Systematic molecular genetic analysis of congenital sideroblastic anemia: evidence for genetic heterogeneity and identification of novel mutations. Pediatr Blood Cancer 54:273–278. https://doi.org/10.1002/pbc.22244
Long Z, Li H, Du Y, Han B (2018) Congenital sideroblastic anemia: advances in gene mutations and pathophysiology. Gene 668:182–189. https://doi.org/10.1016/j.gene.2018.05.074
Ricci A, Di Betto G, Bergamini E, Buzzetti E, Corradini E, Ventura P (2022) Iron metabolism in the disorders of heme biosynthesis. Metabolites 12. https://doi.org/10.3390/metabo12090819
Heeney MM, Berhe S, Campagna DR, Oved JH, Kurre P, Shaw PJ et al (2021) Mutation update SLC25A38. Hum Mutat 42:1367–1383. https://doi.org/10.1002/humu.24267
Le Rouzic M-A, Fouquet C, Leblanc T, Touati M, Fouyssac F, Vermylen C et al (2017) Non syndromic childhood onset congenital sideroblastic anemia: A report of 13 patients identified with an ALAS2 or SLC25A38 mutation. Blood Cells Mol Dis 66:11–18. https://doi.org/10.1016/j.bcmd.2017.07.003
G A-Z, Mt D (2020) Understanding sideroblastic anemia: an overview of genetics, epidemiology, pathophysiology and current therapeutic options. J Blood Med 11. https://doi.org/10.2147/JBM.S232644
Acknowledgements
The authors would like to thank all patients and families participating in the study for their trust and collaboration.
Funding
No funding to declare.
Author information
Authors and Affiliations
Contributions
S.R. designed the study and wrote the manuscript; I.BF contributed to the data collection and interpretation, R.H. participated in writing the manuscript, H.J, and H.Z contributed to data collection, M.O reviewed the final version, M.O, A.BT, AM, I.Z, R.K, M.B, F.M, M.BK participated in the data interpretation. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
Informed consent was obtained from the patients for the publication of any potentially identifiable data included in this article.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rekaya, S., Ben Fraj, I., Hamdi, R. et al. Sideroblastic anemia in children: challenges in diagnosis and management in three cases. Ann Hematol 104, 2537–2543 (2025). https://doi.org/10.1007/s00277-025-06266-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-025-06266-5