
Abstract
Background
The methylation of N6-methyladenosine (m6A) RNA plays a crucial role in the genetic regulation of various cancers. While m6A modifications have been extensively studied in the tumor microenvironment (TME) of several malignancies, their role in cutaneous melanoma (CM) remains unexplored.
Methods
Using Non-negative matrix factorization (NMF) analysis on single-cell RNA-seq data (GSE215121) from three CM samples obtained from public databases, 26 m6A RNA methylation regulators were utilized to determine TME subclusters, their expression, and function.
Results
Six distinct TME cell types were identified and NMF clustering further revealed unique m6A-based subpopulations of cancer-associated fibroblasts and T cells. The prognostic model demonstrated strong predictive capabilities, particularly for fibroblast and T cell m6A clusters, and highlighted COL3A1 as a critical regulator of melanoma-fibroblast interactions.
Conclusion
Highlighting the COL3A1 gene as a critical link and potential therapeutic target in melanoma could offer new avenues for targeted therapies and improve prognostic assessments in cutaneous melanoma.
Similar content being viewed by others


Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
Introduction:
Cutaneous melanoma (CM) is the most fatal and life-threatening form of malignant skin cancer. According to the World Health Organization, melanoma is responsible for approximately 57,000 deaths annually, making it one of the deadliest skin cancers (Arnold et al. 2022). Melanoma incidence has been increasing over the last 50 years. In 2020, more than 1.5 million new cases of skin cancer were reported. Malignant melanomas were responsible for one-fifth of these cancers. (Erdmann et al. 2013; Arnold et al. 2014). Its high metastatic capability is largely attributed to genetic and epigenetic alterations, including dysregulation of key oncogenic pathways (Rebecca et al. 2012). The incidence of melanoma is higher among the older generation, and the risk of the disease generally increases with age, with young adults being the most affected group (Fidler et al. 2017). In predominantly white populations, CM is responsible for the vast majority of melanoma cases, estimated at over 90%. Mucosal and uveal melanomas are significantly less common, accounting for less than 1–5% of cases, with variations depending on the country. In populations with darker skin tones, such as black, brown, and East Asian communities, cutaneous melanomas, particularly acral types, are the most common (Elder et al. 2020, 2018). Given these epidemiological findings, studying melanoma at a molecular level is essential for developing target therapies and markers which could improve patient treatment outcomes.
The mainstay of treatment for primary CM is excision with safety surgical margins, but once it metastasizes, treatment becomes more challenging and overall prognosis is poor. A favorable prognosis depends on early detection; however, inadequate treatment outcomes persist due to a lack of knowledge about the features of the TME in CM, as well as the expression and function of its regulators.
Furthermore, melanoma cells have a strong interaction with the tumor microenvironment and the immune system (Leonardi et al. 2018), indicating that the tumor microenvironment plays an important role in cancer initiation and metastasis (Visser and Joyce 2023). m6A is a key post-transcriptional modification that regulates RNA stability, splicing, and translation thereby influencing tumor progression. In melanoma, aberrant m6A modifications have been linked to altered gene expression patterns that promote tumor proliferation, immune evasion, and therapy resistance. This knowledge has led us to investigate post-transcriptional modifications, which have become a new trend in cancer research, with N6-methyladenosine (m6A) being the most common mRNA modification linked to the pathogenesis of many types of cancers (He et al. 2019; Wang et al. 2020). The three types of m6A regulators are writers, erasers, and readers, which are respectively responsible for m6A’s functions, methylation, and demethylation (Boulias and Greer 2023). Notably, m6A has been shown to play a critical role in melanoma oncogenesis and tumorigenicity. Specifically, m6A regulators such as METTL3 and FTO have been shown to modulate melanoma plasticity and drug sensitivity (Yu et al. 2021; Yang et al. 2019).
The function of tumor-infiltrating lymphocytes in the development and outcome of melanoma was recently thoroughly described by Dan Liu et al. (Liu et al. 2021). In the tumor microenvironment, Monica Marzagalli et al. uncovered the interplay between immune cells and melanoma (Marzagalli et al. 2019). Apart from tumor cells, TME cells comprise multiple cell types, including immune cells which have dual roles such as attacking tumor cells or, conversely, supporting tumor growth by creating an immunosuppressive environment. Furthermore, TME consists of stromal cells which signal molecules that promote tumor growth and facilitate metastasis and the non-cellular component which actively participate in cancer progression through angiogenesis and regulating cell behavior (Anderson and Simon 2020). Therefore, strategies that modulate the TME are being explored to enhance treatment outcomes. However, the cell–cell interaction between m6A mRNA modification-associated subtypes of TME cells and tumor cells has been the subject of a limited amount of research until recently. These findings prove that studying m6A modifications can provide novel insights into melanoma pathogenesis and potential therapeutic targets.
In this study, we examined the ramifications of m6A mRNA methylation on the primary TME cells, which include Melanocytes, T cells, B cells, EC, Fibroblasts, and Macrophages. Our analysis was based on 26,532 cells from three CM datasets that were sourced from public databases. 26 m6A RNA methylation regulators (Zaccara et al. 2019; Chong et al. 2021) were analyzed using NMF. It was observed that the m6A mRNA methylation sequences in each CM TME cell type subpopulation exhibited a substantial and distinctive link with tumor epithelial cells, and were associated with distinct immune and transcription characteristics, metabolic pathways, and prognosis. Essentially, our extensive single-cell analysis uncovers how m6A mRNA steers cell to cell interaction of TME cells with tumor cells and contributes to CM progression.
Materials and methods
Data collection
We obtained two skin cutaneous melanoma datasets from the GEO database: one single-cell dataset (GSE215121, N = 3) (Zhang et al. 2022) and one bulk-RNA dataset (GSE54467, N = 79) (Jayawardana et al. 2015). Additionally, we downloaded TCGA-SKCM dataset (N = 472) from UCSC (https://xenabrowser.net). The TCGA-SKCM dataset was used to establish a prognostic model of m6A-related clustering characteristic genes, while the GSE54467 dataset was used to further validate the expression profiles of characteristic genes. Since these public datasets were obtained from public databases, ethical approval was not required.
CM single-cell data analysis
First, the Read10X function in the Seurat package (v4.0.5) (Butler et al. 2018) in R software (v4.0.3) was used to read the data of each sample, and cells with mitochondrial expression greater than 20% were removed. This is because high mitochondrial expression usually indicates poor cell condition, possibly due to compromised cell membrane integrity, leading to a higher proportion of mitochondrial genes. Such cells may interfere with subsequent analyses, so it is necessary to filter them out based on the proportion of mitochondrial genes in the cells. We normalized the raw counts for each sample using Seurat function NormalizeData and identified 2000 variable genes using the FindVariableGenes function. Then, using the FindIntegrationAnchors function with default parameters and canonical correlation analysis (CCA) with 30 dimensions, we identified integration anchors between the three datasets. These anchors were integrated using the IntegrateData function with the first 30 CCA dimensions and default parameters. We scaled the data using the ScaleData function and performed principal component analysis (PCA) with the RunPCA function. Next, we performed unsupervised clustering on the data using the FindNeighbors and FindClusters functions. For the FindNeighbors function, we used the first 30 PCA dimensions to construct a shared nearest neighbor (SNN) graph for our dataset. We then clustered the cells using an SNN modularity optimization-based clustering algorithm with a resolution of 0.5. Finally, for visualization, we used the RunTSNE function with default parameters and 30 PCA dimensions. To identify cell groups, we used the FindAllMarkers function on the integrated dataset, which uses the Wilcoxon rank-sum test to identify representative genes for each cluster. These representative genes were used to determine the identity of each cluster and to identify cell types typically found in human skin based on literature markers. We used the R package BayesPrism (v2.0) (Chu et al. 2022), leveraging scRNA-seq as prior information to predict cell type composition from the TCGA melanoma dataset.
Cell–Cell communication analysis
We conducted an analysis of cell–cell communication analysis using the R CellChat package (v1.6.1) (Jin et al. 2021). This package contains a ligand-receptor interactions database for humans and mice, which allows for the analysis of cell–cell communication networks using scRNA-seq data annotated with various cell clusters. First, we used CellChat to assess the main signal inputs and outputs between cell clusters through CellChatDB.human database. Then, we used the netVisual_circle function to display the strength of cell–cell communication networks from the target cell cluster to all NMF cell clusters among different clusters. Finally, the netVisual_bubble function was employed to display bubble plots of significant ligand-receptor interactions between the target cell cluster and other clusters.
m6A gene set NMF clustering analysis
The 26 m6A gene sets were clustered across all TME cell types using the NMF R package, v0.25 (Gaujoux and Seoighe 2010). The different clusters of these cell types were determined based on the scRNA expression matrix. The execution of each of these steps was in line with previous studies (Chen et al. 2020; Puram et al. 2017).
Pseudotime analysis
We used the Monocle package (v2.22.0) in R to analyze the single-cell RNA data of fibroblast cell types (Qiu et al. 2017). Based on the following standards, highly variable genes were chosen: mean expression ≥ 0.1. The dynamic expression heatmap of the m6A genes was visualized using the plot_pseudotime_heatmap function, and dimensionality reduction was accomplished using the DDRTree method.
Transcription factor analysis of m6A gene set NMF clusters
Python-based SCENIC analysis software called pySCENIC (v0.9.0) was used to investigate the gene regulatory networks of transcription factors (TFs) in CM (Kumar et al. 2021). Two gene motif rankings from the RcisTarget database (hg19-tss-centered-10 kb and hg19-500 bp-upstream) were used to detect transcription start sites (TSS) and gene regulatory networks in the scRNA-seq data of CM. The threshold for transcription factors was set to Benjamini–Hochberg-False Discovery Rate (BH-FDR) < 0.05.
Construction of prognostic model of m6A cluster characteristic genes
Based on the FindAllMarkers function in the Seurat package, we generated characteristic genes between m6A gene set NMF clusters. Then, using multivariate Cox analysis based on overall survival, we further identified characteristic genes associated with patient prognosis and constructed a prognostic model with these genes. We used the survminer package (v0.4.9) in R to determine the model’s threshold, dividing all patients into low-risk and high-risk groups according to the threshold. Kaplan–Meier (K–M) survival curves were generated using the log-rank test to confirm prognostic differences between the two groups. The timeROC package (v0.4) in R was used to generate 1-year, 2-year, and 3-year ROC curves, and the corresponding area under the curve (AUC) was calculated.
Verification of prognostic characteristic genes
The GSE54467 dataset was used to externally validate the characteristic genes used to construct the prognostic model. The expression profiles of the characteristic genes included in the GSE54467 dataset were extracted. All patients in the validation cohort were divided into low-risk and high-risk groups using the method used to construct the prognostic model. The accuracy of characteristic genes in the validation cohort was evaluated using K-M survival analysis and ROC curves.
Utilizing the TIGER database for melanoma ICB datasets to investigate gene set behaviour in treatment outcomes
The TIGER database (The Tumor Immunotherapy Gene Expression Resource portal, http://tiger.canceromics.org/#/) (Chen et al. 2023a) contains an immunotherapy response module that includes melanoma data with immunotherapy information. This module was used to analyze the immunotherapy efficacy of different gene sets.
Results
Overall situation of TME in SKCM
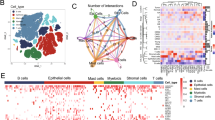
A total of 26,532 cells from 3 CM samples were divided into 6 subgroups (Fig. 1A–C), namely melanocytes accounting for 55.75% (Melanocytes, MLANA, PMEL), T cells accounting for 31.48% (T cells, CD3D), B cells accounting for 4.82% (B cells, MS4A1, CD79A), fibroblasts accounting for 4.56% (Fibroblasts, DCN, COL1A1), endothelial cells accounting for 2.61% (EC, PECAM1, SELE), and macrophages accounting for 0.77% (Macrophages, LYZ, CD68, CD14), with melanocytes being the largest proportion. In the CellChat cell–cell communication network diagram, the thickness of the lines was determined by numerical values, which represented the number of ligand-receptor connections. A more significant number indicated more connections and stronger intercellular interactions. As shown in Fig. 1D, melanoma cells exhibited strong interactions with Fibroblasts, Macrophages, Endothelial Cells (EC), and T cells, suggesting that these four cell types played a crucial role in the tumor microenvironment of skin melanoma. They may directly or indirectly influence the occurrence and progression of melanoma within the tumor. Cell proportions estimated using BayesPrism based on the TCGA melanoma dataset showed that melanoma cells had the highest proportion (Fig. 1E), consistent with the single cell data. The proportions of other cell types also aligned well with the cell composition observed in single-cell dataset. We evaluated the average expression levels of 26 m6A regulatory factors in different cell types (Fig. 1F) and found that some regulatory factors are highly expressed in all cells and could serve as potential therapeutic targets in the TME. Specifically, HNRNPA1 and HNRNPA2B1 were highly expressed in melanocytes compared to other cell types.

Single-cell annotation of skin melanoma and the expression of the m6A gene set in single-cell. A tSNE plot showing the composition of skin melanoma cells; B bar chart of the proportions of cell composition in each sample; C violin plot of cell annotation markers; D interaction network diagram between cells; E TCGA cutaneous melanoma data cell proportions bar chat; F heatmap of the expression of the m6A gene set in various cell types
Contribution of m6A-mediated fibroblasts to TME
Next, we analyzed fibroblasts, known as CAFs (cancer-associated fibroblasts) in tumors. First, we re-clustered them into three subgroups (Fig. 2A, B): pan-myCAFs, myofibroblast-like CAFs (TAGLN, ACTA2, MYLK); pan-dCAFs, desmoplastic CAFs (COL1A1, COL3A1); and pan-mCAFs, melanocytes CAFs (PMEL, MLANA). Then, using the NMF unsupervised clustering method, we clustered 26 m6A regulatory factors, resulting in four m6A-associated fibroblast clusters, namely YTHDF3 + CAF-C1 (n = 331), CBLL1 + CAF-C2 (n = 45), IGF2BP3 + CAF-C3 (n = 774), and METTL4-CAF-C4 (n = 60) (Fig. 2C). The selection criteria for clustering rank values and the heatmap can be found in the attachment (Fig. S1A, Fig. S1B).

Display of fibroblast subpopulations and m6A-related fibroblast clusters. A tSNE plot of fibroblast subpopulations; B heatmap of fibroblast subpopulation markers; C NMF clustering heatmap of m6A regulatory factors; D interaction results between m6A clusters and melanoma cells; E, F pseudotime analysis of m6A clusters; G heatmap of differential genes at various differentiation nodes in pseudotime analysis; H GOBP enrichment analysis of differential genes in m6A clusters; I correlation analysis between fibroblast subpopulations and m6A clusters; J expression of each gene set in m6A clusters
Cell–cell communication analysis using the CellChat method revealed strong interactions between the YTHDF3 + CAF-C1 and IGF2BP3 + CAF-C3 clusters with melanoma cells (Fig. 2D).
Using monocle pseudotime analysis, we found that the cell differentiation direction was from IGF2BP3 + CAF-C3 to YTHDF3 + CAF-C1 (Fig. 2E, F), with m6A regulatory factors playing a key role in the trajectory of CAFs (Fig. 2G). GOBP functional enrichment analysis showed that the IGF2BP3 + CAF-C3 cluster was enriched in various biosynthetic processes, including secondary metabolite biosynthetic process, pigment biosynthetic process, and phenol-containing compound biosynthetic process, while the YTHDF3 + CAF-C1 cluster exhibited cell chemotaxis functions such as eosinophil chemotaxis and T cell chemotaxis (Fig. 2H). We also found that IGF2BP3 + CAF-C3 was closely related to pan-myCAF myofibroblasts, and YTHDF3 + CAF-C1 was closely related to pan-dCAF desmoplastic fibroblasts (Fig. 2I). Common signaling pathway genes such as collagen, extracellular matrix (ECM), matrix metalloproteases (MMPs), transforming growth factor beta(TGFB), Neo-Angio, Contractile, RAS, and pro-inflammatory genes were expressed differently in each m6A-associated fibroblast cluster (Fig. 2J).
Using the single-cell transcription factor analysis method pySCENIC, we found that the transcriptional activities of three transcription factors—ATF4, MXD3, and POLE4—were higher in IGF2BP3 + CAF-C3 compared to the other three m6A cell clusters, while RORA, KDM5B, ATF6, and ELK3 were higher in YTHDF3 + CAF-C1 compared to the other three m6A cell clusters (Fig. S2.A).
Contribution of m6A-mediated T/MAC cells to TME
First, T cells were isolated and subdivided into four subgroups: CD4 + T, CD8 + T, NK, and Treg (Fig. 3A, B). Then, using the NMF method combined with 26 m6A regulatory factors, T cells were subjected to unsupervised clustering, resulting in four clusters (Fig. 3C, D). The selection criteria for clustering rank values and the heatmap can be found in the attachment (Fig. S1C, Fig. S1D). The four clusters are METTL16 + T-C1 (n = 588), YTHDF1 + T-C2 (n = 143), VIRMA + T-C3 (n = 7052), and METTL4 + T-C4 (n = 444). It can be observed that the number of cells in C3 is the largest, while the proportion of each T cell subgroup within each m6A-related cell cluster does not differ significantly.

Display of T/macrophage subpopulations and m6A-related clusters. A tSNE plot of T cell subpopulations; B heatmap of T cell subpopulation markers; C NMF clustering heatmap of T cell m6A regulatory factors; D proportions of T cell subpopulations in each m6A cluster; E expression of each gene set in T cell subpopulations and m6A clusters; F heatmap of transcription factor differences in m6A clusters within T cell subpopulations; G interaction between m6A clusters and melanoma cells; H NMF clustering heatmap of macrophage m6A regulatory factors; I interaction between m6A clusters and melanoma cells; J heatmap of transcription factor differences in macrophage m6A clusters
Through the cellchat cell communication analysis method, it was found that the number of ligand-receptor connections between melanoma cells and m6A-related T cell clusters varies (Fig. 3G).
Using the single-cell transcription factor analysis method pySCENIC, it was discovered that there are significant differences in transcription factor activity (average AUC value) among m6A-related cell clusters C1, C2, C3, and C4 within each T cell subpopulation (Fig. 3F). Additionally, a heatmap showed the average expression levels of these genes in CD4+, CD8+, Treg, and NK cells among the m6A cell clusters, highlighting significant differential characteristics. These include four T cell functional features (T exhaustion score, T effector score, and T escape score), as well as several immune stimulants, inhibitors, and T cell function marker genes (Fig. 3E).
We also isolated macrophages and used NMF clustering analysis to divide them into three classes (Fig. 3H, I): FTO + MAC − C1 (n = 150), METTL16 + MAC − C2 (n = 27), and METTL3 + MAC − C3 (n = 27). The selection criteria for clustering rank values and the heatmap can be found in the attachment (Fig. S1E, Fig. S1F). The pySCENIC analysis revealed that there was a significant difference in transcription factor activity between C1 and the other two m6A-related cell clusters (Kruskal–Wallis test, p < 0.001) (Fig. 3J).
Contribution of m6A-mediated EC Cells to TME
Since strong interactions between EC and melanoma cells were observed in Fig. 1D, we isolated endothelial cells (EC) for separate analysis. Subpopulation analysis was performed, dividing them into three subpopulations: Vascular EC, Melanocytes-like EC, and Fibroblast-like EC (Fig. 4A, B).

Display of endothelial cell subpopulations and m6A-related clusters. A tSNE plot of endothelial cell subpopulations; B heatmap of endothelial cell subpopulation markers; C NMF clustering heatmap of endothelial cell m6A regulatory factors; D interaction between m6A clusters and melanoma cells; E heatmap of transcription factor differences in endothelial cell m6A clusters
Using NMF, EC were clustered into two m6A-related cell clusters (Fig. 4C). The selection criteria for clustering rank values and the heatmap can be found in the attachment (Fig. S1G, Fig. SH). The pySCENIC transcription factor analysis revealed significant differences between C1 and C2 (Wilcox-Wallis, p < 0.001) (Fig. 4E).
Construction of m6A cluster prognostic model
Differential analyses were performed on m6A clusters of fibroblasts, T cells, macrophages, and endothelial cells to identify genes for constructing a prognostic model. This model assesses whether the m6A clusters are meaningful and can positively impact tumor prognosis.
Using the Kruskal–Wallis test for differential analysis of the four fibroblast clusters, differential genes were identified as candidate gene sets. These genes were filtered using multivariate Cox regression to construct a prognostic model. The risk score value was calculated, and combined with survival data from TCGA-SKCM, the model’s prognostic capability was validated. Survival analysis revealed highly significant differences between high-risk and low-risk groups (p < 0.0001) (Fig. 5A), with ROC curve values at multiple time points exceeding 0.7 (Fig. 5D).

Survival analysis of various cell types and multi-timepoint ROC curve graphs. A TCGA data survival analysis for fibroblasts; B TCGA data survival analysis for T cells; C TCGA data survival analysis for macrophage cells; D TCGA data multi-timepoint ROC curve for fibroblasts; E TCGA data multi-timepoint ROC curve for T cells; F TCGA data multi-timepoint ROC curve for macrophage cells; G TCGA data survival analysis for endothelial cells (EC); H GSE54467 data survival analysis for fibroblasts; I GSE54467 data survival analysis for T cells; J TCGA data multi-timepoint ROC curve for endothelial cells (EC); K GSE54467 data multi-timepoint ROC curve for fibroblasts; L GSE54467 data multi-timepoint ROC curve for T cells; M GSE54467 data survival analysis for macrophage cells; N GSE54467 data survival analysis for endothelial cells (EC); O GSE54467 data multi-timepoint ROC curve for macrophage cells; P GSE54467 data multi-timepoint ROC curve for endothelial cells (EC)
Using the Kruskal–Wallis test, differential analyses were conducted separately on the four T cell clusters and three macrophage clusters to identify differential genes as candidate gene sets. These genes were filtered using multivariate Cox regression to construct prognostic models. The risk score values were calculated, and combined with survival data from TCGA-SKCM, the prognostic capabilities of the models were validated.
Analysis revealed that in the prognostic model constructed using the differential genes of the T cell clusters as candidate genes, the differences between the high-risk and low-risk groups were extremely significant (p < 0.0001) (Fig. 5B), with ROC curve values at multiple time points all exceeding 0.65 (Fig. 5E). Similarly, in the prognostic model constructed using the differential genes of the macrophage clusters, the differences between the high-risk and low-risk groups were extremely significant (p < 0.0001) (Fig. 5C), with ROC curve values at multiple time points all exceeding 0.7 (Fig. 5F).
Using the Wilcox-Wallis test, differential analysis was conducted on the two EC cell clusters to identify differential genes as candidate gene sets. These genes were filtered using multivariate Cox regression to construct a prognostic model. The risk score values were calculated, and combined with survival data from TCGA-SKCM, the prognostic capability of the model was validated.
Analysis revealed that the differences between the high-risk and low-risk groups were extremely significant (p < 0.0001) (Fig. 5G), with ROC curve values at multiple time points all exceeding 0.7 (Fig. 5J).
At the same time, the prognostic models constructed for each cell m6A cluster were revalidated using the GEO dataset GSE54467. The results showed that the m6A cluster survival analysis for fibroblasts was significant (p = 0.018) (Fig. 5H), with ROC curve values at multiple time points all above 0.6 (Fig. 5K); the m6A cluster survival analysis for T cells was significant (p = 0.0028) (Fig. 5I), with ROC curve values at multiple time points all above 0.6 (Fig. 5L); the m6A cluster survival analysis for macrophages was not significant (p = 0.065) (Fig. 5M), with ROC curve values at multiple time points all below 0.6 (Fig. 5O); the m6A cluster survival analysis for endothelial cells was significant (p = 0.015) (Fig. 5N) but the 1 and 2 year ROC curve values at multiple time points were below 0.6 (Fig. 5P).
This indicates that utilizing m6A-mediated fibroblast and T cell subpopulations has great prognostic significance for the tumor microenvironment.
Interaction analysis between melanoma cells and fibroblast m6A clusters
Interaction analysis was conducted between melanoma cells and fibroblast m6A clusters (Fig. 6A). Ligand-receptor gene pairs were divided into high and low expression groups based on their median expression levels in the TCGA-SKCM dataset, followed by survival analysis. The results showed that the survival analysis for COL3A1 was notably different (p < 0.05) (Fig. 6B), and COL3A1 expression was higher in fibroblasts compared to melanoma cells (Fig. 6C). This indicates that the COL3A1 gene is likely an important marker linking melanoma cells and fibroblasts and may be a critical target for advancing melanoma treatment.

Interaction between melanocytes and fibroblast m6A clusters. A Scatter plot of ligand-receptor pairs interactions between melanocytes and fibroblast m6A clusters; B Kaplan–Meier curve for the COL3A1 gene; C violin plot showing COL3A1 gene expression in fibroblast m6A clusters and melanoma cells
7. Construction of prognostic models between gene sets of TME cells and TIGER melanoma datasets to probe response group (R) and non-response group (NR) disparity
Prognostic models were constructed for gene sets associated with Fibroblasts, Macrophages, Endothelial Cells (EC), and T cells, and the melanoma dataset from TIGER was used to examine differences between the response group (R) and non-response group (NR). As shown in Fig. S3, there were significant differences in the gene sets of these cell types between the immunotherapy response groups.
Discussion
Thus far, a number of investigations have demonstrated a connection between the pathophysiology of cutaneous melanoma and RNA m6A modification (Liu et al. 2023; Wang et al. 2023a; Ran et al. 2023). Nevertheless, the prospective neoplastic role of m6A-modified single cells has been the subject of only a small number of studies. To our knowledge, this is the first study to systematically investigate m6A modification regulators of primary cell types in the tumor microenvironment (TME) of cutaneous melanoma. Moreover, the composition and characteristics of the TME can profoundly influence treatment responses. For example, a TME rich in immunosuppressive cells like certain macrophages can diminish the effectiveness of immunotherapies. Similarly, dense ECM can hinder the penetration of therapeutic agents, reducing their efficacy (Wang et al. 2023b). We have also discovered, at the single-cell sequence level, the diversity of cell–cell interactions between tumor cells and TME cell subtypes associated with m6A. Through this novel and distinctive viewpoint, we were able to comprehend the ways in which the RNA m6A modification of these various TME cellular constituents impacts the prognosis of individual cutaneous melanoma patients.
The majority of tumor tissue in CM is composed of melanoma cells, which are responsible for tumor expansion. Concurrently, the prognosis of patients and the responsiveness of melanoma cells to treatment are dictated by their heterogeneity. Besides melanoma cells, TME cells, including vascular endothelial cells, infiltrating immune cells, and various types of stromal cells, all contribute to the growth and immune evasion of cutaneous melanoma. Based on the single-cell analysis, we discovered that the TME cells, which included fibroblasts, T cells, macrophages, and endothelial cells, exhibited a wide range of m6A regulatory patterns and a significant amount of communication with melanoma cells. Additionally, the analysis of cellphones revealed that ligand-receptor pairs facilitated communication between melanoma cells and TME cell subtypes associated with m6A.
One of the essential parts of stromal cells, cancer-associated fibroblasts (CAFs), were categorized as pan-myCAFs and pan-dCAFs according to particular molecular characteristics (Galbo et al. 2021). We also discovered melanocytes CAFs, or pan-mCAFs, for the first time. The possible significance of RNA m6A modification in CAFs has not yet been fully explored in many studies. Our study revealed that fibroblasts mediated by m6A had more interactions with melanoma cells than fibroblasts not mediated by m6A. Moreover, fibroblasts exhibited higher expression of desmoplastic CAF Collagen Type III Alpha 1 (COL3A1) in comparison to melanoma cells, and there was a significant difference observed in its survival analysis. Cell interaction analysis revealed that fibroblasts and melanoma cells communicate through the COL3A1-ADGRG1 receptor-ligand pair. Additionally, COL3A1 has a significant prognostic impact on survival in TCGA-SKCM skin melanoma, suggesting that COL3A1 plays a crucial role in tumor progression and metastasis by affecting melanoma cells. It holds potential therapeutic and prognostic value. Future studies could further explore the molecular mechanisms and clinical applications of COL3A1, providing a potential target for melanoma metastasis treatment. Pathogenic variants resulting from COL3A1 mutations are associated with poor prognosis and metastasis of cutaneous melanoma (Zhao and Pritchard 2016). Excessive COL3A1 expression was affiliated with melanoma progression and outcome (Cao et al. 2023; Yuan et al. 2017). Furthermore, COL3A1 is upregulated in Triple-negative breast cancer (TNBC), a rare form of invasive breast cancer, and inhibiting it significantly aided in the metastatic regression of TNBC (Yang et al. 2022). Therefore, it was postulated that RNA m6A modification CAF COL3A1 could lead to an immunosuppressive interaction with melanoma cells, thereby helping the cancer to progress.
In recent years, studies have uncovered the substantial role of RNA m6A methylation in the modulation and development of immune cells (Wu et al. 2024). Obesity-associated protein FTO, an m6A eraser, increased melanoma cell proliferation, migration, and invasion in vitro as well as in vivo in a study by Seungwon Yang et al. and colleagues (Yang et al. 2019). They proposed that the tumorigenicity of melanoma was suppressed by FTO inhibition. Upregulation of FTO also promotes CAF-mediated angiogenesis, a key component in tumor development, according to research by Liao et al. (2024). Molecular clustering of m6A regulators was employed using Non-negative matrix factorization (NMF) which offers advantages over consensus clustering and k-means clustering, including stronger interpretability, sparsity, and greater flexibility. NMF can be combined with other methods incorporating sparse constraints or regularization to enhance clustering performance further. Moreover, since single-cell transcriptomic expression matrices are typically sparse, they align well with the sparsity characteristic of NMF, making it a suitable clustering method. Through NMF analysis, we discovered that all m6A-mediated subtypes of macrophages showed extensive interactions with melanoma cells; however, pySCENIC analysis showed a significant difference in transcription factor activity between FTO + MAC − C3 and the other two m6A-related cell clusters, indicating that m6A-mediated macrophages, particularly for the FTO + MAC − C3 subtype, clearly displayed activation of melanoma tumorigenesis. Furthermore, we discovered that melanoma cells also exhibited extensive interaction with m6A—mediated T cells (CD8+, CD4+, NK, and Treg), B cells, and EC. Moreover, distinct T cell active and inactive features were displayed by m6A-mediated subtypes of the four primary T cell populations. These findings all point to the importance of RNA m6A methylation in immune evasion and the tumor-promoting consequences of macrophages and T cells.
Transcriptional factors (TFs) were analysed at the single-cell level in order to identify gene regulatory networks that are specific to individual cells.In general, each subtype of CAF, T cells, macrophages, B cells, and EC exhibited distinct TF characteristics. For CAFs, IGF2BP3 + CAF-C3 and YTHDF3 + CAF-C1 both exhibited unique TF gene signature, i.e.; ATF4, MXD3, POLE4 and RORA, KDM5B, ATF6, ELK3 respectively. Previous research has demonstrated the connection between m6A modification and the expression of ATF4, RORA, KDM5B, ATF6, and ELK3 (Mo et al. 2023; Han et al. 2021; Huang et al. 2022; Guo et al. 2021; Hua et al. 2021; Zhang et al. 2021), indicating m6A’s function in CAF regulation (Chen et al. 2023b). Moreover, we discovered unique TF traits of m6A-mediated cell subtypes for macrophages, T cells, and EC. To sum up, the TME can be reshaped and reprogrammed by m6A-mediated cell subtypes through their modulation of different TF regulatory networks. Ultimately, melanoma cells were in close communication and connection with these m6A-mediated TME cells, as demonstrated by cell network analysis. Given that either m6A-mediated CAFs or immune cell subtypes demonstrated enhanced interaction with melanoma cells, it is clear that the development of an immunosuppressive tumor microenvironment may be partially impacted by RNA m6A methylation.
With consideration for the complex intrinsic patterns of RNA m6A methylation in TME cells, we compiled in detail the relationships between the scores of these sub-clusters and the immune response and prognosis from the public bulk RNA-seq cohorts. It was evident that patients with varying degrees of TME cell m6A regulator domination had significantly different melanoma prognoses and significantly different immune responses for patients receiving ICB therapy, particularly for macrophages and CAFs. These findings highlight the crucial role that TME m6A plays in the immune response of melanoma patients and warrant further investigation.
To validate these observations, potential wet lab experiments such as MeRip-Seq (m6A RNA immunoprecipitation sequencing) on melanoma cells with perturbed m6A regulators, map changes in m6A methylation sites followed by comparing results to the study’s bioinformatics predictions to confirm overlap in target transcripts. Additionally, utilizing CRISPR-Cas9 to target silencing specific m6A regulators identified as key players in melanoma clusters then validate knockdown efficiency via qRT-PCR (mRNA) and Western blot (protein) can aid in validation of m6A regulator expression and function (Du et al. 2021). In the future, we aim to include wet lab experiments in an effort to comprehensively validate observations.
The low depth of scRNA-seq and the insufficient sample size were the main limitations of our preliminary study, and additional patient verification is required to confirm our findings. A potential source of bias in our study’s clustering method is the fact that, when compared to bulk RNAseq, the scRNA-seq of some m6A regulators in melanoma would mostly be minor and have more zero observations. However, since our study focuses on cutaneous melanoma samples, we utilized three samples from CM single-cell dataset (GSE215121). Single-cell data differs from conventional bulk RNA sequencing, as it allows for gene expression analysis at the single-cell level. Even with a small number of samples, it provides rich information, revealing cellular heterogeneity and overcoming the limitations of averaging cell populations in traditional methods. Additionally, its technical advantages enable efficient single-cell isolation and capture, reducing sample requirements. Through mRNA amplification, sufficient data can be obtained even from minimal RNA amounts. In the future, to broaden our investigation, we aim to include more cutaneous melanoma samples (e.g., GSE81383) in our research. We also plan to expand our sample types to include acral melanoma samples (GSE189889) or melanoma brain metastases and leptomeningeal metastases (GSE174401), enabling a more comprehensive study of melanoma. Moreover, the scRNA-seq analysis still offers us a fresh perspective on the properties of m6A methylation regulators in different TME single cells to lessen melanoma assortment, which is crucial in advanced clinical practice.
Conclusion
Taking into account the intricate patterns of RNA m6A methylation within the cells of the tumor microenvironment (TME), it became evident that variations in m6A regulatory dominance in TME cells resulted in significant differences in the prognosis of cutaneous melanoma (CM) patients and showcased distinct immune responses, particularly among cancer-associated fibroblasts (CAFs) highlighting COL3A1 gene as a critical link and potential therapeutic target in CM. These findings provide new perception on the crucial role of m6A modifications in the TME offering valuable insights into potential biomarkers and targeted treatment strategies for CM.
Data availability
The data that supports the findings are deposited in the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) and The cancer GenomeAtlas-Skin Cutaneous Malenoma (TCGA-SKCM) network from UCSC (https://xenabrowser.net).
References
Anderson NM, Simon MC (2020) The tumor microenvironment. Curr Biol 30(16):R921–R925. https://doi.org/10.1016/j.cub.2020.06.081
Arnold M, Holterhues C, Hollestein LM et al (2014) Trends in incidence and predictions of cutaneous melanoma across Europe up to 2015. J Eur Acad Dermatol Venereol 28(9):1170–1178. https://doi.org/10.1111/jdv.12236
Arnold M, Singh D, Laversanne M et al (2022) Global burden of cutaneous melanoma in 2020 and projections to 2040. JAMA Dermatol 158(5):495–503. https://doi.org/10.1001/jamadermatol.2022.0160
Boulias K, Greer EL (2023) Biological roles of adenine methylation in RNA. Nat Rev Genet 24(3):143–160. https://doi.org/10.1038/s41576-022-00534-0
Butler A, Hoffman P, Smibert P et al (2018) Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36(5):411–420. https://doi.org/10.1038/nbt.4096
Cao X, Zhang Y, Jin F et al (2023) Bioinformatic analysis of the mechanism of action of COL3A1 in pan-cancer and design of specific inhibitors for its conformation. Stud Health Technol Inform 308:199–206. https://doi.org/10.3233/SHTI230840
Chen YP, Yin JH, Li WF et al (2020) Single-cell transcriptomics reveals regulators underlying immune cell diversity and immune subtypes associated with prognosis in nasopharyngeal carcinoma. Cell Res 30:1024–1042
Chen Z, Luo Z, Zhang D, Li H, Liu X, Zhu K, Zhang H, Wang Z, Zhou P, Ren J, Zhao A, Zuo Z (2023a) TIGER: a web portal of tumor immunotherapy gene expression resource. Genom Proteom Bioinformat. 21(2):337–348. https://doi.org/10.1016/j.gpb.2022.08.004. (Epub 2022 Aug 29)
Chen M, Zhang Q, Zheng S et al (2023b) Cancer-associated fibroblasts promote migration and invasion of non-small cell lung cancer cells via METTL3-mediated RAC3 m6A modification. Int J Biol Sci 19(5):1616–1632. https://doi.org/10.7150/ijbs.79467
Chong W, Shang L, Liu J et al (2021) m(6)A regulator-based methylation modification patterns characterized by distinct tumor microenvironment immune profiles in colon cancer. Theranostics 11:2201–2217
Chu T, Wang Z, Pe’er D, Danko CG (2022) Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat Cancer. 3(4):505–517. https://doi.org/10.1038/s43018-022-00356-3. (Epub 2022 Apr 25)
de Visser KE, Joyce JA (2023) The evolving tumor microenvironment: from cancer initiation to metastatic outgrowth. Cancer Cell 41(3):374–403. https://doi.org/10.1016/j.ccell.2023.02.016
Du F, Li H, Li Y, Liu Y, Li X, Dang N, Chu Q, Yan J, Fang Z, Wu H, Zhang Z, Zhu X, Li X (2021) Identification of m6A regulator-associated methylation modification clusters and immune profiles in melanoma. Front Cell Dev Biol 9:761134. https://doi.org/10.3389/fcell.2021.761134
Elder DE, Barnhill RL, Bastian BC, Cook MG, de la Fouchardiere A, Gerami P (2018) Melanocytic tumour classification and the pathway concept of melanoma pathogenesis. In: Elder DE, Massi D, Scolyer RA, Willemze R (eds) WHO classification of skin tumours, 4th edn. International Agency for Research on Cancer, Lyon, pp 66–71
Elder DE, Bastian BC, Cree IA, Massi D, Scolyer RA (2020) The 2018 World Health Organization classification of cutaneous, mucosal, and uveal melanoma: detailed analysis of 9 distinct subtypes defined by their evolutionary pathway. Arch Pathol Lab Med 144:500–522
Erdmann F, Lortet-Tieulent J, Schüz J et al (2013) International trends in the incidence of malignant melanoma 1953–2008—are recent generations at higher or lower risk? Int J Cancer 132(2):385–400. https://doi.org/10.1002/ijc.27616
Fidler MM, Gupta S, Soerjomataram I, Ferlay J, Steliarova-Foucher E, Bray F (2017) Cancer incidence and mortality among young adults aged 20–39 years worldwide in 2012: a population-based study. Lancet Oncol 18(12):1579–1589. https://doi.org/10.1016/S1470-2045(17)30677-0
Galbo PM Jr, Zang X, Zheng D (2021) Molecular features of cancer-associated fibroblast subtypes and their implication on cancer pathogenesis, prognosis, and immunotherapy resistance. Clin Cancer Res 27(9):2636–2647. https://doi.org/10.1158/1078-0432.CCR-20-4226
Gaujoux R, Seoighe C (2010) A flexible R package for nonnegative matrix factorization. BMC Bioinformat 11:367. https://doi.org/10.1186/1471-2105-11-367
Guo JC, Liu Z, Yang YJ et al (2021) KDM5B promotes self-renewal of hepatocellular carcinoma cells through the microRNA-448-mediated YTHDF3/ITGA6 axis. J Cell Mol Med 25(13):5949–5962. https://doi.org/10.1111/jcmm.16342
Han S, Zhu L, Zhu Y et al (2021) Targeting ATF4-dependent pro-survival autophagy to synergize glutaminolysis inhibition. Theranostics 11(17):8464–8479. https://doi.org/10.7150/thno.60028
He L, Li H, Wu A, Peng Y, Shu G, Yin G (2019) Functions of N6-methyladenosine and its role in cancer. Mol Cancer 18(1):176. https://doi.org/10.1186/s12943-019-1109-9
Hua YQ, Zhang K, Sheng J et al (2021) NUCB1 suppresses growth and shows additive effects with gemcitabine in pancreatic ductal adenocarcinoma via the unfolded protein response. Front Cell Dev Biol 9:641836. https://doi.org/10.3389/fcell.2021.641836
Huang Z, Kang W, Zhang Q (2022) N6-methyladenosine methylation-related immune biomarkers correlate with clinicopathological characteristics and prognosis in clear cell renal cell carcinoma. Transl Cancer Res. 11(6):1576–1586. https://doi.org/10.21037/tcr-21-1953
Jayawardana K, Schramm SJ, Haydu L et al (2015) Determination of prognosis in metastatic melanoma through integration of clinico-pathologic, mutation, mRNA, microRNA, and protein information. Int J Cancer 136(4):863–874. https://doi.org/10.1002/ijc.29047
Jin S, Guerrero-Juarez CF, Zhang L et al (2021) Inference and analysis of cell-cell communication using Cell Chat. Nat Commun 12:1088
Kumar N, Mishra B, Athar M, Mukhtar S (2021) Inference of gene regulatory network from single-cell transcriptomic data using pySCENIC. Methods Mol Biol 2328:171–182
Leonardi GC, Falzone L, Salemi R et al (2018) Cutaneous melanoma: from pathogenesis to therapy (review). Int J Oncol 52(4):1071–1080. https://doi.org/10.3892/ijo.2018.4287
Liao Q, Shi H, Yang J et al (2024) FTO elicits tumor neovascularization in cancer-associated fibroblasts through eliminating m6A modifications of multiple pro-angiogenic factors. Cancer Lett 592:216911. https://doi.org/10.1016/j.canlet.2024.216911
Liu D, Yang X, Wu X (2021) Tumor immune microenvironment characterization identifies prognosis and immunotherapy-related gene signatures in melanoma. Front Immunol 12:663495. https://doi.org/10.3389/fimmu.2021.663495
Liu C, Wang X, Yang S, Cao S (2023) Research progress of m6A RNA methylation in skin diseases. BioMed Res Int 2023:3091204. https://doi.org/10.1155/2023/3091204
Marzagalli M, Ebelt ND, Manuel ER (2019) Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin Cancer Biol 59:236–250. https://doi.org/10.1016/j.semcancer.2019.08.002
Mo K, Chu Y, Liu Y et al (2023) Targeting hnRNPC suppresses thyroid follicular epithelial cell apoptosis and necroptosis through m6A-modified ATF4 in autoimmune thyroid disease. Pharmacol Res 196:106933. https://doi.org/10.1016/j.phrs.2023.106933
Puram SV, Tirosh I, Parikh AS et al (2017) Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171:1611–24.e24
Qiu X, Mao Q, Tang Y et al (2017) Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14:979–982
Ran Y, Yan Z, Jiang B, Liang P (2023) N6-methyladenosine functions and its role in skin cancer. Exp Dermatol 32(1):4–12. https://doi.org/10.1111/exd.14696
Rebecca VW, Sondak VK, Smalley KS (2012) A brief history of melanoma: from mummies to mutations. Melanoma Res 22(2):114–122. https://doi.org/10.1097/CMR.0b013e328351fa4d
Wang T, Kong S, Tao M, Ju S (2020) The potential role of RNA N6-methyladenosine in cancer progression. Mol Cancer 19(1):88. https://doi.org/10.1186/s12943-020-01204-7
Wang G, Zeng D, Sweren E et al (2023a) m6A RNA methylation correlates with immune microenvironment and immunotherapy response of melanoma. J Invest Dermatol. https://doi.org/10.1016/j.jid.2023.01.027
Wang Q, Shao X, Zhang Y et al (2023b) Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Med 12(10):11149–11165. https://doi.org/10.1002/cam4.5698
Wu Y, He H, Zheng K et al (2024) RNA M6A modification shaping cutaneous melanoma tumor microenvironment and predicting immunotherapy response. Pigment Cell Melanoma Res 37:496–509. https://doi.org/10.1111/pcmr.13170
Yang S, Wei J, Cui YH et al (2019) m6A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun 10(1):2782. https://doi.org/10.1038/s41467-019-10669-0
Yang F, Lin L, Li X, Wen R, Zhang X (2022) Silencing of COL3A1 represses proliferation, migration, invasion, and immune escape of triple negative breast cancer cells via down-regulating PD-L1 expression. Cell Biol Int 46:1959–1969. https://doi.org/10.1002/cbin.11875
Yu J, Chai P, Xie M et al (2021) Histone lactylation drives oncogenesis by facilitating m6A reader protein YTHDF2 expression in ocular melanoma. Genome Biol 22(1):85. https://doi.org/10.1186/s13059-021-02308-z
Yuan L, Shu B, Chen L et al (2017) Overexpression of COL3A1 confers a poor prognosis in human bladder cancer identified by co-expression analysis. Oncotarget 8(41):70508–70520. https://doi.org/10.18632/oncotarget.19733
Zaccara S, Ries RJ, Jaffrey SR (2019) Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol 20:608–624
Zhang J, Song H, Chen C et al (2021) Methyltransferase-like protein 11A promotes migration of cervical cancer cells via up-regulating ELK3. Pharmacol Res 172:105814
Zhang C, Shen H, Yang T et al (2022) A single-cell analysis reveals tumor heterogeneity and immune environment of acral melanoma. Nat Commun 13(1):7250. https://doi.org/10.1038/s41467-022-34877-3
Zhao B, Pritchard JR (2016) Inherited disease genetics improves the identification of cancer-associated genes. PLoS Genet. https://doi.org/10.1371/journal.pgen.1006081
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by ZY, XX, MK, and LL. The first draft of the manuscript was written by MK and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable since all datasets used are available in public databases.
Consent for publication
All authors agree to publish.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, L., Kishengere, M.A., Xu, X. et al. Revealing tumor microenvironment communication through m6A single-cell analysis and elucidating immunotherapeutic potentials for cutaneous melanoma (CM). J Cancer Res Clin Oncol 151, 135 (2025). https://doi.org/10.1007/s00432-025-06176-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00432-025-06176-z