
Abstract
Purpose
Retinol metabolism is intricately linked to the occurrence and progression of hepatocellular carcinoma (HCC); however, the precise pathogenic relationship between them remains elusive. The aim of this study was to elucidate the characteristics of retinol metabolism in HCC through Mendelian randomization, prognostic model and experimental validation.
Methods
We used transcriptomic data related to HCC in TCGA and GEO databases for a variety of machine learning, including differential gene expression analysis, functional enrichment analysis, protein–protein network interaction, ceRNA regulatory network, and single-cell sequencing analysis. Mendelian randomization analysis was used to elucidate the causal analysis of retinol metabolism and the occurrence of HCC. Consensus cluster analysis was performed based on 11 retinol metabolism-related genes, and the prognostic model was constructed by Lasso regression and Cox regression analysis. The expression level of RDH16 gene was detected in cell lines and clinical samples, and finally the function of RDH16 gene and its regulatory relationship with miR- 665 were verified by in vitro cell experiments.
Results
Differentially expressed genes were mainly concentrated in the retinol metabolic pathway. Mendelian randomization analysis showed that decreased retinol metabolic activity was causally associated with the occurrence of HCC. RDH16 gene was significantly lower expressed in HCC, and inhibition of RDH16 gene expression could promote the proliferation, migration and invasion of HCC cells and inhibit cell apoptosis. miR- 665 is an upstream regulator of RDH16 gene, which can inhibit the expression and function of RDH16.
Conclusion
The decrease of retinol metabolic activity can promote the occurrence and development of HCC. Targeting retinol metabolic pathway may be a new direction for the treatment of HCC.
Similar content being viewed by others

Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
1 Introduction
Hepatocellular carcinoma (HCC) constitutes approximately 80% of primary liver cancer cases and ranks as the third leading cause of cancer-related mortality worldwide [1]. Recently, immune checkpoint inhibitors (ICB) targeting the PD- 1/PD-L1 axis have emerged as a groundbreaking strategy for cancer therapy [2]. Despite advancements in HCC treatment, the overall 5 year survival rate for patients with HCC remains below 20% [3]. Therefore, early screening and diagnosis, along with postoperative prognosis prediction, hold paramount significance [4]. α-fetoprotein (AFP) currently serves as the most practical biomarker for early HCC screening and prognostic assessment of tumor recurrence and patient survival; however, its relatively low sensitivity and specificity are unsatisfactory [5]. Consequently, there is an urgent demand for novel technologies to identify reliable candidate biomarkers that can enhance the diagnosis accuracy and prognosis prediction capability of HCC [6].
Retinol (Vitamin A) and all-trans retinoic acid (atRA) are metabolic byproducts of retinol metabolism, which have been shown to undergo alterations in levels in various diseases [7]. In healthy individuals, retinol can be converted into all-trans retinoic acid, playing diverse roles in regulating normal growth and development [8]. Studies have indicated that all-trans retinoic acid can inhibit activated pancreatic stellate cells and prevent the formation of surrounding connective tissue in pancreatic ductal adenocarcinoma, creating an environment that makes tumor spread more difficult, confirming the anticancer effect of retinol [9]. Jun Han et al. found a significant decrease in the content of retinol and its metabolites in tissues and serum during a metabolomics study of HCC patients, which was also associated with patient survival prognosis [10]. However, it is currently unclear whether there are changes in the expression of genes related to retinol metabolism in HCC or whether these changes affect the survival prognosis of HCC patients.
Currently, Genome-Wide Association Studies (GWAS) have successfully identified numerous genetic variations associated with diseases [11]. Mendelian randomization (MR) is a causal inference method based on genetic variations that leverages the impact of randomly assigned genotypes on phenotypes in nature to infer the influence of biological factors on diseases. Pathway Mendelian Randomization is a methodology that investigates the causal relationship between biological pathways and diseases by utilizing the effect of genetic variations on the activity of biological pathways. It can aid in identifying potential drug targets as well as assessing drug efficacy and safety.
RDH16 belongs to the short-chain dehydrogenase/reductase superfamily and has been implicated in over 70 enzymatic reactions in humans. Members of this protein family are involved in the metabolism of diverse compounds, including steroid hormones, prostaglandins, retinoids, and lipids. It has been reported that RDH16 encodes a 317-amino acid protein located on the peroxisomal membrane. The liver is recognized as the primary storage organ for vitamin A, which serves as a precursor for endogenous synthesis of retinaldehyde and retinoic acid. Notably, RDH16 exhibits robust expression in the liver and is closely associated with retinol metabolism; however, limited research has focused on its role in HCC.
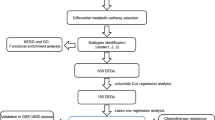
In this study, we demonstrated the association between retinol metabolism and HCC through Mendelian randomization analysis, prognostic model combined with experimental validation. Additionally, we identified RDH16 as a tumor suppressor gene in HCC, which is regulated by miR- 665. Targeting the retinol metaboli6 sm pathway may offer a novel therapeutic approach for HCC. The flowchart of the entire study is shown in Fig. 1.

The flowchart of the entire study
2 Materials and methods
2.1 Data collection and processing
The study incorporated a total of five GEO datasets (GSE76427, GSE84402, GSE101685, GSE164760, and GSE174570) sourced from the NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo). Batch correction was performed using the'limma'R package. The validation dataset consisted of 115 liver cancer samples and 52 adjacent non-tumor samples obtained from the GSE76427 dataset. The remaining four datasets were utilized as training sets, comprising a total of 148 liver cancer samples and 108 adjacent non-tumor samples. Data integration was conducted employing the'sva'R package. Additionally, the single-cell dataset GSE146409 was downloaded from the GEO database. Furthermore, we acquired the TCGA-LIHC dataset from the TCGA database (http://cancergenome.nih.gov/).
2.2 Identification of differentially expressed genes in HCC
Differentially expressed genes (DEGs) between HCC and normal samples were identified using the R package'Limma'[12], with criteria of |log2foldchange|> 1 and an adjusted p-value < 0.05. KEGG functional annotation analysis was performed utilizing the R package'clusterProfiler'.
2.3 Mendelian randomization analysis
A genome-wide association study (GWAS) was conducted to screen for single nucleotide polymorphisms (SNPs) associated with retinol in a cohort of 1447 individuals of South Asian ancestry. HCC-related SNPs were selected from a GWAS involving 197,611 individuals of East Asian ancestry, including 1866 cases of HCC and 195,745 controls. The main Mendelian randomization (MR) analysis employed the inverse variance weighted method (IVW). Sensitivity analyses included MR-Egger regression, heterogeneity tests, multiplicity tests, and leave-one-out sensitivity testing. Additionally, the hypothesis that exposure leads to outcomes was validated using the MR Steiger test. For the MR analysis, we employed instrumental variables (IVs) in the form of single nucleotide polymorphisms (SNPs) that exhibited a strong correlation with the exposure factor (P < 5 × 10 − 6). The selection of IVs ensured their independence from each other, and linkage disequilibrium (LD) was assessed by clustering within a 10 Mb window using an R2 value threshold of < 0.001.
2.4 Protein–protein interactions, drug-gene interactions, and ceRNA network analysis
We constructed a protein–protein interaction network comprising 11 differentially expressed genes (DEGs) associated with retinol metabolism using the String platform (https://cn.string-db.org/). The DGIdb database (https://dgidb.genome.wustl.edu/) was utilized to identify drugs interacting with these DEGs. To predict miRNAs associated with DEGs, we employed four databases in combination: miRanda, miRDB, miRWalk, and TargetScan. Additionally, the spongeScan database (http://spongescan.rc.ufl.edu/) was employed to predict lncRNAs associated with the identified miRNAs.
2.5 Identification of retinol metabolism-related molecular subtypes in HCC
We employed the non-negative matrix factorization (NMF) algorithm, utilizing 11 differentially expressed genes (DEGs), to stratify the merged cohorts of TCGA-LIHC and GSE76427 into distinct subtypes. The determination of optimal subtype numbers was based on comprehensive evaluation criteria including correlation, dispersion, variation, residuals, rss, silhouette score, and sparsity. Subtype-specific differences in overall survival (OS) were compared. To quantify immune activity and levels of immune cell infiltration, single-sample gene set enrichment analysis (ssGSEA) was conducted. Furthermore, we performed enrichment analysis using the R package"GSVA"to investigate disparities in biological processes related to retinol metabolism subtypes [13]. Kyoto Encyclopedia of Genes and Genomes (KEGG) gene sets from the MSigDB database (http://software.broadinstitute.org/gsea/msigdb/) were downloaded for GSVA analysis. Heatmaps with a corrected p-value < 0.05 were utilized to visualize pathways associated with DEGs.
2.6 Development prognosis features and independent prognosis analysis
Initially, we conducted lasso—univariate Cox analysis to identify prognostic variables for HCC using a screening threshold of p < 0.05. Subsequently, independent prognostic factors were determined through multivariate Cox analysis to establish the prognostic features of HCC. Risk scores for each HCC sample were then calculated based on coefficients obtained from multivariate Cox analysis and expression profiles of each independent prognostic gene. The TCGA-LIHC cohort was used as the training dataset, while the GSE76427 cohort served as the validation dataset. Finally, time-dependent ROC curves were generated using the"survivalROC"R package to estimate the area under curve (AUC) for 1 year, 3 year, and 5 year survival rates. Considering both risk scores and clinical features, a nomogram for HCC was developed using the"rms"R package.
2.7 Human protein atlas (HPA)
Cancer tissue microarray sections were obtained from HPA (https://www.proteinatlas.org/) for immunohistochemical staining, and the corresponding slides were digitally scanned to generate images. Immunohistochemistry staining images of retinol dehydrogenase, encoded by the RDH16 gene, in HCC and normal tissues were acquired from the HPA website.
2.8 HCC clinical samples
Tumor tissue specimens, as well as adjacent non-tumor tissue specimens or normal tissue samples, were collected from 16 patients with HCC who underwent primary liver cancer surgery at Xiamen University Affiliated Zhongshan Hospital between 2017 and 2021. Prior to surgery, none of the patients received chemotherapy or radiotherapy. All samples were stored at – 80 ℃. Our study was ethically approved by the Ethics Committee of Xiamen University Affiliated Zhongshan Hospital, and informed consent was obtained from all participants (approval number: XMZSYYKY-2024 - 621).
2.9 Cell culture and transfection.
The LO2 normal human liver cell line and three HCC cell lines—HepG2, Hep3B, and Huh7 -were procured from the School of Life Sciences at Xiamen University in Xiamen City, China. The cells were cultured in a CO2 incubator maintained at 37 °C using DMEM medium (Gibco), supplemented with 10% fetal bovine serum (Gibco), and 1% penicillin–streptomycin solution (Invitrogen). Prior to transfection, the cells were seeded into culture dishes. Once reaching approximately 60% confluency, the miR- 665 inhibitor, RDH16 siRNA, and their respective controls, were transfected using the Genepharm Technologies'siRNA/miRNA-mate plus transfection reagent kit (Shanghai China).The miR- 665 inhibitor (B03001)and RDH16 siRNA(A01001)were purchased from Genepharm Technologies. All experimental procedures followed the manufacturer's instructions.
2.10 Real-time PCR analysis
The TRIzol reagent (Invitrogen, Carlsbad, CA) was utilized for the isolation of total RNA from tissue samples and cell lines following the manufacturer's instructions. Reverse transcription was performed using the ReverTra Ace Qpcr RT Kit (TOYOBO, Japan). Real-time quantitative PCR detection was conducted using SYBR Green Master (Yeasen, China). Finally, data analysis was carried out employing the 2 − ΔΔCt method. RDH16 and GAPDH primers were procured from Beijing Qingke Biotechnology Co., Ltd. in China. miR- 665 and U6 primers were obtained from RiboBio (Guangzhou, China). The primer sequences used are provided below: RDH16 forward: 5′-GAAGGAGTGCGTGAGAGACAAAG- 3', RDH16 reverse: 5’-CCAACAAGTTCACGTCCAGTATG- 3’, GAPDH forward: 5′-TGTCAAGCTCATTTCCTGGTATG- 3’, GAPDH reverse: 5′-TCTCTCTTCCTCTTGTGCTCTTG- 3’.
2.11 Western blot
The protein was transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA) following 10% SDS-PAGE electrophoresis. After blocking with 5% skim milk for 1 h, the membrane was incubated overnight at 4℃ with RDH16 antibody (diluted to 1:1000; #ab224163, Abcam, USA) and GAPDH antibody (diluted to 1:1000; #5174, Cell Signaling Technology Inc., USA). Subsequently, the membrane was incubated at room temperature for 1 h with secondary antibodies conjugated to enzymes. The blot signal was detected using an enhanced chemiluminescence reagent (Advansta, USA).
2.12 Cell counting Kit- 8 assay
The growth of HCC cells was assessed using the CCK- 8 assay kit (APExBIO, USA). A total of 2000 cells were seeded per well in a 96-well plate with 100 μL of culture medium and incubated for a duration of 3 days.
2.13 Wound-healing assay
Cells were seeded in a six-well plate and cultured overnight until confluence was achieved. Subsequently, the cells were gently scratched using a 200 μl pipette tip to create a wound, and the process of wound healing was monitored at both 0 and 48 h.
2.14 Cell migration and invasion assays
After 48 h of cell transfection, the cells were seeded in Transwell chambers (8 mm pore size, Corning, USA) containing 100 μl of serum-free DMEM medium. The lower chamber was filled with 600 μl of medium supplemented with 20% FBS. To evaluate cellular invasion capability, the Transwell chambers were pre-coated with matrigel and after a further incubation period of 48 h, the invaded cells that traversed the membrane were stained using a solution of 0.1% crystal violet and subsequently quantified.
2.15 Luciferase reporter assay
The 3′-UTR and mutated sequence of RDH16 were synthesized and inserted into the pmiR-GLO dual-luciferase vector (GenePharma, Shanghai, China). Cells miR- 665 inhibitor or its control were transfected with RDH16–3′-UTR-Wt and PTPRB–3′-UTR-Mut. After 48 h of transfection, cells were collected and lysed using lysis buffer (Promega, Madison, WI, USA).
2.16 Flow cytometry analysis of the cell apoptosis
Following a 48-h period of cell transfection and incubation, the cells were carefully washed and subsequently collected in flow cytometry tubes. Staining was performed in strict accordance with the instructions provided by the reagent manufacturer (YEASEN, #40305ES20, China), followed by detection using an advanced analytical flow cytometer (BD LSRFortessa™ X- 20, USA).
2.17 Statistical analysis
The bioinformatics analyses were conducted using R 4.3.3, while data analysis was performed with Graphpad Prism 10 software. All data are presented as mean ± standard deviation. Statistical methods employed in this study included Student's t-test, one-way analysis of variance, chi-square test, Kaplan–Meier method, log-rank test, and Pearson correlation coefficient analysis. A significance level of P < 0.05 was considered statistically significant for observed differences.
3 Results
3.1 The differentially expressed genes in HCC are significantly enriched in retinol metabolism pathway
Initially, we retrieved four datasets related to HCC from the GEO database (Fig. 2A), comprising a total of 233 samples, including 85 normal cases and 148 cancer cases. Subsequently, we employed the R package"SVA"to integrate these four datasets and mitigate batch effects that could introduce confounding variations. Principal component analysis demonstrated successful elimination of batch effects across all samples (Fig. 2B). Next, differential expression analysis was conducted between normal and cancer samples, revealing a set of 254 genes with significant differential expression (Fig. 2C). The heatmap visualized the distribution pattern of these differentially expressed genes (Fig. 2D). The GO enrichment analysis revealed that the differentially expressed genes (DEGs) were implicated in diverse biological processes, including small molecule catabolic process, α-amino acid metabolism, and steroid metabolism (Fig. 2E). KEGG enrichment analysis demonstrated significant enrichment of DEGs in the retinol metabolism pathway, with a total of 11 DEGs participating in this process. Additionally, other pathways such as complement pathway, chemical carcinogenesis—DNA adducts, and drug metabolism—cytochrome P450 also exhibited active enrichment (Fig. 2F). These findings provide preliminary evidence for a close association between retinol metabolism and the initiation and progression of HCC.

Retinol metabolism-related genes are enriched in hepatocellular carcinoma. A and B Principal component analysis (PCA) shows the gene expression distribution in 4 hepatocellular carcinoma cohort samples (GSE101685, GSE164760, GSE174570 and GSE84402) before (A) and after (B) batch effect correction. C Volcano plots showing the differentially expressed genes between hepatocellular carcinoma samples and normal samples in merged cohort. Red dots represent upregulated genes, blue dots indicate genes that are downregulated, and gray dots represent genes for which the differences are not statistically significant. D The expression of differentially expressed genes in the normal group and the tumor group was characterized by high expression in red and low expression in blue. E, F GO and KEGG enrichment analysis of differentially expressed genes
3.2 Causal relationship between retinol metabolism and HCC
In our previous analysis, we observed a significant enrichment of differentially expressed genes within the retinol metabolism pathway. To gain further insights into the causal relationship between retinol metabolism and HCC, we employed a two-sample Mendelian randomization approach to explore the potential association from a genetic variation perspective. In this analysis, three SNPs were extracted with retinol as the exposure factor and HCC as the outcome. Figure 3A presents specific forest plots using different MR analysis methods to estimate each causal effect. The IVW analysis revealed that lower retinol levels were associated with higher HCC risk (OR = 0.765; 95%CI 0.634–0.924; p = 0.005), which was also supported by the weighted median method (OR = 0.762; 95%CI 0.603–0.962; p = 0.022). Although the result of weighted mode analysis did not support a causal correlation with HCC (OR = 0·737; 95%CI 0·565–0·963; P = 0154), IVW was considered as the primary outcome due to its high statistical efficiency and reliable results (Bowden & Holmes, 2019). The heterogeneity of outcomes was analyzed using MR-Egger and IVW methods, both indicating no heterogeneity among selected instrumental variables (IVs) (Table 1). MR-Egger was used to assess horizontal pleiotropy of selected IVs, and the result (intercept = 0128; SE = 0171; p = 059) suggested that there was no pleiotropy affecting the results (Table 1; Fig. 3B). The funnel plot displayed an approximate funnel shape for instrumental variables while ignoring directional pleiotropy (Figure S1). Leave-one-out sensitivity analysis demonstrated unbiased estimates of causal effects without being influenced by individual instrumental variables, thus confirming their reliability (Fig. 3C). These findings provide evidence for a protective role of retinol in reducing HCC risk, suggesting a causal relationship between retinol metabolism and HCC.

Mendelian randomized analysis between hepatocellular carcinoma and retinol metabolism. A Estimation of retinol with hepatocellular carcinoma causal effect forest plot. B MR test scatter plot of five methods. The x-axis is the SNP effect on retinol. The y-axis is the SNP effect on hepatocellular carcinoma. C Forest plot for the Mendelian randomization leave-one-out sensitivity analysis of single SNP on HCC. MR, Mendelian randomization; SNP, single-nucleotide polymorphism; OR, odds ratio; CI, confidence interval
3.3 Analysis of the protein–protein interaction (PPI) network, competing endogenous RNA (ceRNA) network, drug-gene interaction network, and correlation with immune cells was performed for differentially expressed genes involved in the retinol metabolism pathway
The volcano plot (Fig. 4A) revealed significant downregulation of all 11 differentially expressed genes involved in retinol metabolism within cancer tissues, consistent with the results obtained from Mendelian randomization analysis. This observation suggests that the retinol metabolism pathway plays a protective role in HCC. To gain further insights into the interactions among these genes, we constructed a protein–protein interaction (PPI) network using data from the STRING database (Fig. 4B). The results demonstrated close interactions among these eleven differentially expressed genes within this specific pathway. Furthermore, we conducted a search on the DGIdb database to identify potential drugs targeting retinol metabolism associated with these differential gene expressions. The identification of 20 drugs targeting CYP1 A2, 2 drugs targeting CYP26 A1, 20 drugs targeting CYP2B6, 20 drugs targeting CYP2 C8, 20 drugs targeting CYP3 A4, 20 drugs targeting CYP2 C9, 20 drugs targeting CYP2 A6, 1 drug targeting ADH4 and 5 drugs targeting ADH1 C is demonstrated in Fig. 4C. Subsequently, utilizing cytoscape software generated a gene-drug network comprising of a total of124 nodes (9 genes and 115 drugs). These potential medications offer novel therapeutic avenues for HCC by specifically modulating retinol metabolism. Subsequently, we employed four well-established miRNA databases (miRanda, miRDB, miRWalk, and TargetScan) to predict the binding of target miRNAs with 11 differentially expressed genes in specific pathways. A total of 39 mRNA-miRNA pairs were identified. Subsequently, utilizing these 39 validated miRNAs as a basis, we conducted a comprehensive search for relevant lncRNAs in the spongeScan database and successfully obtained 117 potential miRNA-lncRNA pairs. Finally, employing Cytoscape software, we constructed a ceRNA network comprising of 122 nodes (6 mRNAs, 18 miRNAs, and 98 lncRNAs) as depicted in Fig. 4D. The tumor immune microenvironment plays a pivotal role in the initiation and progression of tumors. Therefore, we investigated the association between differentially expressed genes involved in retinol metabolism and immune cell infiltration, leading to the generation of a corresponding heatmap (Fig. 4E). Our findings revealed a strong positive correlation between γδT cells and most pathway differentially expressed genes. Furthermore, we observed that the RDH16 gene exhibited a robust negative correlation with regulatory T cells and a significant positive correlation with monocytes. Subsequently, in the validation cohort, we conducted further analysis on the expression of 11 differentially expressed genes associated with retinol metabolism pathway. Remarkably, these results demonstrated substantial downregulation of these genes in cancer tissues, consistent with our previous observations (Fig. 4F). Collectively, our data highlight significant downregulation of retinol metabolism pathway-related genes in HCC, suggesting their potential as therapeutic targets or prognostic biomarkers.

Comprehensive analysis of differentially expressed genes. A The volcano diagram shows 11 differentially expressed genes in the retinol metabolism pathway. The red dots indicate up-regulated genes, the green dots indicate down-regulated genes, and the gray dots indicate genes with no significant differences. B Protein–protein interaction of 11 differentially expressed genes (C) Constructed the network of drug-gene interactions. Red represents differentially expressed genes, and blue represents potential drugs. D Construction of the ceRNA networks and the potential RNA regulatory pathways of 11 differentially expressed genes. Red represents differentially expressed genes, green represents miRNA, and blue represents lncRNA. E The correlation between 11 differentially expressed genes and 21 types of immune infiltrating cells. F The expression levels of 11 differentially expressed genes were examined in the validation set (GSE76427). “*” indicates P < 0.01 and “***” indicates P < 0.001
3.4 Comprehensive analysis of RM-related DEGs in HCC
In order to determine the molecular subtypes of HCC based on retinol metabolism, we collected 115 HCC samples from GSE76427 and 363 HCC samples from the TCGA-LIHC dataset. Consensus clustering analysis was performed on the 11 differentially expressed genes (DEGs), revealing two distinct gene clusters (Fig. 5A). Notably, downregulation of retinol metabolism-related genes was observed in subtype B (Fig. 5B), which correlated with a more advanced clinical stage in HCC patients belonging to this subtype. Importantly, there was a significant difference in clinical prognosis between the two RM-based subtypes, with RM-cluster B showing poorer prognosis compared to RM-cluster A (p = 0.036, Fig. 5C). The differential KEGG pathway analysis further confirmed enrichment of the retinol metabolism pathway in HCC, with significantly decreased activity observed in subtype B (Fig. 5D). Additionally, single-sample gene set enrichment analysis (ssGSEA) algorithm was applied to explore immune infiltration levels between the two subgroups. While most immune cells exhibited high infiltration levels in RM-cluster B, it is noteworthy that this subgroup also displayed an abundance of immunosuppressive cells such as dendritic cells, myeloid-derived suppressor cells (MDSCs), and macrophages (Fig. 5E). These findings suggest that reduced activity of retinol metabolism is associated with poor prognosis and immune suppression among HCC patients.

Identification of RM-related subgroups. A Consensus matrix heatmap defining two subgroups. TCGA-LIHC and GSE76427 cohorts were divided into two subgroups based on gene expression profiles. The variable k is a parameter used to determine the number of clusters and k = 2 indicates that two subgroups are clustered. B A complex heatmap showing the differential gene expression levels related to retinol metabolism between two subgroups. Blue represents the gene expression of subgroup A, while orange represents the gene expression of subgroup B. Clinical pathological features, including clinical stage, gender, and age are displayed on top of the complex heatmap. C The overall survival of the two subgroups. Blue represents patients in subgroup A and orange represents patients in subgroup B. The p-value is 0.036. D KEGG pathway analyses of GSVA in the two subgroups. Blue represents subgroup A and orange represents subgroup B. E Boxplot of the abundance of immune cells in the two subgroups. The x-axis represents the type of immune cells and the y-axis represents the level of immune infiltration. Blue represents subgroup A and orange represents subgroup B. RM, retinol metabolism, *p < 0.05, **p < 0.01, ***p < 0.001
3.5 Construction of prognostic signature based on RM-related DEGs
We conducted an analysis to assess the prognostic significance of differentially expressed genes (DEGs) related to regenerative medicine in HCC. The TCGA-LIHC dataset was utilized as the training set, while GSE76427 served as the testing set. Single-factor Cox regression analysis was employed to identify seven prognostic factors (p < 0.05), all of which exhibited a disease-protective effect (Fig. 6A). Subsequently, utilizing the'glmnet'R package, we performed LASSO analysis and selected three crucial variables from these seven prognostic factors (Fig. 6B). A prognostic risk model comprising three independent prognostic factors was established using multivariate Cox analysis, and the risk scores for HCC samples were calculated accordingly. The formula for calculating the risk score is as follows: Risk Score = RDH16 * (− 0.0028) + ADH4 * (− 0.068) + CYP2 C9 * (− 0.058). Based on the median value of the risk score, patients were categorized into two groups. In the training dataset, patients with high-risk scores exhibited lower survival rates compared to those with low-risk scores (Fig. 6C). Moreover, consistent results were observed in terms of overall survival rates among different risk groups in the testing dataset (Fig. 6D). Furthermore, the AUC values on the ROC curves for patient survival at 1 year, 3 years, and 5 years exceeded 0.6, indicating enhanced predictive accuracy (Fig. 6E). By integrating clinical data from TCGA-LIHC and GSE76427 datasets, we developed a nomogram based on the risk score and clinical variables to assess the survival potential of HCC samples at different time points (1 year, 3 years, and 5 years) (Fig. 6F). The calibration curve demonstrated close alignment between predicted outcomes and ideal expectations across all three time intervals (Fig. 6G). Overall, our successful establishment of a prognostic risk model utilizing RM-related DEGs showcases its robust performance in predicting liver cancer patient survival rates.

Constructing a prognostic model based on RM-related DEGs. A The univariate Cox analysis of RM-related DEGs. B Identification of feature prognostic variables via LASSO analysis. C Clinical survival curve analysis of HCC samples in the risk subtypes in the training cohorts (TCGA-LIHC). D Clinical survival curve analysis of HCC samples in the risk subtypes in the test cohorts (GSE76427). E One- three- and 5 year’s AUC in the TCGA-LIHC cohorts. FNomogram construction of risk score and clinical variables. G Correction curve for survival prediction. RM, retinol metabolism; DEGs, differentially expressed genes. *p < 0.05, ***p < 0.001
3.6 RDH16 was identified as a key gene in the disease based on the retinol metabolism pathway
To further investigate the key genes associated with the disease, we employed the random forest algorithm to predict the significance of 11 differentially expressed genes in the retinol metabolism pathway. The error rates for both treatment and control groups, as well as overall samples, were depicted on a tenfold cross-validation curve. Additionally, we ranked these genes based on their Gini coefficients (Fig. 7A). Among all 11 genes, RDH16 exhibited the highest importance in relation to the disease, underscoring its substantial impact and confirming its status as a pivotal gene. According to analysis results from the GEPIA database, the mRNA expression level of RDH16 in HCC tissues increased significantly (p < 0.05) (Fig. 7B). Furthermore, patients with diminished RDH16 expression displayed markedly reduced overall survival rates (Fig. 7C). The immunohistochemical results from the HPA database revealed a significant decrease in the expression level of RDH16 protein in HCC tissues compared to adjacent non-cancerous tissues (Fig. 7D, E). Analysis of the GSE146409 single-cell dataset also demonstrated a significant upregulation of the RDH16 gene in normal liver cells, while it was downregulated in other cell types including malignant cells (Fig. 7F, G). Furthermore, qPCR analysis on four cell lines and sixteen paired clinical samples of liver cancer confirmed that the RDH16 gene was significantly downregulated in cancer cell lines and tumor tissues (Fig. 7H, I). Western Blotting results further supported these findings by showing a substantial downregulation of retinol dehydrogenase 16 encoded by the RDH16 gene in three liver cancer cell lines and six clinical tissue samples from HCC patients (Fig. 7J, K). Collectively, these observations suggest that there is a significant downregulation of the RDH16 gene in HCC which correlates with poor prognosis for patients.

RDH16 is identified as a key gene for disease based on RM. A Random Forests Identifying key genes for disease. B Box line plots of mRNA expression levels for RDH16 in the GEPIA database. C Kaplan–Meier curves showed overall survival difference between high and low RDH16 expression groups (p = 0.011). D, E Immunohistochemical analysis of RDH16 in HCC by HPA database. F Identification of cell subgroups in the tumor microenvironment of HCC. G Expression pattern of RDH16 in the tumor microenvironment. H Real time-PCR revealed that the expression of RDH16 was notably decreased in HCC cell lines. I Real time-PCR was conducted to explore the expression of RDH16 in HCC tissues (n = 16) and normal tissues (n = 16). J Western blot analysis was performed to examine the expression of RDH16 in one normal liver cell line (LO2) and three HCC cell lines, with GAPDH expression used as an internal control. K Western blot analysis of RDH16 expression levels in 6 paired HCC and non-tumor specimens. GAPDH was used as a loading control. T, tumor tissue; N, non-tumor tissue.RM, retinol metabolism; HCC, hepatocellular carcinoma; HPA, Human Protein Atlas. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
3.7 Inhibiting the expression of RDH16 can promote proliferation, migration, and invasion of liver cancer cells while suppressing cell apoptosis.
To investigate the potential role of RDH16 in HCC cells, we employed siRNA to modulate the expression of RDH16 in HepG2 and Huh- 7 cells. The successful transfection was confirmed by qPCR and WB results (Fig. 8A, B). CCK- 8 experiments revealed that inhibition of RDH16 expression significantly enhanced the proliferative ability of HCC cells (Fig. 8C, D). Moreover, wound healing assays demonstrated a significant increase in the migratory ability of HCC cells upon inhibiting RDH16 expression (Fig. 8E, F). Furthermore, Transwell experiments were conducted to assess changes in migration and invasion abilities of both HCC cell lines following inhibition of RDH16 expression. The results showed a substantial increase in the number of cells passing through the membrane when RDH16 was inhibited (Fig. 8G, H). Finally, we examined the effect of RDH16 gene on HCC cell apoptosis by flow cytometry. The results showed that the apoptosis rate of HCC cells was significantly reduced when the expression of RDH16 was inhibited (Fig. 8I, J). Collectively, these findings suggest that RDH16 exerts suppressive effects on proliferation, migration, invasion while promoting apoptosis in HCC cells.

The effects of RDH16 on the proliferation, migration, invasion, and apoptosis of HCC cells. A, B Detection of the inhibitory effect of siRNA on RDH16 expression in HepG2 and Huh- 7 cell lines using Real time-PCR and Western blot analysis. C, D CCK- 8 assay indicated that inhibiting RDH16 markedly enhanced viability of HepG2 and Huh- 7 cells. E, F Wound healing assay revealed that inhibiting RDH16 obviously strengthened mobility of HepG2 and Huh- 7 cells. G, H Transwell assays revealed that inhibiting RDH16 notably promoted migration and invasion of HepG2 and Huh- 7 cells. I, J Suppression of RDH16 expression resulted in a significant reduction of apoptosis in HepG2 and Huh- 7 cells. *p < 0.05, **p < 0.01, ***p < 0.001
3.8 RDH16 is a downstream target of miR- 665 in HCC cells
In our previous analysis, we predicted the ceRNA regulatory network of differentially expressed genes (DEGs) involved in retinol metabolism and identified miR- 665 as a potential regulator targeting RDH16 directly in HCC. Previous studies have reported that miR- 665 promotes HCC progression. Therefore, we quantified the expression levels of miR- 665 using qPCR in four cell lines and sixteen pairs of clinical liver cancer samples. The results revealed a significant upregulation of miR- 665 both in cancer cell lines and liver tumor tissues (Fig. 9A). Moreover, a negative correlation was observed between the expression levels of miR- 665 and RDH16 (r = − 0.6966, p < 0.01; Fig. 9B). Furthermore, inhibition of miR- 665 through qPCR and Western blot analyses resulted in increased expression of RDH16 specifically in HepG2 and Huh- 7 cells (Fig. 9C, D). These findings suggest that miR- 665 binds to the 3'UTR region of RDH16 (Fig. 9E). Additionally, fluorescence activity assays demonstrated enhanced activity upon inhibition of miR- 655 on WT-RDH16 − 3'UTRs but no effect on MUT-RDH16 − 3'UTRs (Fig. 9F), indicating that RDH16 is indeed targeted by miR- 655 and may mediate its effects on HCC cells.

RDH16 is a direct target of miR- 665 in HCC. A The mRNA levels of miR- 665 in HCC cell lines, normal L02 cells and16-paired human HCC and adjacent normal tissues. B A negative correlation between the levels of miR- 665 and RDH16 expression in HCC specimens. C, D The mRNA and protein levels of RDH16 in silencing miR- 665 HepG2 and Huh- 7 cells. E The potential miR- 665 binding site in the RDH16 3′-UTR was predicted by TargetScan. F Dual-luciferase reporter assay was used to analyze the effects of miR- 665 expression on the activities of WT and MUT RDH16 3′-UTR. *P < 0.05, **P < 0.01, ***p < 0.001, ****p < 0.0001, nsp > 0.05
3.9 miR- 665 mediates the effects of RDH16 on migration, invasion, and proliferation of HCC cells
To validate the hypothesis that RDH16 functions as a mediator of miR- 665 in HCC cells, we conducted rescue experiments by co-transfecting distinct plasmids harboring miR- 665 and RDH16 into HepG2 or Huh- 7 cells. qPCR and Western blot analyses revealed that individual transfection of RDH16-siRNA significantly reduced the expression of RDH16 in both HepG2 and Huh- 7 cells, whereas sole transfection of the miR- 665 inhibitor increased the expression level of RDH16 in these cells. However, when anti-miR- 665 was combined with RDH16-siRNA, the decline in the mRNA and protein expression of RDH16 induced by RDH16-siRNA was partially counteracted by the presence of the miR- 665 inhibitor (Fig. 10A, B). The results obtained from CCK- 8 assay-based rescue experiments demonstrated that the proliferative effect exerted by RDH16-siRNA on HepG2 and Huh- 7 cell lines was attenuated upon addition of the miR- 665 inhibitor (Fig. 10C, D). Moreover, Transwell assay and wound healing assay-based rescue experiments further substantiated that migration, invasion, and motility enhancement mediated by RDH16-siRNA were mitigated upon inhibition of miR- 665 in HepG2 and Huh- 7 cells (Fig. 10E–H). In conclusion, our findings provide evidence supporting a role for miR- 665 as a mediator through which RDH16 influences migration, invasion, and proliferation dynamics within HCC cells.

Rescue experiments confirmed the interaction between miR- 665 and RDH16 in HCC progression. A, B RDH16 expression in HepG2 and Huh- 7 cells co-transfected with RDH16 siRNA, miR- 665 inhibitors, and corresponding controls, was detected by qPCR and Western blot. C, D CCK- 8 assay revealed that miR- 665 reversed the effect of RDH16 on viability of HCC cells. E, F Transwell assays revealed that miR- 665 reversed the effect of RDH16 on migration and invasion of HCC cells. (G, H) Wound healing assay revealed that miR- 665 reversed the effect of RDH16 on mobility of HCC cells. *P < 0.05, **P < 0.01, ***p < 0.001, ****p < 0.0001
4 Discussion
Retinol, the biologically active form of vitamin A, plays a pivotal role in numerous biological processes encompassing normal growth and development [14], maintenance of tissue homeostasis [15], and disease prevention [16]. Functioning as nuclear DNA-binding receptors, retinol regulates gene transcription through RARs and RXRs [16]. Extensive studies have demonstrated the inhibitory effects of retinol on various tumor types including breast cancer, skin cancer, lung cancer, and prostate cancer [17]. Notably, a published study has reported an elevated risk of liver cell carcinoma associated with high serum retinol levels [10].
Emerging evidence has increasingly underscored the intricate involvement of retinol metabolism in cancer biology. Notably, retinol and its derivatives, particularly atRA, have been demonstrated to play pivotal roles in regulating cellular differentiation, proliferation, and apoptotic processes, which are fundamental mechanisms underlying cancer pathogenesis and progression [17]. Specifically in HCC, retinol metabolism has been identified as a crucial modulator of tumor microenvironment dynamics and immune regulation. A recent investigation has revealed significant dysregulation of retinol metabolic pathways in HCC, which consequently affects immune cell infiltration patterns and facilitates tumor advancement [18]. These findings are consistent with our observations indicating that diminished retinol metabolism activity correlates with enhanced immune suppression and unfavorable clinical outcomes in HCC patients.
In this study, we further demonstrated the strong correlation between decreased activity in the retinol metabolism pathway and the risk of HCC, as well as poor prognosis in HCC patients, through batch RNA sequencing, Mendelian randomization analysis, and experimental validation. Firstly, significant enrichment of differentially expressed genes in the retinol metabolism pathway was identified in HCC patients using bulk RNA sequencing from public datasets. Subsequently, a causal relationship between retinol levels and HCC susceptibility at the genetic variation level was confirmed, indicating that lower retinol levels are associated with a higher risk of developing HCC. Furthermore, an extensive investigation on 11 differentially expressed genes involved in the retinol metabolism pathway was conducted. Close interactions among these differential genes were discovered and specific drugs targeting these genes were predicted using DGIdb data to provide insights for precise treatment of HCC patients based on the retinol metabolism pathway. Additionally, a ceRNA regulatory network was constructed to explore the functions and regulatory mechanisms of these genes at a deeper level; it was found that miR- 665 can bind to RDH16's 3'-UTR.
Our findings align with prior research investigating the role of retinol metabolism in hepatic pathophysiology. Previous investigation has demonstrated that retinol metabolism plays a pivotal role in maintaining hepatic homeostasis, with its dysregulation being implicated in the pathogenesis of chronic liver diseases, including HCC [19]. Furthermore, emerging evidence suggests that genes associated with retinol metabolism may serve as potential biomarkers for early HCC detection, which corroborates our identification of RDH16 as a critical prognostic marker in HCC [20]. These collective findings not only reinforce the significance of retinol metabolism in hepatic oncogenesis but also provide a comprehensive scientific framework for interpreting our results within the broader context of liver cancer biology.
Based on the expression levels of 11 differentially expressed genes in the retinol metabolism pathway, we performed subtype analysis on patients with HCC. We observed a significant reduction in overall survival rates among patients with low expression of these differential genes (RMcluter B). Immune infiltration analysis revealed that RMcluter B patients exhibited abundant immune infiltration, primarily composed of inhibitory cells such as MDSCs, activated dendritic cells, and tumor-associated macrophages. MDSCs possess potent immunosuppressive activity and can hinder the function of T cells and NK cells, thereby promoting immune evasion [21]. Previous studies have suggested that systemic disruption of dendritic cell differentiation and development within the tumor microenvironment is a crucial factor contributing to tumor immune escape [22]. Recent studies have increasingly emphasized the pivotal role of tumor microenvironment in hepatobiliary cancer progression and patient outcomes. Particularly, emerging evidence has highlighted the prognostic significance of tumor-associated lymphatic vessel density in these malignancies, underscoring the critical influence of immune and stromal components on tumor behavior and clinical prognosis [23]. These findings are consistent with our observations regarding immune infiltration patterns in HCC subtypes, reinforcing the importance of tumor microenvironment in HCC progression. The identification of novel biomarkers and prognostic indicators for HCC has become a focal point in current research. A recent investigation has demonstrated that portal venous and hepatic arterial coefficients serve as reliable predictors for post-hepatectomy overall and recurrence-free survival in HCC patients, offering valuable prognostic tools for clinical application [24]. Furthermore, the impact of environmental factors on HCC progression has been increasingly recognized, with evidence suggesting that high-altitude hypoxic stress may significantly influence HCC development, providing new perspectives for future biomarker research [25].These collective findings highlight the necessity for continued exploration of novel biomarkers and prognostic indicators in HCC, particularly those associated with metabolic pathways such as retinol metabolism. The integration of microenvironmental factors and metabolic pathway analysis may offer more comprehensive insights into HCC progression and potential therapeutic targets.
Subsequently, we developed a prognostic model based on the expression levels of RDH16, ADH4, and CYP2 C9 genes which demonstrated excellent predictive value. The ADH4 gene is an important member located at 4q22 encoding class II ADH4 pi subunit within the ADH family consisting of 9 exons [26]. Studies have indicated that polymorphisms in the ADH4 gene are associated with decreased risk of HCC and ovarian cancer [27], suggesting its involvement in HCC development. CYP2 C9 is one among the most abundant and vital xenobiotic-metabolizing enzymes whose substrates include commonly prescribed drugs like warfarin, nonsteroidal anti-inflammatory drugs (NSAIDs), tolbutamide, phenytoin, and torasemide [28]. Furthermore,some studies have reported reduced expression of CYP2 C9 in HCC tissues [29,30,31],suggesting its potential involvement in detoxification processes contributing to HCC pathogenesis.
RDH16 belongs to the short-chain dehydrogenase/reductase superfamily and is considered a tumor suppressor in glioblastoma [32]. Although the catalytic activity of RDH16 has been indirectly investigated, its biological function in HCC remains elusive. In this study, we observed downregulation of RDH16 in HCC patients, and low expression of RDH16 was associated with poor prognosis. Single-cell analysis and cytological levels further validated this finding. Functionally, we discovered that inhibition of RDH16 gene expression promoted proliferation, migration, and metastasis of liver cancer cells while suppressing tumor cell apoptosis. Additionally, we provided substantial evidence demonstrating the interaction between miR- 665 and RDH16. Firstly, miR- 665 negatively regulated both mRNA and protein levels of RDH16 in HCC cells. Secondly, miR- 665 influenced luciferase activity on WT 3'-UTR but not on MUT 3'-UTR of RDH16. Thirdly, there was a negative correlation between miR- 665 expression and RDH16 in HCC tissues. The functional role of miR- 665 has been proposed in malignant tumors; for instance, Chen et al., reported upregulation of miR- 665 in gastric adenocarcinoma but downregulation in signet ring cell carcinoma [33]. This suggests that miR- 665 may exert different biological functions depending on the specific type of tumor involved. However, our study still has limitations as it does not elucidate the precise functional role played by the RDH16 gene within an organism; furthermore, we were unable to determine the specific mechanism underlying interactions between RDH16 and miR- 665 which requires further investigation.
5 Conclusions
In conclusion, our study provides the first molecular and genetic evidence indicating a reduction in retinol metabolism activity among HCC patients, which is causally associated with HCC development. Moreover, we have identified RDH16 as a characteristic gene involved in retinol metabolism that acts as an anti-cancer gene by suppressing the migration, invasion, and proliferation of HCC cells. Additionally, we have discovered miR- 665 as a novel upstream regulator of the RDH16 gene in HCC cells. Therefore, we propose that retinol metabolism holds significant potential for clinical diagnosis, treatment, and prognosis of HCC.
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49. https://doi.org/10.3322/caac.21660.
Thomas H. Liver cancer: lenvatinib non-inferior to sorafenib for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2018;15(4):190. https://doi.org/10.1038/nrgastro.2018.20.
Luo P, Yin P, Hua R, Tan Y, Li Z, Qiu G, et al. A Large-scale, multicenter serum metabolite biomarker identification study for the early detection of hepatocellular carcinoma. Hepatology. 2018;67(2):662–75. https://doi.org/10.1002/hep.29561.
Brown ZJ, Tsilimigras DI, Ruff SM, Mohseni A, Kamel IR, Cloyd JM, et al. Management of hepatocellular carcinoma: a review. JAMA Surg. 2023;158(4):410–20. https://doi.org/10.1001/jamasurg.2022.7989.
Chaiteerakij R, Addissie BD, Roberts LR. Update on biomarkers of hepatocellular carcinoma. Clin Gastroenterol Hepatol. 2015;13(2):237–45. https://doi.org/10.1016/j.cgh.2013.10.038.
Kimhofer T, Fye H, Taylor-Robinson S, Thursz M, Holmes E. Proteomic and metabonomic biomarkers for hepatocellular carcinoma: a comprehensive review. Br J Cancer. 2015;112(7):1141–56. https://doi.org/10.1038/bjc.2015.38.
Wu Y, Huang T, Li X, Shen C, Ren H, Wang H, et al. Retinol dehydrogenase 10 reduction mediated retinol metabolism disorder promotes diabetic cardiomyopathy in male mice. Nat Commun. 2023;14(1):1181. https://doi.org/10.1038/s41467-023-36837-x.
Szymański Ł, Skopek R, Palusińska M, Schenk T, Stengel S, Lewicki S, et al. Retinoic acid and its derivatives in skin. Cells. 2020. https://doi.org/10.3390/cells9122660.
Chronopoulos A, Robinson B, Sarper M, Cortes E, Auernheimer V, Lachowski D, et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat Commun. 2016;7:12630. https://doi.org/10.1038/ncomms12630.
Han J, Han ML, Xing H, Li ZL, Yuan DY, Wu H, et al. Tissue and serum metabolomic phenotyping for diagnosis and prognosis of hepatocellular carcinoma. Int J Cancer. 2020;146(6):1741–53. https://doi.org/10.1002/ijc.32599.
Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47(D1):D1005-d12. https://doi.org/10.1093/nar/gky1120.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. https://doi.org/10.1093/nar/gkv007.
Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7. https://doi.org/10.1186/1471-2105-14-7.
Zile MH. Function of vitamin A in vertebrate embryonic development. J Nutr. 2001;131(3):705–8. https://doi.org/10.1093/jn/131.3.705.
Tyson-Capper AJ, Cork DM, Wesley E, Shiells EA, Loughney AD. Characterization of cellular retinoid-binding proteins in human myometrium during pregnancy. Mol Hum Reprod. 2006;12(11):695–701. https://doi.org/10.1093/molehr/gal070.
Niles RM. Signaling pathways in retinoid chemoprevention and treatment of cancer. Mutat Res. 2004;555(1–2):81–96. https://doi.org/10.1016/j.mrfmmm.2004.05.020.
Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer. 2001;1(3):181–93. https://doi.org/10.1038/35106036.
Melis M, Tang XH, Trasino SE, Gudas LJ. Retinoids in the pathogenesis and treatment of liver diseases. Nutrients. 2022. https://doi.org/10.3390/nu14071456.
Cortes E, Lachowski D, Rice A, Chronopoulos A, Robinson B, Thorpe S, et al. Retinoic acid receptor-β is downregulated in hepatocellular carcinoma and cirrhosis and its expression inhibits myosin-driven activation and durotaxis in hepatic stellate cells. Hepatology. 2019;69(2):785–802. https://doi.org/10.1002/hep.30193.
Zhao Y, Wan K, Wang J, Wang S, Chang Y, Du Z, et al. DNA methylation and gene expression profiling reveal potential association of retinol metabolism related genes with hepatocellular carcinoma development. PeerJ. 2024;12:e17916. https://doi.org/10.7717/peerj.17916.
Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol. 2006;176(1):284–90. https://doi.org/10.4049/jimmunol.176.1.284.
Li R, Fang F, Jiang M, Wang C, Ma J, Kang W, et al. STAT3 and NF-κB are simultaneously suppressed in dendritic cells in lung cancer. Sci Rep. 2017;7:45395. https://doi.org/10.1038/srep45395.
Li J, Liang YB, Wang QB, Li YK, Chen XM, Luo WL, et al. Tumor-associated lymphatic vessel density is a postoperative prognostic biomarker of hepatobiliary cancers: a systematic review and meta-analysis. Front Immunol. 2024;15:1519999. https://doi.org/10.3389/fimmu.2024.1519999.
Li YK, Wu S, Wu YS, Zhang WH, Wang Y, Li YH, et al. Portal venous and hepatic arterial coefficients predict post-hepatectomy overall and recurrence-free survival in patients with hepatocellular carcinoma: a retrospective study. J Hepatocell Carcinoma. 2024;11:1389–402. https://doi.org/10.2147/jhc.S462168.
Liang Y, Li L, Liu B, Gao J, Chen X, Li J, et al. [Research Advances in the Roles of High-Altitude Hypoxic Stress in Hepatocellular Carcinoma]. Sichuan Da Xue Xue Bao Yi Xue Ban. 2024;55(6):1436–45. https://doi.org/10.12182/20241160605.
Luo X, Kranzler HR, Zuo L, Yang BZ, Lappalainen J, Gelernter J. ADH4 gene variation is associated with alcohol and drug dependence: results from family controlled and population-structured association studies. Pharmacogenet Genom. 2005;15(11):755–68. https://doi.org/10.1097/01.fpc.0000180141.77036.dc.
Goode EL, White KL, Vierkant RA, Phelan CM, Cunningham JM, Schildkraut JM, et al. Xenobiotic-metabolizing gene polymorphisms and ovarian cancer risk. Mol Carcinog. 2011;50(5):397–402. https://doi.org/10.1002/mc.20714.
Miners JO, Birkett DJ. Cytochrome P4502C9: an enzyme of major importance in human drug metabolism. Br J Clin Pharmacol. 1998;45(6):525–38. https://doi.org/10.1046/j.1365-2125.1998.00721.x.
Chen X, Cheung ST, So S, Fan ST, Barry C, Higgins J, et al. Gene expression patterns in human liver cancers. Mol Biol Cell. 2002;13(6):1929–39. https://doi.org/10.1091/mbc.02-02-0023.
Kim JW, Ye Q, Forgues M, Chen Y, Budhu A, Sime J, et al. Cancer-associated molecular signature in the tissue samples of patients with cirrhosis. Hepatology. 2004;39(2):518–27. https://doi.org/10.1002/hep.20053.
Yu D, Green B, Marrone A, Guo Y, Kadlubar S, Lin D, et al. Suppression of CYP2C9 by microRNA hsa-miR-128–3p in human liver cells and association with hepatocellular carcinoma. Sci Rep. 2015;5:8534. https://doi.org/10.1038/srep08534.
Hu PS, Xia QS, Wu F, Li DK, Qi YJ, Hu Y, et al. NSPc1 promotes cancer stem cell self-renewal by repressing the synthesis of all-trans retinoic acid via targeting RDH16 in malignant glioma. Oncogene. 2017;36(33):4706–18. https://doi.org/10.1038/onc.2017.34.
Chen J, Sun D, Chu H, Gong Z, Zhang C, Gong B, et al. Screening of differential microRNA expression in gastric signet ring cell carcinoma and gastric adenocarcinoma and target gene prediction. Oncol Rep. 2015;33(6):2963–71. https://doi.org/10.3892/or.2015.3935.
Acknowledgements
We would like to express our sincere gratitude to Professor Zhou Jianyin for his invaluable guidance, assistance, and unwavering support throughout this research endeavor. Additionally, we extend our heartfelt appreciation to the team of Academician Lin Shengcai at Xiamen University's School of Life Sciences for providing us with access to their exceptional experimental platform and various forms of assistance.
Funding
This study was supported by Fujian Provincial Natural Science Foundation (No.2024 J011331) and Xiamen Natural Science Foundation(No.3502Z20227269).
Author information
Authors and Affiliations
Contributions
Fuqing Chen: Writing- -original draft, Methodology, Formal analysis, Validation. Yifan Cai: Formal analysis, Software, Investigation. Jianyin Zhou: Writing-review & editing, Supervision, Project administration, Funding acquisition, Conceptualization. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The current study was approved (approval number: XMZSYYKY (2024–621)) by the Institutional Review Board (or Ethics Committee) of Medical Ethics Committee of Zhongshan Hospital of Xiamen University. Written informed consent for the utilization of tissues in scientific research was duly obtained from all patients.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, F., Cai, Y. & Zhou, J. Relationship between retinol metabolism and hepatocellular carcinoma: a comprehensive analysis of Mendelian randomization, prognostic characteristic and experiment. Discov Onc 16, 513 (2025). https://doi.org/10.1007/s12672-025-02295-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-025-02295-8