
Abstract
In the last decades the survival of metastatic gastrointestinal (GI) cancer patients could have been significantly extended due to the introduction of targeted- and immunotherapy. However, only the minority of patients will experience long-lasting survival. Hence, novel therapeutics are clearly necessary for GI cancer patients. Molecular high-throughput profiling techniques have revealed potential novel targetable molecular alterations, emphasizing the necessity for tailored therapeutic approaches. Nuclear export proteins, particularly Exportin-1 (XPO1), have emerged as promising targets in cancer therapy due to their crucial role in cellular homeostasis and regulation of key cellular functions. Dysregulation of XPO1-mediated nuclear export leads to the functional loss of tumor suppressors and pro-apoptotic factors, facilitating cancer progression. Selinexor, a XPO1 inhibitor, has shown promising activity in preclinical and clinical studies, particularly in hematological malignancies. However, its efficacy in GI cancers remains underexplored. This review aims to elucidate the functional and pathophysiological role of XPO1 in GI cancers. Despite the potential of XPO1 inhibitors in suppressing cell proliferation and inducing apoptosis, comprehensive molecular landscape data and validation of selective inhibitors in GI cancers are lacking. Targeting XPO1 presents a significant therapeutic potential for the treatment of GI cancer patients. Further research is necessary to fully elucidate the molecular landscape according to XPO1 expression in GI tumors and to validate the efficacy of selective XPO1 inhibitors. These efforts are expected to contribute to the development of more effective and personalized therapeutic strategies for GI cancer patients.
Similar content being viewed by others

Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
Introduction
Despite growing insights into tumor biology and therapeutic improvements in recent decades, the identification of novel targets for personalized cancer therapy remains of great interest. This has provided a significant boost to the field of cancer genetics, which focuses primarily on identifying genes that cause cancer, facilitate its progression, or alter the response to treatments [1, 2]. In recent years, molecular profiling of tumor tissues using novel technologies, such as next-generation sequencing, has revolutionized the therapeutic management of cancer patients, and led to the discovery of new targetable molecular alterations [3]. The resulting challenge is to integrate these alterations into a personalized treatment approach that targets key molecules driving cancer growth and progression, while also offering more precise therapies based on the unique genomic changes of each individual`s tumor.
Targeting nuclear export proteins has emerged as a viable strategy for cancer therapy [4, 5]. These proteins are integral to the transport of macromolecules from the nucleus to the cytoplasm, thereby maintaining cellular homeostasis and regulating essential cellular processes, including gene expression, cell cycle progression, and signal transduction. Dysregulation or aberrant localization of nuclear export proteins often leads to the loss of their functional integrity, contributing to oncogenesis and tumor progression [6,7,8,9]. The localization of tumor suppressor proteins (TSPs) is regulated by the nuclear export protein Exportin-1, also known as chromosome region maintenance 1 (CRM1) or XPO1 [10, 11]. XPO1 is a member of the importin-β superfamily of karyopherins and the major exporter from the nucleus for the majority of proteins, some mRNAs, rRNAs, and snRNAs [10]. Aberrant function of XPO1 (via overexpression or mutations) can enhance the nuclear export of tumor suppressors and pro-apoptotic factors, while retaining oncogenic factors in the nucleus. Thus, inhibiting XPO1-mediated nuclear export can restore the normal function of these proteins and induce cell cycle arrest, apoptosis, and senescence in cancer cells [12]. Several small-molecule inhibitors targeting XPO1 have been developed and tested in preclinical and clinical studies. The most advanced XPO1 inhibitor is selinexor, which has been approved by the FDA for the treatment of relapsed or refractory multiple myeloma (MM) and diffuse large B-cell lymphoma (DLBCL), and is currently being studied in multiple clinical trials for various other malignancies [13,14,15]. Preclinical data support the potential role of XPO1 as a promising therapeutical target also in gastrointestinal (GI) malignancies. Especially in pancreatic ductal adenocarcinoma (PDAC), the inhibition of XPO1 may suppress cell proliferation and induce apoptosis [16, 17]. However, the biological and clinical role of XPO1 in GI cancers is widely unclear and the efficacy of selective inhibitors of nuclear export (also called SINEs or SINE compounds) has yet to be validated.
In this review, we will discuss functional, pathophysiological and clinical implications of XPO1 in GI malignancies.
Main text
Structure of XPO1 gene and protein
The human XPO1 gene is located on chromosome 2p15 [18]. It consists of 62,710 bases and is oriented on the minus strand [19]. The XPO1 gene contains 25 exons and 24 introns [18], and consists of 1071 amino acids [19, 20], which are organized in 20 or 21 HEAT motifs (H1–H21), depending on numbering convention [21,22,23,24]. A HEAT motif comprises two antiparallel α helices, which are linked by a short loop. The XPO1 protein, which has a molecular weight of 120 kDa, is composed of three main domains: the importin-beta N-terminal domain, the exportin-1-like protein domain and the C-terminal domain (Fig. 1a, b) [25]. Since the close proximity of the N-terminal and the C-terminal HEAT, XPO1 has a ring shaped protein form (Fig. 1b) [26]. Between H11 and H12 repeats, there is a hydrophobic groove to bind to nuclear export signal (NES) peptides [24, 26, 27]. XPO1 cargo proteins carry a so-called leucine-rich NES motif [11, 26]. Especially, Cys528 plays a pivotal role in the structural integrity of XPO1 and its ability to anchor cargo proteins during the export process. Furthermore, it is also the target of specific XPO1 inhibitors [28, 29].

a Domains of XPO1 protein: Importin-beta N-terminal domain, Exportin-1-like protein, XPO1 C terminal domain. b Ring shaped structure of XPO1, domains and Cys528 residue. (orange: Importin-beta N-terminal domain, yellow: Exportin-1-like protein, blue: C terminal domain)
Biological and physiological function of XPO1
In eukaryotic cells, ions and small molecules (< 40 kDa) diffuse across the nuclear membrane following a diffusion gradient [30]. Larger molecules (> 40 kDa) require active transport for their translocation between the nucleus and cytoplasm [31]. This process is mediated by the nuclear pore complex (NPC) that facilitates the exchange of hundreds of macromolecules per minute using various transport receptors [11, 30, 32,33,34]. The karyopherin family predominately governs nuclear transport, encompassing two main sub-families: importins and exportins [35, 36]. To date, more than ten different importins and seven distinct exportins have been identified [37], including XPO1, which recognizes leucine-rich hydrophobic NES [13, 36].
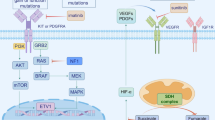
XPO1, initially identified in the yeast Schizosaccharomyces pombe in 1989, was subsequently established as a critical exporter in Schizosacchiaromyces cerevisiae, responsible for protein and mRNA transport from the nucleus to the cytoplasm [30, 33, 35, 38]. XPO1 belongs to the importin ß superfamily of nuclear transport receptors and mediates the nuclear export of NES-containing proteins and RNAs (Fig. 2) [32, 35].

Transport mechanism of XPO1-mediated nuclear export of protein and RNA cargoes Left: Schematic representation of the transport of RNA cargoes by XPO1. Right: Schematic representation of the transport of protein cargoes by XPO1. In both cases, XPO1 forms a complex with RanGTP and the protein cargoes (or RNA cargoes and adapter protein), which is transported through the NPC. The complex then disassembles in the cytoplasm, converting RanGTP to RanGDP and releasing the XPO1 cargoes into the cytoplasm. XPO1 then migrates back into the nucleus and repeats the whole process. Created with BioRender.com
The transport process is powered by the Ran-GTPase, which maintains a Ran-GTP-Ran-GDP gradient across the nuclear envelope [26, 32]. XPO1 binds NES-tagged cargo proteins or RNA adaptor proteins along with Ran-GTP, forming a complex that translocates through the NPC [26, 32]. Once in the cytoplasm, Ran-GTP is hydrolyzed to Ran-GDP by GTPase-activating protein (RanGAP), while RCC1 converts Ran-GDP back to Ran-GTP, allowing XPO1 to dissociate from its cargo and return to the nucleus [26, 30, 32, 33, 35]. XPO1 also has a vital role in spindle assembly, ensuring microtubule stability at kinetochores and maintaining chromosome integrity [39,40,41]. It is essential for microtubule formation from kinetochores and interacts with pericentrin at centrosomes during the cell cycle [41]. The functionality of XPO1 is crucial for cell homeostasis and mitotic regulation [40], and any dysfunction or aberration in XPO1 activity can significantly impair normal cellular operations.
Structural insights into XPO1 alterations and their functional impact
Several studies have focused on the identification of molecular alterations of XPO1 and their clinical implication. For instance, missense substitutions in XPO1 have been reported in chronic lymphocytic leukemia (CLL) [42, 43] and esophageal squamous cell carcinoma (ESCC) [44], and moreover, a heterozygous mutation hotspot (XPO1E571K) was detected in various malignancies, such as primary mediastinal diffuse large B-cell lymphoma (PMBL) and classical Hodgkin's lymphoma (cHL) [45, 46]. So far, the role of this mutation has not been determined and it has not been found to alter the nuclear export function. Of note, no differences in the respond to SINEs were observed in wild-type or XPO1E571K-mutant cells [46, 47].
In a recent study, whole-exome and -genome sequencing was conducted on a large cohort of 42,793 cancer patients, comprising 322 different cancer entities. This comprehensive analysis revealed hotspot mutations in XPO1, with notable mutations at positions XPO1E571, XPO1R749 and XPO1D624 [48]. Among them, the XPO1E571K mutation (NM_003400, chr2:g61718472C > T) was found to be the most common XPO1 mutation in PMBL (33%), cHL (14%), DLBCL (2%) and CLL (3%) [48]. These observations indicate that this gain-of-function mutations transform XPO1 into an oncogenic driver [26, 32]. Although the exact mechanism underlying its oncogenic properties has not been fully understood, the E571K amino acid substitution has been shown to modify the binding preferences of XPO1 for specific nuclear export cargo, possibly mediated by changes in the hydrophobic NES-binding groove [49, 50].
Of note, in ESCC a specific missense mutation (XPO1D624G) has been described [44]. The XPO1D624G mutation leads to a loss of the salt bridge and thus presumably to a decrease in the affinity of interactions. As a result, XPO1 changes its transition from the bound to the unbound state, increasing its export efficiency [44].
Currently, more clinical studies are necessary to determine the clinical potential of XPO1 alterations and its use as a novel biomarker for diagnosis and prognosis.
The role of XPO1 in malignancies
In healthy cells, the nuclear export of TSPs is crucial since their prolonged retention in the nucleus can lead to transcriptional repression in the absence of DNA damage or oncogenic stimuli [51]. TSPs are pivotal in preventing cancer development, progression, and drug resistance when appropriately localized within in the nucleus [52]. They achieve this through multiple mechanisms, including the suppression of cell division, induction of apoptosis, facilitation of DNA damage repair, and inhibition of metastasis [53].
Overexpression of XPO1 leads to an increased inactivation of TSPs that correlates with poor prognosis in various human hematologic and solid malignancies [32, 34, 51], including GI cancers, such as PDACs [54], gastric (GC) [55], and colorectal cancers (CRC) [56]. In silico analysis for XPO1 mRNA expression has also been shown to be increased in all GI cancer entities compared to non-tumor tissues as shown in Fig. 3a [57].

a XPO1 mRNA expression in gastrointestinal cancer tissues compared to non-tumor tissues. b Selective inhibitor of nuclear export (SINE) mechanism. SINE compounds selectively inhibit XPO1, disrupting the nuclear export of cargo proteins and RNAs and thus preventing their transport to the cytoplasm. (abbreviations: ESCA: Esophageal carcinoma, STAD: Stomach adenocarcinoma, COAD: Colon adenocarcinoma, LIHC: Liver hepatocellular carcinoma, CHOL: Cholangiocarcinoma, PAAD: Pancreatic adenocarcinoma) Created with BioRender.com
Consequently, cancer cells proliferate rapidly due to the absence of TSPs and growth-regulating proteins in the nucleus, emphasizing the critical role of XPO1 in maintaining cellular homeostasis.
XPO1 overexpression enhances the export of cargo to the cytoplasm leading to the mislocalization and subsequent inactivation of TSPs. This mislocalization promotes oncogenesis by facilitating cell proliferation, cell division, and inhibition of apoptosis induction [13, 52]. Consequently, cancer cells proliferate rapidly due to the absence of TSPs in maintaining cellular homeostasis [58]. Correlation between increased copy number and XPO1 overexpression was reported in some leukemia and lymphoma subtypes [59], although the mechanisms driving XPO1 overexpression in other malignancies remain largely unexplored.
It has been demonstrated that c-Myc induces the transcriptional activation of XPO1 [60]. Furthermore, the suppression of XPO1 has been evidenced to attenuate the protein expression levels of both c-Myc and epidermal growth factor receptor (EGFR) in diverse malignancies [61,62,63]. This regulatory cascade offers insights into the observed XPO1 overexpression in neoplasms lacking a concurrent increase in genomic copy number. Elevated XPO1 expression fosters tumorigenesis and is correlated with adverse clinical outcomes including poor prognosis, disease progression, and resistance to therapeutic interventions, thereby resulting in diminished overall survival (OS) or progression-free survival (PFS) across various cancer phenotypes [13, 32, 33]. However, the precise molecular mechanisms governing the upregulation of XPO1 remain a subject of ongoing investigation, emphasizing the imperative of elucidating the functional role and molecular alterations associated with this gene [50].
Therapeutics targeting XPO1
The research involving the development of XPO1 inhibitors has started several decades ago, in the beginning of the 90’s when the antifungal antibiotic leptomycin B isolated from a strain of Streptomyces, was identified as a potential XPO1 inhibitor [64] that covalently and irreversibly binds to C528 and occupies its NES-binding groove [65]. The first clinical trial with leptomycin B was conducted in patients with advanced refractory cancers, but the trial demonstrated severe toxicities without significant clinical response. Thus, no further studies were conducted [66]. After this, several researcher started to develop semisynthetic and synthetic inhibitors of XPO1. SINE compounds are molecules that promote the intranuclear accumulation of TSPs [51]. This restoration of TSP function disrupts cancer cell growth. Both leptomycin B and SINE inhibitors bind to the cysteine residue Cysteine-528 in the cargo binding pocket of the XPO1 protein, thereby inhibiting its activity [28, 64]. Unlike leptomycin B, the binding of XPO1 by SINE inhibitors is reversible, which reduces toxicity and enhances treatment tolerability [67].
SINE compounds are a group of structurally related small molecule inhibitors (Fig. 3b), including KPT-185, KPT-276, KPT-335 (verdinexor), KPT-330 (selinexor), KPT-8602 (eltanexor), KPT-251 and SL-801 (felezonexor). Hindered by poor pharmacokinetics, only a few SINEs (KPT-335, KPT-330, KPT-8602 and SL-801) have entered clinical trials and the most evaluated component to this date is KPT-330 (also known as selinexor).
Selinexor is an orally administered, first-in-class, highly specific, slowly reversible SINE [68]. Numerous clinical trials have demonstrated the efficacy of selinexor in the treatment of hematological malignancies, including MM, DLBCL and acute myeloid leukemia [69,70,71]. Moreover, it has also displayed efficacy in solid tumors, such as prostate, ovarian, and CRC [72,73,74]. The efficacy of selinexor was demonstrated in patients with relapsed and refractory MM by achieving significant responses and prolonged survival rates [75]. Subsequently, FDA and EMA approved selinexor for the treatment of refractory and relapsed MM. Moreover, selinexor has also been approved for patients with relapsed and refractory DLBCL by the FDA [15, 76].
The therapeutic potential of SINEs and their application are continuously evolving. Identification of prognostic biomarkers, refining dose regimens and understanding potential mechanisms of synergy are crucial for successful integration of these drugs into standard cancer treatment algorithms. Another important aspect to be considered is the safety and adverse effects of SINE inhibitors. For instance, among the most common reported side effects of selinexor included gastrointestinal (i.e. nausea, diarrhea) and hematological (i.e. thrombocytopenia, anemia) toxicities [77,78,79]. Clinical trials including close monitoring, dose modifications and detailed investigation of side effects of SINE's alone or in combination regimes are crucial to optimize patients care and ensure patient well-being during treatment. At the time of writing, a search for clinical studies that are investigating the efficacy of various SINE inhibitors and were registered in the ClinicalTrials.gov database yielded a total of 142 trials (Table 1). The majority of clinical trials listed are predominantly conducted in hematological malignancies, confirming the research emphasis on their therapeutic potential within these oncological malignances. Out of the listed studies, 12 trials are dedicated to investigate the role of SINE inhibitors in GI tumors. This research imbalance indicates an urgent need for expanded studies into GI and other solid tumors to truly determine the efficacy of SINE inhibitors in cancer therapy, both individually and in synergy with other therapeutic agents, and their ability to surmount the challenge of drug resistance.
XPO1 in cancer chemoresistance and overcoming drug resistance through SINE Drug resistance is a constant challenge for cancer patients and is often considered the main barrier to cure. Many cancer patients develop resistance to traditional chemotherapy or targeted therapies, thereby limiting further treatment options, which underlines the need for investigating and implementing combinational therapies in early stages of the disease [80,81,82]. In most cases, an initial positive response to one or multiple chemotherapies tends to diminish within several months to years, due to the emergence of secondary resistance mechanisms [13, 34]. Among the various factors contributing to resistance, modification of nuclear proteins was found to play an important role [34, 83, 84]. Several investigators have shown that XPO1 overexpression facilitates the trigger of drug resistance mechanisms by mislocalization of proteins that function as drug targets [34, 83, 84]. The potential of SINE inhibitors to overcome the resistance upon systemic therapeutics was reported in various studies [13, 85]. For instance, topoisomerases are important targets in many cancer chemotherapy regimens due to their distinct cellular functions [34]. Resistance to the topoisomerase II α inhibitor anthracycline is conferred by the misplacement of the transcription inhibitor E2F7 and the topoisomerase topo II α into the cytosol. Inhibition of XPO1 keeps E2F7 and topo II alpha in the cell nucleus, thereby restoring anthracycline sensitivity [34, 86]. A significant correlation between XPO1 overexpression and platinum resistance in ovarian cancer patients has also been observed [87]. Targeting XPO1 by using KPT-185 or selinexor has demonstrated the ability to enhance tumor killing, inhibit cellular proliferation, and overcome platinum resistance [13, 88].
In a recent publication, Wang et al. found that XPO1 was overexpressed in hepatocellular carcinoma (HCC) and was associated with worse survival outcomes [89]. When XPO1 was knocked down, it inhibited the migration and proliferation of HCC cells. It also reduced their resistance to the multi-kinase inhibitor sorafenib and effectively suppressed tumor growth [89].
Importantly, several studies demonstrated that XPO1 inhibition is effective independently of the TP53 mutational status [90,91,92]. Deng et al. found that TP53 mutations predict resistance to selinexor and that the resistance was overcome after combined treatment with selinexor and BET inhibitor in DLBCL [93], while in triple negative breast cancer preclinical model [94] the synergistic effect of selinexor with olaparib was confirmed regardless of the mutation status of BRCA1 [95]. However, in clinical practice, combining multiple drugs for intensive cancer treatment faces various challenges due to inconsistent pharmacokinetics among the therapeutic agents. The recent achievements in nanomedicine provide an innovative solution by encapsulating multiple drugs within a nano-carrier [96, 97]. In a proof-of-concept study, researchers created a targeted therapy by inhibiting contemporary both XPO1 and ataxia telangiectasia mutated-Rad3-related (ATR). This treatment demonstrated extraordinary biocompatibility and enhanced the therapeutic effect, thus, providing a co-delivery strategy for liver cancer treatment [98]. These data indicate the urgent need for further investigations to enable more precise and personalized selinexor-based/combination cancer treatment strategies. However, until now, the functional and prognostic role of XPO1 in GI cancers remains somewhat unclear. In the next section we will summarize the current state of knowledge regarding XPO1 in the context of GI cancers.
The role of XPO1 in GI cancers
Esophageal cancer (EC)
Several studies have shown that both XPO1 mRNA and protein are overexpressed in EC [44, 52, 58, 99]. Interestingly, the localization of XPO1 expression was altered in tumor compared to healthy tissue. While in healthy tissue XPO1 is solely expressed in the nucleus, in tumors, the expression was found in the nucleus and in the cytoplasm [58]. While, Van der Watt et al. described no correlation between XPO1 expression and tumor stage [58], Yang et al. reported a significant association between XPO1 overexpression and histological differentiation, lymph node metastasis, tumor size [52]. The inhibition of XPO1 resulted in decreased cell proliferation as well as apoptosis in various EC models [44, 52, 58]. Knockdown of XPO1 revealed a decrease in cellular viability and an induction of apoptosis [52]. Moreover, high XPO1 expression also impacts the survival of EC patients. According to data from The Cancer Genome Atlas (TCGA), patients with EC with high XPO1 mRNA expression have a significantly higher survival probability (Fig. 4). However, other studies have reported that XPO1 overexpression is associated with shorter OS and event-free survival [52, 58]. Therefore, further studies are clearly needed to answer the question whether XPO1 is a prognostic marker in EC patients.

Data (mRNA & clinical data) was acquired from TCGA and cBioportal using the R packages TCGA biolinks and cbioportalR, respectively. Counts (mRNA) were normalized using the within & between lane normalization from the EDASeq package. Ideal cut-offs for XPO1 high and low tumors were determined iteratively using the survival package to choose the cut-off with the largest difference in survival. P values reported concern the difference in survival and are from a logrank-test. Differential gene expression was analyzed using the limma package – plots show the log2-fold change between groups on the x-axis and the log10(p-value) on the y-axis. The large blue dot represents XPO1. The results shown here are in whole based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. (Abbreviations: TCGA: The Cancer Genome Atlas, COAD: colon adenocarcinoma, ESCA: esophageal carcinoma, STAD: stomach adenocarcinoma, LIHC: liver hepatocellular carcinoma, PAAD: pancreatic adenocarcinoma, CHOL: cholangiocarcinoma, LogFC: log2-fold change)
So far, only a few reports have investigated XPO1 inhibitors in clinical and preclinical studies in patients with EC (Table 1). In a multi-arm phase Ib study (NCT02419495) 6 patients with advanced or metastatic solid tumors (two patients with EC) were included [100]. The safety and tolerability of selinexor was validated when given in combination with the standard chemotherapy drugs carboplatin and paclitaxel. One patient showed a partial response, whereas the other patient was excluded from the study before treatment start [100]. Two further clinical trials (NCT02283359, NCT02213133) have been investigating the safety and efficacy of selinexor in combination with standard of care chemotherapy in EC patients. The first trial (NCT02283359) examined the effect of selinexor in combination with irinotecan in adenocarcinoma of stomach and distal esophagus. However, the study enrolled only three patients and was terminated without any results presented or published. The other study (NCT02213133) investigated selinexor treatment in cancer patients with squamous cell histologies (including EC). However, none of the three EC patients enrolled completed the study.
Gastric cancer (GC)
Numerous studies have shown that XPO1 is overexpressed in GC samples compared to normal tissue [55, 101,102,103]. A correlation between XPO1 expression and TNM stage, Her-2 status, metastasis and tumor progression has also been demonstrated. Moreover, a high XPO1 expression correlated with poor survival in GC [102, 103]. For instance, Zhou et al. reported that 4-year survival rates were better for XPO1-low compared to XPO1-high expressors [103]. This is also confirmed by an analysis conducted using the TCGA. We observed that a higher XPO1 mRNA expression correlated significantly with a lower OS in GC patients compared to low XPO1 mRNA expressors (Fig. 4). Sexton et al. investigated the impact of inhibiting XPO1 mRNA via siRNA (siXPO1) and revealed that inhibiting XPO1 leads to a significant decrease of cancer cell proliferation and colony formation [101]. Thus, XPO1 might represent a potential target for GC patients.
In one of the first reports regarding the efficacy of SINEs in GC cell lines, Subhash et al. showed that selinexor had the greatest efficacy compared to other SINE components tested [55]. They demonstrated that selinexor activity is dependent on nuclear accumulation of p53. Nuclear retention of p53 is followed by an upregulation of p21, suggesting a restoration of p53 tumor suppressor function. In GC, in vivo and in vitro models, Sexton and colleagues established synergism of selinexor and other SINE compounds enhancing the efficacy of nab-paclitaxel [101]. They also demonstrated that SINE compounds can alter the expression of non-coding RNAs and reported a significant downregulation of oncogenic miR-33b-3p by SINE treatment.
We identified two studies involving patients with GC (Table 1). The first one, a phase I trial of selinexor (NCT02078349) was designed to evaluate the safety, pharmacokinetics, and pharmacodynamics of selinexor. This study has been terminated, but the outcomes concerning the GC patients who were enrolled have not yet been published. The second trial (NCT02283359) aimed to assess the efficacy of selinexor in combination with irinotecan for the treatment of GC and distal EC. Regrettably, none of the three GC patients enrolled have completed the study.
The lack of published data from these trials underscores the inherent challenges in progressing clinical research for GC patients. This emphasizes the critical need for ongoing efforts to elucidate the role of XPO1 in the pathogenesis GC, thereby providing essential insights for the development of future therapeutic strategies including XPO1 inhibitors.
Hepatocellular carcinoma (HCC)
Only a few studies have examined the influence of XPO1 expression in HCC. Zheng et al. reported that the expression of XPO1 is increased in HCC compared with normal liver tissue, and that inhibition with SINE compounds leads to growth inhibition and cell cycle arrest [105]. Similarly, Chen et al. showed that mRNA levels of XPO1 are significantly upregulated in HCC tissues compared to normal liver tissues [106]. XPO1 was associated with clinic pathological parameters, such as tumor grading, clinical stage and prognosis of HCC. High levels of XPO1 mRNA were more frequently observed in undifferentiated tumors and correlated with poor prognosis in HCC patients [106, 107]. Data from TCGA indicate that a lower mRNA expression of XPO1 is associated with a worse OS compared to high expressors (Fig. 4).
The potential of selinexor and other SINE in HCC is yet to be explored. In a preclinical study [105], selinexor reduced the viability of HCC cell lines in vitro and inhibited tumor growth in HCC xenograft murine model. Treatment with selinexor up-regulated the expression of the TSPs p53 and p27 and the pro-apoptotic protein PUMA, and reduced the expression of the HCC promoting proteins c-Myc and c-Met [105].
A recent study investigated the effect of KPT-330 and the CDK4/6 inhibitor palbociclib in HCC cell lines [108]. Experiments have revealed that this combination treatment induces cellular senescence. In vivo, combination treatment with palbociclib and KPT-8602 (a structural analog of KPT-330) led to a significant reduction in tumor burden and improved survival in the HCC mouse model [108]. Deutzmann et al. demonstrated in vitro that after XPO1 inhibition with selinexor c-Myc-driven HCC showed an increase in apoptosis [109]. Moreover, XPO1 inhibition decreased the tumor volume in mice about 95% compared to control mice. Of note, Zhang et al. recently found elevated DBF4 expression in HCC that served as an independent prognostic factor [110]. Mechanistically, DBF4 complexed with CDC7 to form a DBF4-dependent kinase (DDK) and activated STAT3 signaling through XPO1-mediated nuclear export. Combining the DBF4 inhibitor XL413 with anti-PD-1 therapy suppressed HCC growth and prolonged survival in mice, demonstrating the involvement of XPO1 in this regulatory network and the potential of targeting DDK to enhance HCC immunotherapy.
A recent study combined selinexor with radiation in a preclinical model in human osteosarcoma and human HCCs [111]. The authors found enhanced radiation response in both cell lines by decreasing the intranuclear HIF-1α protein levels [111]. Nevertheless, more studies, especially clinical trials, are clearly necessary to understand the underlying mechanisms on irradiated cells and the significance of inhibition of DNA damage repair in cells treated with selinexor. Currently, only one clinical trial involving HCC patients and selinexor in combination with bevacizumab and atezolizumab is registered on ClinicalTrials.gov (NCT05093608). However, this study was terminated prematurely, having enrolled just two participants, and consequently, no results have been reported yet.
Gallbladder cancer (GBC) & cholangiocarcinoma (CC)
Data regarding XPO1 in GBC and CC remains limited. To the best of our knowledge, only one study described the role of XPO1 in GBC. Zhao et al. reported that XPO1 mRNA expression was significantly increased in GBC tissue compared to corresponding non-cancerous tissues [112]. Moreover, a correlation of XPO1 expression and clinic-pathological data, such as lymph node metastasis and higher TNM stage, was observed. OS after curative surgery in patients with high XPO1 expression was significantly shorter compared with patients with low XPO1 levels [112]. The investigators also reported that inhibition of XPO1 by selinexor led to a significant reduction of cell proliferation and colony formation as well as an increase of apoptosis. The XPO1 inhibitor KPT-330 induced nuclear accumulation of p53 and activated the p53/mTOR signaling to regulate cell autophagy. The in vivo experiment showed that administration of selinexor suppressed the growth of GBC cells in a mouse xenograft model [112]. These results indicate selinexor as a potential treatment for GBC and highlights the need to further investigate its effects in humans.
Limited data on the role of XPO1 in CC is available [113, 114]. According to the TCGA, higher XPO1 mRNA expression appears to be associated with better OS in CC patients compared to those with lower expression, although this difference is not statistically significant (Fig. 4). Bioinformatic analysis utilizing two public open accessible database revealed high XPO1 expression in various tumors including CC, which was associated with poor prognosis [114]. In CC, XPO1 expression is significantly increased compared to healthy controls. Moreover, XPO1 positively correlated with tumor grade and clinical stage [113]. Treatment with the XPO1 inhibitor KPT-330 demonstrated inhibition effects on CC cells [114]. Furthermore, XPO1 knockdown resulted in significantly impaired colony forming ability, reduced viability and G1 cell cycle arrest indicating apoptosis [113, 114]. Conversely, overexpression of XPO1 led to a significantly higher proliferation of CC cells suggesting XPO1 promotes proliferation and progression. Consequently, XPO1 emerges as a potential novel target for the diagnosis, treatment, and prognosis of this cancer type [113, 114]. However, currently no clinical trial was initiated for patients with CC.
Pancreatic ductal adenocarinoma (PDAC)
In 2009, Huang et al. found that XPO1 is overexpressed in over 55% of PDACs compared to normal tissue [10, 69]. High XPO1 levels were linked to larger tumor size, lymph node involvement, metastasis, and poorer OS and PFS, suggesting XPO1 as a potential prognostic marker for PDACs [115]. Azmi et al. showed that XPO1 regulates the shuttling of mature microRNAs (miRNAs), including tumor-suppressive miR-145 [17]. Inhibiting XPO1 upregulates miR-145, which downregulates its target signaling pathways, thus inhibiting PDAC cell proliferation and migration. The lack of miR-145 in PDAC cells contributes to the cancer's aggressiveness, and XPO1 inhibition mitigates this by increasing miR-145 levels and suppressing its target pathways [17].
Saulino et al. described that in the majority of PDACs (86%) a positive staining for XPO1 was detected [116]. The localization of the XPO1 expression was mainly in the nucleus. A positive correlation between XPO1 and survivin expression was observed. In a separate study, researchers examined the expression of XPO1 in tissues from normal, primary, and metastatic patients [117]. The findings revealed a notable rise in XPO1 expression as the tumor stage advanced. Additionally, a significant correlation was observed between pathological lymph node status and the expression of XPO1. A shorter OS was observed for high vs low XPO1 expressors [117]. This result was confirmed by another study, where survival was also shorter in XPO1 high compared to the XPO1 low group [118]. These contradictory findings from the TCGA indicate that high expression of XPO1 is significantly associated with extended OS in PDAC patients compared to patients with low XPO1 expression (Fig. 4). Altogether, several studies have shown that overexpression of XPO1 in PDACs is associated with poor prognosis, most likely due to the inactivation of TSPs [17, 115,116,117,118]. Thus, it would be very important to analyze how XPO1 expression might predict prognosis and response to clinical SINE compounds. So far, several studies reported promising pre-clinical data using XPO1 inhibitors in PDACs. Azmi et al. reported that SINE compounds (KPT-185, KPT-127, KPT-205, and KPT-227) blocked PDAC cell proliferation, induced apoptosis, and retained important TSPs, such as p53, p73, FOXO and prostate apoptosis response-4 (PAR-4) in the nucleus [16]. Moreover, selinexor disrupted the XPO1-PAR-4 interaction and activated PAR-4 signaling, one of the critical pro-apoptotic pathways in PDAC [119, 120], resulting in enhanced apoptosis. The oral administration of the drug in mice reduced the growth of xenograft tumors without major toxicities revealing a promising novel treatment in PDAC [16].
Further, the combined use of SINE and gemcitabine in PDACs was investigated and their combination resulted in a synergistic effect, drastically reducing the growth of PDAC cells in vitro and reducing tumor volume in vivo in an orthotopic mouse model, through depletion of the anti-apoptotic proteins and induction of apoptosis [121]. Moreover, selinexor in combination with gemcitabine and nab-paclitaxel revealed the suppression of spheroid formation in cancer stem cells (CSC), induced spheroid disintegration and blocked CSC growth in a xenograft model [72]. In line with these results, another study showed that the combined treatment of selinexor with gemcitabine and paclitaxel resulted in a prolonged survival time of mice [122].
Since the tumor progression from normal ductal epithelium to PDAC requires sequential genetic alterations initiated by the KRAS mutation, and considering that nearly 90% of conventional PDACs show an oncogenic KRAS mutation, the selinexor effect was analyzed in combination with KRASG12C inhibitors in a further PDAC model [123]. Khan et al. demonstrated that in KRASG12C inhibitor-resistant cells, the addition of selinexor to the KRASG12C inhibitor significantly decreased cell colony formation in vitro and inhibited tumor formation in mice [123]. These studies highlight novel combination therapies for PDAC and provide a founded rationale for the use of selinexor in a clinical setting to improve patient outcomes and to prevent the development of resistance to the standard monotherapy. Currently, two clinical trials are investigating the utilization of selinexor and its potential for innovative combination therapeutic strategies for PDAC patients (Table 1). In a phase I trial (NCT02178436), nine patients with metastatic PDAC were treated with the combination of selinexor, gemcitabine and nab-paclitaxel and a partial response and a stable disease were observed in two PDAC patients [72]. A phase IB study is currently ongoing (NCT02419495) with the objective of assessing the safety profile of selinexor when used in combination with various established chemotherapy or immunotherapy in patients with advanced malignant solid neoplasms, including colorectal cancer, EC and PDAC. Nonetheless, the cohort size of PDAC patients enrolled in this trial is still very low.
Despite the encouraging outcomes derived from pre-clinical investigations and the initial clinical phase, further validation of these results, alongside comprehensive studies to evaluate the safety and therapeutic efficacy of selinexor in PDAC patients, are still pending.
Colorectal "Cancer" instead of "carcinoma" (CRC)
Colorectal cancer (CRC) is another tumor in which XPO1 is frequently overexpressed. However, data on the role of XPO1 overexpression in CRC remains unclear [56, 124, 125]. Elevated XPO1 activity can increase oncogenic activity through mislocalization of proteins like p27, p53 and survivin and, thus dysfunction in cancer cells and a general association with poor outcome [56, 125]. According to the TCGA, high mRNA expression of XPO1 leads to a significantly better OS compared to low expression of XPO1 in CRC patients (Fig. 4). No correlation was observed between XPO1 overexpression and tumor size [56]. XPO1 was strongly expressed in more than 50% of samples (n = 40). Especially nuclear expression was detected in more than half and cytoplasmic expression in 37% [56]. Inhibition of XPO1 leads to impaired proliferation in 2D and 3D cell cultures [56, 124]. Moreover, after inhibition of XPO1, apoptosis was induced in these cells [56, 124, 126]. Inoue and colleagues reported that cell cycle arrest was due to expression of specific DNA damage response (DDR) genes [124]. Conversely, it has been demonstrated that XPO1 overexpression and loss of p53 was mostly observed in poorly differentiated tumors, advanced tumor stage and lymph node metastasis [56, 125].
XPO1 is also responsible for the transportation and subcellular distribution of survivin between the nucleus and cytoplasm [104]. Survivin is an inhibitor of the apoptosis protein (IAP) and is overexpressed in most human tumors [104, 127]. Survivin is usually undetectable or expressed at very low levels in normal cells and mainly produced by cancer cells, therefor making it a putative tumor marker [127]. A significant positive correlation between XPO1 and survivin expression was seen in CRC [104]. Overexpression of survivin in cytoplasm may protect cancer cells by inhibiting apoptosis and thus promotes tumor progression [104].
Several preclinical and clinical studies have demonstrated the therapeutic potential of XPO1 inhibition mediated by selinexor in CRC. Aladhraei et al. reported that selinexor reduced cancer cells proliferation in a dose- and time-dependent manner and significantly inhibited-colon cancer growth in vitro [56]. A synergistic effect was observed when combining selinexor with the proteasome inhibitor bortezomib on wild-type p53 CRC cells, as reported by Wu et al. [128]. The results were confirmed in vivo, indicating that p53 is a major mediator of the synergistic cytotoxic effect of bortezomib and selinexor.
Moreover, selinexor synergistically enhanced the radiation response in preclinical models of CRC [129]. Both in CRC cell lines and xenograft tumor models, combination treatment of selinexor and radiotherapy resulted in a decrease of proliferation and reduction of the tumor size by promoting nuclear survivin accumulation and induction of apoptosis [129].
Building on the foundation of these promising preclinical studies, a path has been established for the initiation of clinical trials targeting CRC patients. Employing the previously delineated search parameters, our investigation on ClinicalTrials.gov identified seven clinical studies involving CRC patients (Table 1). In a phase I trial (NCT01607905), the safety and efficacy of selinexor were assessed in a cohort of 189 pretreated tumor patients, including 59 CRC patients [130]. The phase II dose of selinexor established at 35 mg/m2 and administered bi-weekly was deemed safe and well-tolerated [130]. Notably, selinexor showed a disease control rate of 44% [130]. It was established based on this study, that selinexor dosing interruptions alone are likely sufficient for treating selinexor-induced thrombocytopenia [131]. The treatment of selinexor in combination with multiple standard chemotherapy or immunotherapy agents is under investigation in a phase IB trial (NCT02419495) in patients with advanced malignancies, including CRC patients. Another clinical trial (NCT04854434) is currently investigating the efficacy and safety of selinexor, both as a monotherapy and in conjunction with pembrolizumab, in participants with advanced or metastatic colorectal cancer. Additionally, the pharmacokinetics and pharmacodynamics of selinexor have been validated in Asian populations through a further phase I trial (NCT02078349). This study involved screening patients for approximately 2800 COSMIC mutations across 50 cancer genes, including those in the RAS/RAF and PIK3CA/AKT/mTOR pathway. The results indicated that 40% of CRC patients harboring mutations in the RAS/AKT pathway achieved a disease control rate exceeding three months, in contrast to 12.5% of CRC patients with no mutations in the respective genes [132].
Lastly, the investigation of safety, tolerability, and efficacy of KPT-8602 (eltanexor), another SINE, is currently ongoing. This study (NCT02649790) includes participants with relapsed/refractory cancer indications, among them also patients with metastatic CRC.
In light of these findings, it remains crucial to advance additional clinical investigations in CRC, aiming to validate the promising therapeutic potential of selinexor and refine treatment approaches for better patient outcomes.
Gastrointestinal stromal tumors (GIST) & neuroendocrine neoplasia (NEN) of the gastrointestinal tract (GIT)
The relevance of nuclear exporters in GIST or gastroenteropancreatic NEN is unclear and only a handful of research articles have been published on this topic. Nakayama et al. investigated the efficacy of selinexor in vitro and in vivo using 17 cell lines and 9 sarcoma xenograft models including GIST [90]. In GIST cells with cKIT mutations, selinexor induced G1-arrest independent of attenuation of phosphorylation of KIT, AKT, or MAPK, indicating the potential of selinexor and thereby also highlighting the need for further investigation, potentially also for the use of selinexor in clinical trials targeting various sarcoma subtypes. A further clinical trial (NCT04138381) is analyzing the maximum tolerated dose and the recommended phase II doses of selinexor in combination with imatinib among unresectable and/or metastatic GIST patients with prior failure to at least imatinib for advanced/metastatic disease. However, there are no data regarding the use of selinexor in GIST patients.
Regarding NEN and XPO1, the list of publications is even more limited. The ten-eleven translocation (TET) protein family influences epigenetics, initiates demethylation pathways and is associated with tumorigenesis [133, 134]. In 2018, Barazheghi et al. showed a loss of nuclear localization of TET2 in small intestine neuroendocrine tumors (SI-NETs) and demonstrated in a cell culture experiment with SI-NET cell lines (KRJ-I and CNDT2.5) that incubation of these cell lines with leptomycin B led to a retention of TET2 in the nucleus and a reduction of it in the cytoplasm [135], implicating XPO1 as a potential therapeutic target.
Conclusion
Disruption of nuclear export processes compromises various cellular functions crucial for inhibiting oncogenesis and restraining tumor growth. Extensive investigations have been undertaken to identify and characterize the pivotal molecular entities governing the bidirectional exchange between the nucleus and cytoplasm [4]. These endeavors have yielded invaluable insights, laying the groundwork for targeted drug development initiatives aimed at modulating key factors orchestrating nuclear export, prominently exemplified by XPO1, as delineated in this review. The emergent body of knowledge not only elucidates novel facets of cancer pathogenesis but also revolutionizes therapeutic paradigms, facilitating the design of precision medicines tailored to individual patient profiles. Selective inhibition of XPO1 holds promise in rectifying intracellular imbalances, thereby impeding tumorigenesis. The versatility of XPO1 inhibitors is underscored by their capacity to synergistically potentiate the anticancer efficacy of extant therapies and potentially surmount resistance to conventional modalities, thereby fostering improved clinical outcomes [121, 136].
The preeminent XPO1 inhibitor under investigation is selinexor, which has shown promising outcomes in both hematologic and solid malignancies. While other SINEs, such as verdinexor, eltanexor, KPT-9274, and KPT-8602, are undergoing clinical trials at various stages, the focus on GI tumors in these trials remains limited. This paucity of clinical research poses a barrier to comprehensively understanding selinexor's efficacy, safety, and optimal dosing and combination regimens in the realm of GI malignancies, necessitating further targeted investigation. The establishment of ideal conditions for achieving maximal anti-tumor effects with minimal toxicity is paramount for delineating the role of SINEs in GI cancers. This imperative is underscored by the recognition that treatment optimization will necessitate tailored regimens based on the utilization of XPO1 inhibitors across diverse patient cohorts. Furthermore, the identification of specific prognostic markers holds potential for discerning patient subsets likely to derive the greatest benefit from XPO1 inhibition. Achieving this objective mandates a nuanced comprehension of the genetic and molecular profiles characterizing tumors, facilitating the precision matching of patients with their most efficacious treatment modalities.
Data Availability
No datasets were generated or analysed during the current study.
Abbreviations
- ATR:
-
Ataxia telangiectasia mutated-Rad3-related
- BCR:
-
B-cell receptor
- BTK:
-
Bruton tyrosine kinase
- CC:
-
Cholangiocarcinoma
- cHL:
-
Classical Hodgkin's lymphoma
- CLL:
-
Lymphocytic leukemia
- CRC:
-
Colorectal cancer
- CRM1:
-
Chromosome region maintenance 1
- CSC:
-
Cancer stem cells
- DCR:
-
Disease control rates
- DDK:
-
DBF4-dependent kinase
- DDR:
-
DNA damage response
- DLBCL:
-
Diffuse large b-cell lymphoma
- EC:
-
Esophageal cancer
- EGFR:
-
Epidermal growth factor receptor
- ESCC:
-
Esophageal squamous cell carcinoma
- GBC:
-
Gallbladder cancer
- GC:
-
Gastric cancer
- GI:
-
Gastrointestinal
- GIT:
-
Gastrointestinal tract
- GIST:
-
Gastrointestinal stromal tumors
- HCC:
-
Hepatocellular carcinoma
- IAP:
-
Inhibitor of the apoptosis protein
- MM:
-
Multiple myeloma
- miRNAs:
-
MicroRNAs
- NES:
-
Nuclear export signal
- NEN:
-
Neuroendocrine neoplasia
- NLS:
-
Nuclear localization signal
- NPC:
-
Nuclear pore complex
- OS:
-
Overall survival
- PAR-4:
-
Prostate apoptosis response-4
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PFS:
-
Progression-free survival
- PMBL:
-
Primary mediastinal diffuse large b-cell lymphoma
- Ran:
-
Ras-related nuclear protein
- RanGAP:
-
GTPase-activating protein
- RCC1:
-
Regulator of chromosome condensation 1
- SINE:
-
Selective inhibitors of nuclear export
- SI-NETs:
-
Small intestine neuroendocrine tumors
- TET:
-
Ten-eleven translocation
- TSPs:
-
Tumor suppressor proteins
- XPO1:
-
Exportin-1
References
Chen HZ, Bonneville R, Roychowdhury S (2019) Implementing precision cancer medicine in the genomic era. Semin Cancer Biol 55:16–27. https://doi.org/10.1016/j.semcancer.2018.05.009
Hyman DM, Taylor BS, Baselga J (2017) Implementing genome-driven oncology. Cell 168(4):584–599. https://doi.org/10.1016/j.cell.2016.12.015
Zimmer K, Kocher F, Spizzo G, Salem M, Gastl G, Seeber A (2019) Treatment according to molecular profiling in relapsed/refractory cancer patients: a review focusing on latest profiling studies. Comput Struct Biotechnol J 17:447–453. https://doi.org/10.1016/j.csbj.2019.03.012
Gravina GL, Senapedis W, McCauley D, Baloglu E, Shacham S, Festuccia C (2014) Nucleo-cytoplasmic transport as a therapeutic target of cancer. J Hematol Oncol 7(85):9. https://doi.org/10.1186/s13045-014-0085-1
Kosyna FK, Depping R (2018) Controlling the gatekeeper: therapeutic targeting of nuclear transport. Cells 7(11). ARTN 221. https://doi.org/10.3390/cells7110221.
Nguyen KT, Holloway MP, Altura RA (2012) The CRM1 nuclear export protein in normal development and disease. Int J Biochem Mol Biol 3(2):137–151
Yang Y, Guo L, Chen L, Gong B, Jia D, Sun Q (2023) Nuclear transport proteins: structure, function, and disease relevance. Signal Transduct Target Ther 8(1):425. https://doi.org/10.1038/s41392-023-01649-4
Azmi AS (2014) The evolving role of nuclear transporters in cancer. Semin Cancer Biol 27:1–2. https://doi.org/10.1016/j.semcancer.2014.04.011
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. https://doi.org/10.1016/j.cell.2011.02.013
Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M et al (1997) CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature 390(6657):308–311. https://doi.org/10.1038/36894
Stade K, Ford CS, Guthrie C, Weis K (1997) Exportin 1 (Crm1p) is an essential nuclear export factor. Cell 90:10
Dickmanns A, Monecke T, Ficner R (2015) Structural basis of targeting the exportin CRM1 in cancer. Cells 4(3):538–568. https://doi.org/10.3390/cells4030538
Azmi AS, Uddin MH, Mohammad RM (2021) The nuclear export protein XPO1—from biology to targeted therapy. Nat Rev Clin Oncol 18(3):152–169. https://doi.org/10.1038/s41571-020-00442-4
Kim E, Mordovkina DA, Sorokin A (2022) Targeting XPO1-dependent nuclear export in cancer. Biochemistry (Mosc) 87(Suppl 1):S178–S170. https://doi.org/10.1134/S0006297922140140
Kasamon YL, Price LSL, Okusanya OO, Richardson NC, Li RJ, Ma L et al (2021) FDA approval summary: selinexor for relapsed or refractory diffuse large B-cell lymphoma. Oncologist 26(10):879–886. https://doi.org/10.1002/onco.13859
Azmi AS, Aboukameel A, Bao B, Sarkar FH, Philip PA, Kauffman M et al (2013) Selective inhibitors of nuclear export block pancreatic cancer cell proliferation and reduce tumor growth in mice. Gastroenterology 144(2):447–456. https://doi.org/10.1053/j.gastro.2012.10.036
Azmi AS, Li Y, Muqbil I, Aboukameel A, Senapedis W, Baloglu E et al (2017) Exportin 1 (XPO1) inhibition leads to restoration of tumor suppressor miR-145 and consequent suppression of pancreatic cancer cell proliferation and migration. Oncotarget 8(47):12
The human genome browser at UCSC (2002). Genome Res.
Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S et al (2016) The GeneCards Suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics 54(1):30-31. https://doi.org/10.1002/cpbi.5.
Cunningham F, Allen JE, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM et al (2022) Ensembl 2022. Nucleic Acids Res 50(D1):D988–D995. https://doi.org/10.1093/nar/gkab1049
Monecke T, Güttler T, Neumann P, Dickmanns A, Görlich D, Ficner R (2009) Crystal structure of the nuclear export receptor CRM1 in complex with Snurportin1 and RanGTP. Science 324:6
Dian C, Bernaudat F, Langer K, Oliva MF, Fornerod M, Schoehn G et al (2013) Structure of a truncation mutant of the nuclear export factor CRM1 provides insights into the auto-inhibitory role of its C-terminal helix. Structure 21(8):1338–1349. https://doi.org/10.1016/j.str.2013.06.003
Dong X, Biswas A, Chook YM (2009) Structural basis for assembly and disassembly of the CRM1 nuclear export complex. Nat Struct Mol Biol 16(5):558–560. https://doi.org/10.1038/nsmb.1586
Dong X, Biswas A, Suel KE, Jackson LK, Martinez R, Gu H et al (2009) Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature 458(7242):1136–1141. https://doi.org/10.1038/nature07975
Paysan-Lafosse T, Blum M, Chuguransky S, Grego T, Pinto BL, Salazar GA et al (2023) InterPro in 2022. Nucleic Acids Res 51(D1):D418–D427. https://doi.org/10.1093/nar/gkac993
Camus V, Miloudi H, Taly A, Sola B, Jardin F (2017) XPO1 in B cell hematological malignancies: from recurrent somatic mutations to targeted therapy. J Hematol Oncol 10(1):47. https://doi.org/10.1186/s13045-017-0412-4
Liu S, Qiao W, Sun Q, Luo Y (2021) Chromosome region maintenance 1 (XPO1/CRM1) as an anticancer target and discovery of its inhibitor. J Med Chem 64(21):15534–15548. https://doi.org/10.1021/acs.jmedchem.1c01145
Lapalombella R, Sun Q, Williams K, Tangeman L, Jha S, Zhong Y et al (2012) Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood 120(23):4621–4634. https://doi.org/10.1182/blood-2012-05-429506
Van Neck T, Pannecouque C, Vanstreels E, Stevens M, Dehaen W, Daelemans D (2008) Inhibition of the CRM1-mediated nucleocytoplasmic transport by -azolylacrylates: structure-activity relationship and mechanism of action. Bioorg Med Chem 16(21):9487–9497. https://doi.org/10.1016/j.bmc.2008.09.051
Muqbil I, Azmi AS, Mohammad RM (2018) Nuclear export inhibition for pancreatic cancer therapy. Cancers (Basel) 10(5). https://doi.org/10.3390/cancers10050138.
Görlich D, Mattaj IW (1996) Nucleocytoplasmic transport. Science 271:6
Azizian NG, Li Y (2020) XPO1-dependent nuclear export as a target for cancer therapy. J Hematol Oncol 13(1):61. https://doi.org/10.1186/s13045-020-00903-4
Mathew C, Ghildyal R (2017) CRM1 inhibitors for antiviral therapy. Front Microbiol 8:1171. https://doi.org/10.3389/fmicb.2017.01171
Turner JG, Sullivan DM (2008) CRM1-mediated nuclear export of proteins and drug resistance in cancer. Curr Med Chem 15(26):8
Hutten S, Kehlenbach RH (2007) CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol 17(4):193–201. https://doi.org/10.1016/j.tcb.2007.02.003
Soniat M, Chook YM (2015) Nuclear localization signals for four distinct karyopherin-β nuclear import systems. Biochem J 468:353–362. https://doi.org/10.1042/Bj20150368
Mahipal A, Malafa M (2016) Importins and exportins as therapeutic targets in cancer. Pharmacol Ther 164:135–143. https://doi.org/10.1016/j.pharmthera.2016.03.020
Adachi Y, Yanagida M (1989) Higher order chromosome structure is affected by cold-sensitive mutations in a Schizosaccharomyces pombe gene crm1+ which encodes a 115-kD protein preferentially localized in the nucleus and its periphery. J Cell Biol 108(4):1195–1207. https://doi.org/10.1083/jcb.108.4.1195
Arnaoutov A, Azuma Y, Ribbeck K, Joseph J, Boyarchuk Y, Karpova T et al (2005) Crm1 is a mitotic effector of Ran-GTP in somatic cells. Nat Cell Biol 7(6):626–632. https://doi.org/10.1038/ncb1263
Liu Q, Jiang Q, Zhang C (2009) A fraction of Crm1 locates at centrosomes by its CRIME domain and regulates the centrosomal localization of pericentrin. Biochem Biophys Res Commun 384(3):383–388. https://doi.org/10.1016/j.bbrc.2009.04.154
Torosantucci L, De Luca M, Guarguaglini G, Lavia P, Degrassi F (2008) Localized RanGTP accumulation promotes microtubule nucleation at kinetochores in somatic mammalian cells. Mol Biol Cell 19(5):1873–1882. https://doi.org/10.1091/mbc.e07-10-1050
Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N et al (2011) Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 475(7354):101–105. https://doi.org/10.1038/nature10113
Jeromin S, Weissmann S, Haferlach C, Dicker F, Bayer K, Grossmann V et al (2014) SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia 28(1):108–117. https://doi.org/10.1038/leu.2013.263
Lin DC, Hao JJ, Nagata Y, Xu L, Shang L, Meng X et al (2014) Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet 46(5):467–473. https://doi.org/10.1038/ng.2935
Camus V, Stamatoullas A, Mareschal S, Viailly PJ, Sarafan-Vasseur N, Bohers E et al (2016) Detection and prognostic value of recurrent exportin 1 mutations in tumor and cell-free circulating DNA of patients with classical Hodgkin lymphoma. Haematologica 101(9):1094–1101. https://doi.org/10.3324/haematol.2016.145102
Jardin F, Pujals A, Pelletier L, Bohers E, Camus V, Mareschal S et al (2016) Recurrent mutations of the exportin 1 gene (XPO1) and their impact on selective inhibitor of nuclear export compounds sensitivity in primary mediastinal B-cell lymphoma. Am J Hematol 91(9):923–930. https://doi.org/10.1002/ajh.24451
Garcia-Santisteban I, Arregi I, Alonso-Marino M, Urbaneja MA, Garcia-Vallejo JJ, Banuelos S et al (2016) A cellular reporter to evaluate CRM1 nuclear export activity: functional analysis of the cancer-related mutant E571K. Cell Mol Life Sci 73(24):4685–4699. https://doi.org/10.1007/s00018-016-2292-0
Taylor J, Sendino M, Gorelick AN, Pastore A, Chang MT, Penson AV et al (2019) Altered nuclear export signal recognition as a driver of oncogenesis. Cancer Discov 9(10):1452–1467. https://doi.org/10.1158/2159-8290.CD-19-0298
Maracaja DLV, Puthenpura V, Pels SG, O’Malley DP, Sklar JL, Finberg KE et al (2020) EBV-positive primary large b-cell lymphoma: the role of immunohistochemistry and XPO1 in the diagnosis of mediastinal lymphomas. Appl Immunohistochem Mol Morphol 28(10):725–730. https://doi.org/10.1097/PAI.0000000000000820
Balasubramanian SK, Azmi AS, Maciejewski J (2022) Selective inhibition of nuclear export: a promising approach in the shifting treatment paradigms for hematological neoplasms. Leukemia 36(3):601–612. https://doi.org/10.1038/s41375-021-01483-z
Parikh K, Cang S, Sekhri A, Liu D (2014) Selective inhibitors of nuclear export (SINE)—a novel class of anti-cancer agents. J Hematol Oncol 7(78):8. https://doi.org/10.1186/s13045-014-0078-0
Yang X, Cheng L, Yao L, Ren H, Zhang S, Min X et al (2014) Involvement of chromosome region maintenance 1 (CRM1) in the formation and progression of esophageal squamous cell carcinoma. Med Oncol 31(9):155. https://doi.org/10.1007/s12032-014-0155-9
Sun W, Yang J (2010) Functional mechanisms for human tumor suppressors. J Cancer 1:136–140. https://doi.org/10.7150/jca.1.136
Saulino DM, Younes PS, Bailey JM, Younes M (2018) CRM1/XPO1 expression in pancreatic adenocarcinoma correlates with survivin expression and the proliferative activity. Oncotarget 9(30):21289–21295. https://doi.org/10.18632/oncotarget.25088.
Subhash VV, Yeo MS, Wang L, Tan SH, Wong FY, Thuya WL et al (2018) Anti-tumor efficacy of Selinexor (KPT-330) in gastric cancer is dependent on nuclear accumulation of p53 tumor suppressor. Sci Rep 8(1):12248. https://doi.org/10.1038/s41598-018-30686-1
Aladhraei M, Kassem Al-Thobhani A, Poungvarin N, Suwannalert P (2019) Association of XPO1 overexpression with NF-kappaB and Ki67 in colorectal cancer. Asian Pac J Cancer Prev 20(12):3747–3754. https://doi.org/10.31557/APJCP.2019.20.12.3747.
Tang ZF, Li CW, Kang BX, Gao G, Li C, Zhang ZM (2017) GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 45(W1):W98–W102. https://doi.org/10.1093/nar/gkx247
van der Watt PJ, Zemanay W, Govender D, Hendricks DT, Parker MI, Leaner VD (2014) Elevated expression of the nuclear export protein, Crm1 (exportin 1), associates with human oesophageal squamous cell carcinoma. Oncol Rep 32(2):730–738. https://doi.org/10.3892/or.2014.3231
Cosson A, Chapiro E, Bougacha N, Lambert J, Herbi L, Cung HA et al (2017) Gain in the short arm of chromosome 2 (2p+) induces gene overexpression and drug resistance in chronic lymphocytic leukemia: analysis of the central role of XPO1. Leukemia 31(7):1625–1629. https://doi.org/10.1038/leu.2017.100
Golomb L, Bublik DR, Wilder S, Nevo R, Kiss V, Grabusic K et al (2012) Importin 7 and exportin 1 link c-Myc and p53 to regulation of ribosomal biogenesis. Mol Cell 45(2):222–232. https://doi.org/10.1016/j.molcel.2011.11.022
Gao J, Azmi AS, Aboukameel A, Kauffman M, Shacham S, Abou-Samra AB et al (2014) Nuclear retention of Fbw7 by specific inhibitors of nuclear export leads to Notch1 degradation in pancreatic cancer. Oncotarget 5(11):3444–3454. https://doi.org/10.18632/oncotarget.1813.
Liu Y, Azizian NG, Dou Y, Pham LV, Li Y (2019) Simultaneous targeting of XPO1 and BCL2 as an effective treatment strategy for double-hit lymphoma. J Hematol Oncol 12(1):119. https://doi.org/10.1186/s13045-019-0803-9
Ranganathan P, Kashyap T, Yu X, Meng X, Lai TH, McNeil B et al (2016) XPO1 inhibition using selinexor synergizes with chemotherapy in acute myeloid leukemia by targeting DNA repair and restoring topoisomerase iialpha to the nucleus. Clin Cancer Res 22(24):6142–6152. https://doi.org/10.1158/1078-0432.CCR-15-2885
Nishi K, Yoshida M, Fujiwara D, Nishikawa M, Horinouchi S, Beppu T (1994) Leptomycin B targets a regulatory cascade of crm1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. J Biol Chem 269(9):6320–6324
Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B et al (1999) Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci U S A 96(16):9112–9117. https://doi.org/10.1073/pnas.96.16.9112
Newlands ES, Rustin GJ, Brampton MH (1996) Phase I trial of elactocin. Br J Cancer 74(4):648–649. https://doi.org/10.1038/bjc.1996.415
Sun Q, Carrasco YP, Hu Y, Guo X, Mirzaei H, Macmillan J et al (2013) Nuclear export inhibition through covalent conjugation and hydrolysis of Leptomycin B by CRM1. Proc Natl Acad Sci U S A 110(4):1303–1308. https://doi.org/10.1073/pnas.1217203110
Fung HY, Chook YM (2014) Atomic basis of CRM1-cargo recognition, release and inhibition. Semin Cancer Biol 27:52–61. https://doi.org/10.1016/j.semcancer.2014.03.002
Chen CI, Gutierrez M, Brown PD, Gabrail N, Baz R, Reece DE et al (2013) Anti tumor activity of selinexor (KPT-330), a first-in-class oral selective inhibitor of nuclear export (SINE) XPO1/CRM1 antagonist in patients (pts) with relapsed/refractory multiple myeloma (MM) Or Waldenstrom's macroglobulinemia (WM). Blood 122(21).
Kalakonda N, Maerevoet M, Cavallo F, Follows G, Goy A, Vermaat JSP et al (2020) Selinexor in patients with relapsed or refractory diffuse large B-cell lymphoma (SADAL): a single-arm, multinational, multicentre, open-label, phase 2 trial. Lancet Haematol 7(7):E509–E522
Savona M, Garzon R, Brown PD, Yee K, Lancet JE, Gutierrez M et al (2013) Phase I Trial of Selinexor (KPT-330), a first-in-class oral selective inhibitor of nuclear export (SINE) in patients (pts) with advanced acute myelogenous leukemia (AML). Blood 122(21).
Azmi AS, Khan HY, Muqbil I, Aboukameel A, Neggers JE, Daelemans D et al (2020) Preclinical assessment with clinical validation of selinexor with gemcitabine and nab-paclitaxel for the treatment of pancreatic ductal adenocarcinoma. Clin Cancer Res 26(6):1338–1348. https://doi.org/10.1158/1078-0432.CCR-19-1728
Wei XX, Siegel AP, Aggarwal R, Lin AM, Friedlander TW, Fong L et al (2018) A Phase II Trial of selinexor, an oral selective inhibitor of nuclear export compound, in abiraterone- and/or enzalutamide-refractory metastatic castration-resistant prostate cancer. Oncologist 23(6):656-+. https://doi.org/10.1634/theoncologist.2017-0624.
Westin SN, Fu SQ, Tsimberidou A, Piha-Paul S, Akhmedzhanov F, Yilmaz B et al (2023) Selinexor in combination with weekly paclitaxel in patients with metastatic solid tumors: Results of an open label, single-center, multi-arm phase 1b study with expansion phase in ovarian cancer. Gynecol Oncol 168:76–82. https://doi.org/10.1016/j.ygyno.2022.11.004
Grosicki S, Simonova M, Spicka I, Pour L, Kriachok I, Gavriatopoulou M et al (2020) Once-per-week selinexor, bortezomib, and dexamethasone versus twice-per-week bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): a randomised, open-label, phase 3 trial. Lancet 396(10262):1563–1573
XPO1 Inhibitor Approved for Multiple Myeloma (2019). Cancer Discovery 9(9):1150–1151. https://doi.org/10.1158/2159-8290.Cd-nb2019-085.
Chen CX, Miao XY, Guo XJ, Xu JF, Liang JZ, Zheng Y et al (2023) Safety of selinexor as the only exportin 1 (XPO1) inhibitor so far: a post-marketing study based on the world Health Organization pharmacovigilance database (Vigibase). Expert Opin Drug Saf. https://doi.org/10.1080/14740338.2023.2248888
Liu Y, Yang R, Feng H, Du Y, Yang B, Zhang M et al (2024) Adverse events reporting of XPO1 inhibitor—selinexor: a real-word analysis from FAERS database. Sci Rep 14(1):12231. https://doi.org/10.1038/s41598-024-62852-z
Vogl DT, Dingli D, Cornell RF, Huff CA, Jagannath S, Bhutani D et al (2018) Selective inhibition of nuclear export with oral selinexor for treatment of relapsed or refractory multiple myeloma. J Clin Oncol 36(9):859-+. https://doi.org/10.1200/Jco.2017.75.5207.
Chatterjee N, Bivona TG (2019) Polytherapy and targeted cancer drug resistance. Trends Cancer 5(3):170–182. https://doi.org/10.1016/j.trecan.2019.02.003
Dias MP, Moser SC, Ganesan S, Jonkers J (2021) Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat Rev Clin Oncol 18(12):773–791. https://doi.org/10.1038/s41571-021-00532-x
Vasan N, Baselga J, Hyman DM (2019) A view on drug resistance in cancer. Nature 575(7782):299–309. https://doi.org/10.1038/s41586-019-1730-1
Engel R, Valkov NI, Gump JL, Hazlehurst L, Dalton WS, Sullivan DM (2004) The cytoplasmic trafficking of DNA topoisomerase IIalpha correlates with etoposide resistance in human myeloma cells. Exp Cell Res 295(2):421–431. https://doi.org/10.1016/j.yexcr.2004.01.012
Turner JG, Dawson J, Sullivan DM (2012) Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol 83(8):1021–1032. https://doi.org/10.1016/j.bcp.2011.12.016
Newell S, van der Watt PJ, Leaner VD (2024) Therapeutic targeting of nuclear export and import receptors in cancer and their potential in combination chemotherapy. IUBMB Life 76(1):4–25. https://doi.org/10.1002/iub.2773
Sellin M, Berg S, Hagen P, Zhang J (2022) The molecular mechanism and challenge of targeting XPO1 in treatment of relapsed and refractory myeloma. Transl Oncol 22:101448. https://doi.org/10.1016/j.tranon.2022.101448
Agarwal R, Kaye SB (2003) Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer 3(7):502–516. https://doi.org/10.1038/nrc1123
Chen Y, Camacho SC, Silvers TR, Razak AR, Gabrail NY, Gerecitano JF et al (2017) Inhibition of the nuclear export receptor XPO1 as a therapeutic target for platinum-resistant ovarian cancer. Clin Cancer Res 23(6):1552–1563. https://doi.org/10.1158/1078-0432.CCR-16-1333
Wang ZB, Pan BL, Yao YX, Qiu JC, Zhang XX, Wu XX et al (2023) XPO1 intensifies sorafenib resistance by stabilizing acetylation of NPM1 and enhancing epithelial-mesenchymal transition in hepatocellular carcinoma. Biomed Pharmacotherapy, 160. ARTN 114402. https://doi.org/10.1016/j.biopha.2023.114402.
Nakayama R, Zhang Y, Czaplinski JT, Anatone AJ, Sicinska ET, Fletcher JA et al (2016) Preclinical activity of selinexor, an inhibitor of XPO1, in sarcoma. Oncotarget 7(13):12
Tai YT, Landesman Y, Acharya C, Calle Y, Zhong MY, Cea M et al (2014) CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia 28(1):155–165. https://doi.org/10.1038/leu.2013.115
De Cesare M, Cominetti D, Doldi V, Lopergolo A, Deraco M, Gandellini P et al (2015) Anti-tumor activity of selective inhibitors of XPO1/CRM1-mediated nuclear export in diffuse malignant peritoneal mesothelioma: the role of survivin. Oncotarget 6(15):13119–13132
Deng MM, Zhang MZ, Xu-Monette ZJY, Pham LV, Tzankov A, Visco C et al (2020). XPO1 expression worsens the prognosis of unfavorable DLBCL that can be effectively targeted by selinexor in the absence of mutant p53. J Hematol Oncol 13(1). ARTN 148. https://doi.org/10.1186/s13045-020-00982-3.
Arango NP, Yuca E, Zhao M, Evans KW, Scott S, Kim C et al (2017) Selinexor (KPT-330) demonstrates anti-tumor efficacy in preclinical models of triple-negative breast cancer. Breast Cancer Res 19. ARTN 93. https://doi.org/10.1186/s13058-017-0878-6.
Marijon H, Gery S, Chang H, Landesman Y, Shacham S, Lee DH et al (2021) Selinexor, a selective inhibitor of nuclear export, enhances the anti-tumor activity of olaparib in triple negative breast cancer regardless of BRCA1 mutation status. Oncotarget 12(18):1749–1762. https://doi.org/10.18632/oncotarget.28047.
He WX, Wang SM, Yan J, Qu YP, Jin L, Sui F et al (2019) Self-assembly of therapeutic peptide into stimuli-responsive clustered nanohybrids for cancer-targeted therapy. Adv Funct Mater 29(10). ARTN 1807736. https://doi.org/10.1002/adfm.201807736.
Yan J, Ji FP, Yan SQ, You WM, Ma F, Li FN et al (2020) A general-purpose nanohybrid fabricated by Polymeric Au(I)-peptide precursor to wake the function of peptide therapeutics. Theranostics 10(19):8513–8527. https://doi.org/10.7150/thno.47243
Gong LY, Lu YL, Wang J, Li XY, Zhao J, Chen YT et al (2023) Cocktail hepatocarcinoma therapy by a super-assembled nano-pill targeting XPO1 and ATR synergistically. J Pharmaceut Anal 13(6):603–615. https://doi.org/10.1016/j.jpha.2023.04.017
Ou L, Wang X, Cheng S, Zhang M, Cui R, Hu C et al (2022) Verdinexor, a selective inhibitor of nuclear exportin 1, inhibits the proliferation and migration of esophageal cancer via XPO1/c-Myc/FOSL1 Axis. Int J Biol Sci 18(1):276–291. https://doi.org/10.7150/ijbs.66612
Thein KZ, Fu S, Janku F, Tsimberidou AM, Piha-Paul SA, Karp DD et al (2022) Selinexor in combination with carboplatin and pemetrexed in patients with advanced or metastatic solid tumors results of an open-label, single-center, multi-arm phase 1b study. J Immunother Precis Oncol 5(1):10–12. https://doi.org/10.36401/JIPO-21-20.
Sexton R, Mahdi Z, Chaudhury R, Beydoun R, Aboukameel A, Khan HY et al (2019) Targeting nuclear exporter protein XPO1/CRM1 in gastric cancer. Int J Mol Sci 20(19). https://doi.org/10.3390/ijms20194826.
Zhou F, Chen E, You D, Song Y, Sun Z, Yue L (2016) Both high expression of nucleophosmin/B23 and CRM1 predicts poorer prognosis in human gastric cancer. APMIS 124(12):1046–1053. https://doi.org/10.1111/apm.12604
Zhou F, Qiu W, Yao R, Xiang J, Sun X, Liu S et al (2013) CRM1 is a novel independent prognostic factor for the poor prognosis of gastric carcinomas. Med Oncol 30(4):726. https://doi.org/10.1007/s12032-013-0726-1
Shintani M, Tashiro A, Sangawa A, Yamao N, Kamoshida S (2016) Expression of chromosomal regional maintenance protein-1 may be associated with subcellular survivin expression in human gastric and colorectal carcinoma. Oncol Lett 12(6):4630–4634. https://doi.org/10.3892/ol.2016.5220
Zheng Y, Gery S, Sun H, Shacham S, Kauffman M, Koeffler HP (2014) KPT-330 inhibitor of XPO1-mediated nuclear export has anti-proliferative activity in hepatocellular carcinoma. Cancer Chemother Pharmacol 74(3):487–495. https://doi.org/10.1007/s00280-014-2495-8
Chen L, Huang Y, Zhou L, Lian Y, Wang J, Chen D et al (2019) Prognostic roles of the transcriptional expression of exportins in hepatocellular carcinoma. Biosci Rep 39(8). https://doi.org/10.1042/BSR20190827.
Xie QL, Liu Y, Zhu Y (2016) Chromosome region maintenance 1 expression and its association with clinical pathological features in primary carcinoma of the liver. Exp Ther Med 12(1):59–68. https://doi.org/10.3892/etm.2016.3283
Wang H, Yuan S, Zheng Q, Zhang S, Zhang Q, Ji S et al (2024) Dual Inhibition of CDK4/6 and XPO1 induces senescence with acquired vulnerability to CRBN-based PROTAC drugs. Gastroenterology. https://doi.org/10.1053/j.gastro.2024.01.025
Deutzmann A, Sullivan DK, Dhanasekaran R, Li W, Chen XY, Tong L et al (2024) Nuclear to cytoplasmic transport is a druggable dependency in MYC-driven hepatocellular carcinoma. Nature Commun 15(1). ARTN 963. https://doi.org/10.1038/s41467-024-45128-y.
Zhang L, Hong JW, Chen W, Zhang W, Liu X, Lu JH et al (2023) DBF4 dependent kinase inhibition suppresses hepatocellular carcinoma progression and potentiates anti-programmed cell death-1 therapy. Int J Biol Sci 19(11):3412–3427. https://doi.org/10.7150/ijbs.80351
von Fallois M, Kosyna FK, Mandl M, Landesman Y, Dunst J, Depping R (2021) Selinexor decreases HIF-1alpha via inhibition of CRM1 in human osteosarcoma and hepatoma cells associated with an increased radiosensitivity. J Cancer Res Clin Oncol 147(7):2025–2033. https://doi.org/10.1007/s00432-021-03626-2
Zhao C, Yang ZY, Zhang J, Li O, Liu SL, Cai C et al (2022) Inhibition of XPO1 with KPT-330 induces autophagy-dependent apoptosis in gallbladder cancer by activating the p53/mTOR pathway. J Transl Med 20(1):434. https://doi.org/10.1186/s12967-022-03635-w
Luo J, Chen Y, Li Q, Wang B, Zhou Y, Lan H (2016) CRM-1 knockdown inhibits extrahepatic cholangiocarcinoma tumor growth by blocking the nuclear export of p27Kip1. Int J Mol Med 38(2):381–390. https://doi.org/10.3892/ijmm.2016.2628
Zhao C, Ma B, Yang ZY, Li O, Liu SL, Pan LJ et al (2023) Inhibition of XPO1 impairs cholangiocarcinoma cell proliferation by triggering p53 intranuclear accumulation. Cancer Med 12(5):5751–5763. https://doi.org/10.1002/cam4.5322
Huang W, Yue L, Wang L, Zhou X, Sun Y (2009) Prognostic value of CRM1 in pancreas cancer. Clin Invest Med 32(6).
Saulino DM, Younes PS, Bailey JM, Younes M (2018) CRM1/XPO1 expression in pancreatic adenocarcinoma correlates with survivin expression and the proliferative activity. Oncotarget 9(30):7
Birnbaum DJ, Finetti P, Birnbaum D, Mamessier E, Bertucci F (2019) XPO1 expression is a poor-prognosis marker in pancreatic adenocarcinoma. J Clin Med 8(5). https://doi.org/10.3390/jcm8050596.
Zhu JH, Yan QL, Wang JW, Chen Y, Ye QH, Wang ZJ et al (2020) The key genes for perineural invasion in pancreatic ductal adenocarcinoma identified with Monte-Carlo feature selection method. Front Genet 11:554502. https://doi.org/10.3389/fgene.2020.554502
Azmi AS, Ahmad A, Banerjee S, Rangnekar VM, Mohammad RM, Sarkar FH (2008) Chemoprevention of pancreatic cancer: characterization of Par-4 and its modulation by 3,3’ diindolylmethane (DIM). Pharm Res 25(9):2117–2124. https://doi.org/10.1007/s11095-008-9581-8
Azmi AS, Wang Z, Burikhanov R, Rangnekar VM, Wang G, Chen J et al (2008) Critical role of prostate apoptosis response-4 in determining the sensitivity of pancreatic cancer cells to small-molecule inhibitor-induced apoptosis. Mol Cancer Ther 7(9):2884–2893. https://doi.org/10.1158/1535-7163.MCT-08-0438
Kazim S, Malafa MP, Coppola D, Husain K, Zibadi S, Kashyap T et al (2015) Selective nuclear export inhibitor KPT-330 enhances the antitumor activity of gemcitabine in human pancreatic cancer. Mol Cancer Ther 14(7):1570–1581. https://doi.org/10.1158/1535-7163.MCT-15-0104
Uddin MH, Al-Hallak MN, Khan HY, Aboukameel A, Li YW, Bannoura SF et al (2023) Molecular analysis of XPO1 inhibitor and gemcitabine-nab-paclitaxel combination in KPC pancreatic cancer mouse model. Clin Transl Med 13(12). ARTN e1513. https://doi.org/10.1002/ctm2.1513.
Khan HY, Nagasaka M, Li Y, Aboukameel A, Uddin MH, Sexton R et al (2022) Inhibitor of the nuclear transport protein XPO1 enhances the anticancer efficacy of KRAS G12C inhibitors in preclinical models of KRAS G12C-mutant cancers. Cancer Res Commun 2(5):342–352. https://doi.org/10.1158/2767-9764.crc-21-0176
Inoue A, Robinson FS, Minelli R, Tomihara H, Rizi BS, Rose JL et al (2021) Sequential administration of XPO1 and ATR inhibitors enhances therapeutic response in TP53-mutated colorectal cancer. Gastroenterology 161(1):196–210. https://doi.org/10.1053/j.gastro.2021.03.022
Aladhraei M, Al-Salami E, Poungvarin N, P (2019) The roles of p53 and XPO1 on colorectal cancer progression in Yemeni patients. J Gastrointest Oncol 10(3):437–444. https://doi.org/10.21037/jgo.2019.01.17.
Niu M, Chong Y, Han Y, Liu X (2015) Novel reversible selective inhibitor of nuclear export shows that CRM1 is a target in colorectal cancer cells. Cancer Biol Ther 16(7):1110–1118. https://doi.org/10.1080/15384047.2015.1047569
Shintani M, Sangawa A, Yamao N, Kamoshida S (2013) Immunohistochemical expression of nuclear and cytoplasmic survivin in gastrointestinal carcinoma. Int J Clin Pathol 6(12):2919–2927
Wu T, Chen W, Zhong Y, Hou X, Fang S, Liu CY et al (2017) Nuclear export of ubiquitinated proteins determines the sensitivity of colorectal cancer to proteasome inhibitor. Mol Cancer Ther 16(4):717–728. https://doi.org/10.1158/1535-7163.MCT-16-0553
Ferreiro-Neira I, Torres NE, Liesenfeld LF, Chan CH, Penson T, Landesman Y et al (2016) XPO1 inhibition enhances radiation response in preclinical models of rectal cancer. Clin Cancer Res 22(7):1663–1673. https://doi.org/10.1158/1078-0432.CCR-15-0978
Abdul Razak AR, Mau-Soerensen M, Gabrail NY, Gerecitano JF, Shields AF, Unger TJ et al (2016) First-in-class, first-in-human phase I study of selinexor, a selective inhibitor of nuclear export, in patients with advanced solid tumors. J Clin Oncol 34(34):4142–4150. https://doi.org/10.1200/JCO.2015.65.3949
Machlus KR, Wu SK, Vijey P, Soussou TS, Liu ZJ, Shacham E et al (2017) Selinexor-induced thrombocytopenia results from inhibition of thrombopoietin signaling in early megakaryopoiesis. Blood 130(9):1132–1143. https://doi.org/10.1182/blood-2016-11-752840
Heong VYM, Koe P, Yong WP, Soo R, Chee CE, Wong A et al (2016) RAS/AKT pathway mutations as predictive biomarkers in patients with colorectal cancer treated with the exportin 1 (XPO1) inhibitor selinexor (SEL) - inhibition of nuclear-cytoplasmic translocation of p27 as a mechanism of anti-tumour activity. Ann Oncol, 27. https://doi.org/10.1093/annonc/mdw368.26.
Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y (2010) Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466(7310):1129–1133. https://doi.org/10.1038/nature09303
Rasmussen KD, Helin K (2016) Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 30(7):733–750. https://doi.org/10.1101/gad.276568.115
Barazeghi E, Prabhawa S, Norlen O, Hellman P, Stalberg P, Westin G (2018) Decrease of 5-hydroxymethylcytosine and TET1 with nuclear exclusion of TET2 in small intestinal neuroendocrine tumors. BMC Cancer 18(1):764. https://doi.org/10.1186/s12885-018-4579-z
Fragomeni RAS, Chung HW, Landesman Y, Senapedis W, Saint-Martin JR, Tsao HS et al (2013) CRM1 and BRAF Inhibition synergize and induce tumor regression in BRAF-mutant melanoma. Mol Cancer Ther 12(7):1171–1179. https://doi.org/10.1158/1535-7163.Mct-12-1171
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
V.S., R.G., L.M.P., F.K., E.K., A.A., R.P., M.G., S.O., D.N. and A.S. wrote the main manuscript text and prepared the figures and tables. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publish
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sokolova, V., Gruber, R., Pammer, L.M. et al. Prognostic and functional role of the nuclear export receptor 1 (XPO1) in gastrointestinal cancers: a potential novel target?. Mol Biol Rep 52, 87 (2025). https://doi.org/10.1007/s11033-024-10169-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-024-10169-5