
Abstract
Background
Pathogenic variants in QARS1 (MIM:603727; Glutaminyl-TRNA Synthetase 1), which encodes Glutaminyl-tRNA synthetase 1, have been associated with rare progressive microcephaly with seizures and cerebral and cerebellar atrophy (MSCCA MIM:615760). Only a handful of MSCCA patients have been reported in the literature mostly associated with compound heterozygous QARS1 variants. In the current study, we aimed molecular characterization of a large consanguineous Pakistani family affected with microcephaly, severe intellectual and developmental disability.
Methods
We isolated genomic DNA from blood samples collected from the two affected and three normal individuals of the family. We employed whole exome sequencing, homozygosity mapping, Sanger sequencing and in silico protein modelling tools to characterize the pathogenic variant causing the disease phenotype. Moreover, we collected data of 26 MSCCA patients previously reported in the literature.
Results
The phenotype in the affected individuals of the family was characterized by microcephaly, severe intellectual and developmental disability, but no epilepsy. We found a rare QARS1 variant NM_005051.3:c.1133G > A, p.(Arg378His) in homozygous state in the family. This variant was recently cited in a patient of Turkish ethnicity, the only MSCCA patient reported with nonepileptic phenotype. This variant lying in the catalytic domain of glutaminyl-tRNA synthetase 1 showed deleterious structure and functional impacts on the protein predicted by in silico tools. The variant was classified as ‘likely pathogenic’ following ACMG guidelines.
Conclusions
We present first report of QARS1 associated developmental encephalopathy from Pakistan. Our study adds to the restricted clinical and mutational database of this rare disorder supporting the growing evidence that homozygous missense QARS1 genotypes may lead to the milder phenotype. The reports of more patients with molecular studies will enhance the understanding of the genotype-phenotype correlations.
Similar content being viewed by others
Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
Introduction
Progressive microcephaly, seizures, and cerebral and cerebellar atrophy (MSCCA; MIM: 615760) is a neurodegenerative and neurodevelopmental disorder with onset in the first days or months of life. MSCCA is an extremely rare autosomal recessive disease characterized by progressive microcephaly, intractable seizures, cerebral and cerebellar atrophy, moderate to severe developmental delay, and hypotonia. For the first time, Zhang et al. (2014) unpinned MSCCA in 4 patients from 2 unrelated families associated with the QARS1 gene. In vitro experiments demonstrated a significant impairment of QARS aminoacetylation in cell lines, while homozygous loss of QARS function in zebrafish exhibited widespread cell death in the brain with reduced brain and eye size [1]. QARS1 or glutaminyl-tRNA synthetase 1 (ENST00000306125.12) comprised 24 exons (775 amino acids) with chromosomal position 3p21.31 (https://asia.ensembl.org). So far, few cases of MSCCA have been reported in the literature caused by QARS1 [1,2,3,4,5,6,7,8,9,10,11]. Animal models including C. elegans, Drosophila melanogaster, Danio rerio and Mus musculus provide insight into the pathogenic mechanism of QARS1-related disorders. Knockdown of QARS-1 through RNAi was generated by Zheng et al. (2022) to assess the functional role of QARS-1 deficiency at cellular and developmental levels [12]. To confirm the role of QARS1 in neuronal development and structure, Chihara et al. (2007) generated Drosophila melanogaster as a model for the variant of GlnRS (glutaminyl-tRNA synthetase). This study confirmed that the variant had affected the dendritic and axonal development [13]. The loss of function in brain development was confirmed by Zhang et al. (2014) in Danio rerio [1]. QARS1 knockout was achieved in zebrafish embryos by inserting a gene-trap cassette through retroviral infection.
A large consanguineous family affected with nonepileptic phenotype of MSCCA is presented in this report. Moreover, we summarized clinical and molecular findings of the MSCCA patients harboring the QARS1 variants previously reported in the literature (Table 1).
Materials and methods
Ascertainment of patients
All experiments involving human subjects or related data were conducted in accordance with relevant ethical regulations. Informed consent was signed by the affected family members and ethical approval for this study was obtained from the Institutional Review Board (IRB) of Quaid-I-Azam University, Islamabad. Detailed clinical history and venous blood samples were collected from the available healthy and affected individuals at the time of ascertainment. Peers of the affected family were interviewed to construct pedigree and collect disease history. Through standard protocol, DNA was extracted from peripheral blood lymphocytes.
Genetic analysis by next-generation sequencing
Whole exome sequencing for the subject V:5 was performed at Macrogen (Korea) by using the Agilent SureSelect Human All Exome V6 Kit (Agilent Technologies, Santa Clara, CA, USA) as described [14]. High-throughput sequencing system, Illumina NovaSeq 6000 (Illumina, Santa Clara, CA, USA) was used for paired-end sequencing or PE150. The obtained sequencing reads were aligned to the reference genome via Burrows-Wheeler Aligner v0.7.17 (BWA; http://bio-bwa.sourceforge.net/). For the enrolled family, assembly hg19 was selected, while hg38 genome assembly was utilized for the sequencing reads. BAM files were sorted through SAMtools (v1.8) while duplicated reads were identified with the help of Picard (v2.18.9). Genotyping was conducted through the Genome Analysis Toolkit (GATK; http://www.broadinstitute.org/gatk/) v4.0. Functional annotation and filtration of variants were performed using ANNOVAR (http://www.openbioinformatics.org/annovar/) and FILTUS, respectively. After annotation, the obtained file was saved in CSV format and filtered to find the most likely pathogenic variants. Our filtration strategy concentrated on exploring the coding regions as well as the splice acceptor and donor site. Based on the clinical history and pedigree, we chose the rarest variants with minor allele frequency (MAF < 0.01%) in public databases including the Genome Aggregation Database (https://gnomad.broadinstitute.org/) http://evs.gs.washington.edu) and 1000 Genomes project (http://www.1000genomes.org/). Different in silico tools were also used to predict the functional effect of the filtered variants. According to the ACMG guidelines, the identified variants were classified aspathogenic, likely pathogenic or variant of uncertain significance (VUS).
Homozygosity mapping and in Silico tools
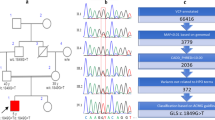
AutoMap (https://automap.iob.ch/process) tool with the default settings on the VCF (Variant Call Format) file of whole exome sequencing was used for homozygosity mapping (shown in Fig. 1) because the identified variant was found in the region shared by ancestors [15]. The pathogenicity of our selected variant was confirmed by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), MutationTaster (www.mutationtaster.org), SIFT (http://sift.jcvi.org/), CADD (Combined Annotation Dependent Depletion (https://cadd.gs.washington.edu), DANN, VarSome (https://varsome.com/), PROVEAN, EIGEN and VARITY. The detected variant was further classified) based on ACMG guidelines through the Franklin variant assessment tool (https://franklin.genoox.com/clinical-db/home).

(A) Pedigree of the consanguineous Pakistani family affected with MSCCA. (B) Sanger sequencing of the healthy and affected individuals. (C) Domains and reported mutations of QARS1 protein. (D) Evolutionary conservation of the mutated QARS1 residue p.Arg378. (E) Homozygosity mapping using AutoMap tool
Sanger sequencing validation
To validate c.1133G > A, p.(Arg378His) in the QARS1, we amplified genomic DNA of the affected and normal individuals of the family (Fig. 1) with a pair of primers (Forward: CCAGGGCCATGACGTAGAG and Reverse: CCAAACCCAGATGCTCCTCT) and sequenced with Sanger method by forward primer. These primers were designed with the Primer3 tool (https://primer3.ut.ee/). Although NGS has considerably improved accuracy, it is usually recognized that the identified variant be validated with Sanger sequencing prior to reporting [16]. To date, using whole-exome sequencing, 188 variants in 143 distinct genes have been reported in the Pakistani population as described previously [17].
Results
QARS1 biallelic variant and algorithms
Whole exome sequencing in our enrolled family was used to identify a single genetic variant that fulfilled the criteria for causality based on established protocols [18]. Family had homozygous QARS1: NM_005051.3: c.(1133G > A) p.(Arg378His) variant. The candidate variant was validated by Sanger sequencing amongst family members following the recessive mode of inheritance (Fig. 1). Minor allele frequencies; exomes: ƒ = 0.0000137 (cov: 57.9) genomes: ƒ = 0.00000657 were available in VarSome (https://varsome.com/). Algorithms such as SIFT (deleterious (0), Polyphen2 (probably damaging (0.996), MutationTaster (disease-causing), DANN (deleterious (1), PROVEAN (pathogenic supporting), EIGEN (pathogenic supporting (0.7135), MutPred (pathogenic supporting (0.671) and VARITY (deleterious (0.94) predicted our variant as disease-causing or deleterious. Furthermore, the CADD score for the identified variant was also high (28.4), indicating a pivotal role in the pathogenesis of the disease.
Biallelic variant associated with MSCCA phenotypes
Our family comprised of seven patients (III:4, IV:4, V:4, V:5, V:6, V:7 and VI:1) as shown in Fig. 1. Two of them, V:5 (15 years old) and V:7 (12 years old) were born without pregnancy complications. Symptoms of both were consistent with MSCCA, except for lack of epilepsy. Their clinical features were microcephaly, poor weight gain, severe intellectual disabilities, and slurry speech. They also exhibited symptoms such as mild facial dysmorphia including a sloping forehead, broad flat nasal bridge and deep-set eyes. Other features were mood instabilities, anxiety reactions, learning disabilities (writing and reading), less responsiveness to tasks or questions, short-term memory, poor judgment or decision-making ability and poor communication to maintaining or initiating conversations. Symptoms of the affected individual III:4 were mild alive at 90 years of age, while his affected son (IV:4) died at 50 years of age. The affected individual V:4 had moderate symptoms and died at the age of 5 years due to complications from measles, while the affected individual V:6 died at the age of 8 years following an accident. Biochemical, metabolic and karyotyping evaluations were normal in the proband (V:5). Retinal examination, hearing tests, electroencephalogram, and brain MRI scans were normal for the affected individuals (V:5 and V:7).
QARS1 variant alters evolutionarily conserved amino acids
Our reference amino acid (arginine at 378 position) is highly conserved among different species (Homo sapiens, Mus musculus, Danio rerio, Gallus gallus, Pan troglodytes, Macaca mulatta, Canis lupus familiaris, Sus scrofa, Rattus norvegicus) as presented in Fig. 1. Functionally confirmed variants are highly conserved as examined by Zhang et al. (2014) [1]. Therefore, conservation across diverse taxa highlights the evolutionary importance of our mutated catalytic domain, indicating further investigation into its functional role in related pathologies.
Impact of variant on protein function or structure
Protein modeling of the mutated QARS1: p.Arg378His was done by the SWISS Model (https://swissmodel.expasy.org, accessed on 14th October 2024), visualized with Pymol (https://www.pymol.org, accessed on 14th October 2024) and compared with the wild -type protein present in AlphaFold 3 database (https://alphafold.ebi.ac.uk/entry/P47897, accessed on 14th October 2024). Comparison depicts that Agr378 makes H-bonds with Trp375, Val357 and Glu382 whilst the mutant His378 makes H-bond with Trp375 only (Fig. 2). Both Arg and His are polar amino acids and positively charged but the imidazole ring in His indicates weak and conditional charge in both protonated and deprotonated forms which can contribute to less bonding (Fig. 2B) and protein structure instability. Mutational analysis by the DynaMut (https://biosig.lab.uq.edu.au/dynamut/, accessed on 14th October 2024) also suggests destabilization of protein with Δ ΔG stability = -1.24 kcal/mol score. Figure 2 indicates that Arg378 (wild) has a guanidinium side chain that favors interactions and bonding, stabilizes the structure and contains clashes with Leu386 and Ser383 (purple dotted lines in Fig. 2). On the other hand, His378 makes steric hindrance with Trp375, Val 357 and Ser383 which is not present in wild-type protein. The size difference in both Arg378 and His378 further provides evidence of instability of the protein’s active site conformation, making it unable to precisely bind with the tRNA.

(A). Wild-type QARS1 protein where Arg378 (red) makes H-bonds with Trp375 (blue) 2.2Å, Val357 (magenta) 1.2 Å and Glu382 (orange) 1.8 Å. (B). Mutant QARS1 with His378 (red) depicts H-bond with Trp375 (blue) 2.3 Å. (C). Wild-type protein with H-bonds interactions of Arg378 with neighboring amino acids where dotted red lines depict H-bonds. D. Mutant protein His378 where red dotted lines show the H-bonds and purple dotted lines depict the protein structure clashes in both (C) and (D)
Discussion
So far, approximately 28 MSCCA patients have been reported in the literature caused by QARS1 including the two patients in our conducted study. Only five patients are reported in homozygous condition and the rest are compound heterozygotes. This report presents the study of Pakistani family affected with MSCCA caused by a missense variant NM_005051.3:c.1133G > A, p.(Arg378His)classified as likely pathogenic (PM2, PM5, PP1, PP4, PM3). Clinical findings were progressive microcephaly, poor weight gain, severe intellectual disabilities, minor facial dysmorphia, and slurry speech. The 3D analysis of both mutant and wild-type GlnRS protein revealed potential structural changes induced by the homozygous mutation in the catalytic domain (Fig. 1).
MSCCA patients generally exhibit a triad of symptoms such as primary or secondary microcephaly, epilepsy with neonatal or infantile-onset and moderate to severe intellectual disabilities as shown in Table 1. Our patients are extremely rare in the literature presenting with non-epileptic symptoms [5, 7], slurry speech and normal brain MRI while the single female patient reported with the same homozygous mutation NM_005051.3:c.1133G > A, p.(Arg378His) [7] had no speech and an abnormal MRI scan (hypoplasia of the corpus callosum, white matter atrophy and enlarged ventricles).
Most of the patients with homozygous variants are reported with milder phenotypes [5, 7]. indicating homozygosity might be protective for the risk of developing epilepsy [7]. The same is the case in our patients in the context of homozygosity and milder phenotype. In contrast, compound heterozygous mutations showed severe phenotypic manifestations, with some exceptions. Two patients were mildly affected by the compound heterozygous mutation, possibly because of null and hypomorphic allele phenomena [7].
Anxiety and mood instabilities were seen in a patient reported by Zhang et al. (2014), who had sensitivity to sound and severe agitation [1]. These symptoms are consistent with our proband (V:5) phenotype. Craniofacial anomalies including facial dysmorphia were also reported by Leshinsky-Silver et al. (2017) [4], Chan et al. (2022) [9] and Sakka et al. (2024) [11] and matched with all patients of our family. The most common feature of severe intellectual disability (ID) was noticed in different studies [1, 3, 4, 7].
Conclusions
Our study diagnosed a familial case consistent with MSCCA, enabled by whole exome sequencing. Notably, just a single patient of Turkish ethnicity was already cited but our study further explores the spectrum in the context of disease severity, zygosity and CP1 domain of glutaminyl-tRNA synthetase 1. Our study supports the potential role of the variant in the mild form of MSCCA because our patients and a single patient of Turkish ethnicity are mildly affected without epilepsy. Functional studies should be encouraged in MSCCA patients exhibiting QARS1 variants, for insights into exploring the disease pathogenesis.
Data availability
No datasets were generated or analysed during the current study.
References
Zhang X, Ling J, Barcia G, Jing L, Wu J, Barry BJ, Mochida GH, Hill RS, Weimer JM, Stein Q, Poduri A (2014) Mutations in QARS, encoding glutaminyl-tRNA synthetase, cause progressive microcephaly, cerebral-cerebellar atrophy, and intractable seizures. Am J Hum Genet 94(4):547–558
Salvarinova R, Ye CX, Rossi A, Biancheri R, Roland EH, Pavlidis P, Ross CJ, Tarailo-Graovac M, Wasserman WW, van Karnebeek CD (2015) Expansion of the QARS deficiency phenotype with report of a family with isolated supratentorial brain abnormalities. Neurogenetics 16:145–149
Kodera H, Osaka H, Iai M, Aida N, Yamashita A, Tsurusaki Y, Nakashima M, Miyake N, Saitsu H, Matsumoto N (2015) Mutations in the glutaminyl-tRNA synthetase gene cause early-onset epileptic encephalopathy. J Hum Genet 60(2):97–101
Leshinsky-Silver E, Ling J, Wu J, Vinkler C, Yosovich K, Bahar S, Yanoov-Sharav M, Lerman-Sagie T, Lev D (2017) Severe growth deficiency, microcephaly, intellectual disability, and characteristic facial features are due to a homozygous QARS mutation. Neurogenetics 18:141–146
Alabdullatif MA, Al Dhaibani MA, Khassawneh MY, El-Hattab AW (2017) Chromosomal microarray in a highly consanguineous population: diagnostic yield, utility of regions of homozygosity, and novel mutations. Clin Genet 91(4):616–622
Fuchs SA, Schene IF, Kok G, Jansen JM, Nikkels PG, van Gassen KL, Terheggen-Lagro SW, van der Crabben SN, Hoeks SE, Niers LE, Wolf NI (2019) Aminoacyl-tRNA synthetase deficiencies in search of common themes. Genet Sci 21(2):319–330
Johannesen KM, Mitter D, Janowski R, Roth C, Toulouse J, Poulat AL, Ville DM, Chatron N, Brilstra E, Geleijns K, Born AP (2019) Defining and expanding the phenotype of QARS-associated developmental epileptic encephalopathy. Neurology: Genet 5(6):e373
Ji J, Shen L, Bootwalla M, Quindipan C, Tatarinova T, Maglinte DT, Buckley J, Raca G, Saitta SC, Biegel JA, Gai X (2019) A semiautomated whole-exome sequencing workflow leads to increased diagnostic yield and identification of novel candidate variants. Mol Case Stud 5(2):003756.
Chan DL, Rudinger-Thirion J, Frugier M, Riley LG, Ho G, Kothur K, Mohammad SS (2022) A case of QARS1 associated epileptic encephalopathy and review of epilepsy in aminoacyl-tRNA synthetase disorders. Brain Develop 44(2):142–147
Mao B, Lin N, Guo D, He D, Xue H, Chen L, He Q, Zhang M, Chen M, Huang H, Xu L (2023) Molecular analysis and prenatal diagnosis of seven Chinese families with genetic epilepsy. Front NeuroSci 17:p1165601
Sakka R, Hamida HB, Abdelali M, Chaabane A, Zrig A, M’rad R, Mekki M, Monastiri K (2024) Microcephaly, progressive, seizures, and cerebral and cerebellar atrophy: QARS1 new variants associated with a severe phenotype in a patient
Zheng T, Luo Q, Han C, Zhou J, Gong J, Chun L, Xu XS, Liu J (2022) Cytoplasmic and mitochondrial aminoacyl-tRNA synthetases differentially regulate lifespan in Caenorhabditis elegans. Iscience, 25(11)
Chihara T, Luginbuhl D, Luo L (2007) Cytoplasmic and mitochondrial protein translation in axonal and dendritic terminal arborization. Nat Neurosci 10(7):828–837
Efthymiou S, Salpietro V, Malintan N, Poncelet M, Kriouile Y, Fortuna S, De Zorzi R, Payne K, Henderson LB, Cortese A, Maddirevula S (2019) Biallelic mutations in neurofascin cause neurodevelopmental impairment and peripheral demyelination. Brain 142(10):2948–2964
Quinodoz M, Peter VG, Bedoni N, Bertrand R, Cisarova B, Salmaninejad K, Sepahi A, Rodrigues N, Piran R, Mojarrad M, M. and, Pasdar A (2021) AutoMap is a high performance homozygosity mapping tool using next-generation sequencing data. Nature communications, 12(1), p.518
Arteche-López A, Ávila-Fernández A, Romero R, Riveiro-Álvarez R, López-Martínez MA, Giménez-Pardo A, Vélez-Monsalve C, Gallego-Merlo J, García-Vara I, Almoguera B, Bustamante-Aragonés A (2021) Sanger sequencing is no longer always necessary based on a single-center validation of 1109 NGS variants in 825 clinical exomes. Sci Rep 11(1):5697
Ahmad R, Naeem M (2025) A systematic review of hereditary neurological disorders diagnosed by whole exome sequencing in Pakistani population: updates from 2014 to November 2024. Neurogenetics 26(1):1–19
MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, Adams DR, Altman RB, Antonarakis SE, Ashley EA, Barrett JC (2014) Guidelines for investigating causality of sequence variants in human disease. Nature 508(7497):469–476
Funding
RA was supported by the Higher Education Commission of Pakistan for visiting University College London, UK under International Research Support Initiative Program (IRSIP 52 BMS 22).
Author information
Authors and Affiliations
Contributions
R.A. Conceptualization, Recruitment of subjects, Methodology, Original draft preparation.M.N. Conceptualization, Investigation, Supervision, Review and editing of the manuscript. H.H. Conceptualization, Supervision & Resources. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethical approval
The study was approved by the Institutional Review Board of Quaid-i-Azam University Islamabad (QAU/IRB/505).
Competing interests
The authors declare no competing interests.
Consent to participate
Informed consent was obtained from the enrolled family to participate in the study.
Consent to publish
Informed consent was obtained from the enrolled families to allow publication of the clinical and diagnostic information collected in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ahmad, R., Naeem, M. & Houlden, H. QARS1 associated developmental epileptic encephalopathy: first report of a rare homozygous missense variant from Pakistan causing nonepileptic phenotype in a family of seven patients and a comprehensive review of the literature. Mol Biol Rep 52, 528 (2025). https://doi.org/10.1007/s11033-025-10574-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-025-10574-4