
Abstract
Background
Lung squamous cell carcinoma (LUSC) is a leading cause of cancer-related mortality, and tumor heterogeneity could result in diverse prognostic subtypes. Traditional prognostic factors, like tumor, node, and metastasis (TNM) staging, offer limited predictive accuracy. This study aims to identify LUSC subtypes and develop predictive models that have the potential to improve prognosis prediction accuracy and support personalized treatment.
Methods
Expression and clinical data were collected from three datasets. One dataset (TCGA-LUSC) was used as a training set, while the others (GSE30219 and GSE73403) were independent testing sets. Unsupervised clustering was applied to the training set to identify LUSC subtypes. The relationship between survival outcomes and these identified subtypes was validated in the testing sets using binary machine learning models and survival curve analysis. The impact of chemotherapy on the prognosis for subtypes was also presented. Subsequently, four survival machine learning models were developed to predict LUSC prognosis. These models were validated in the testing sets and integrated into an online tool to assist in survival prediction.
Results
Two subtypes, C1 and C2, were identified in the training set. The C1 subtype was associated with poorer survival outcomes and was enriched in cancer-associated fibroblasts and macrophages. In contrast, the C2 subtype correlated with better outcomes and was enriched in CD8 + T cells. Regarding chemotherapy, the C2 subtype with chemotherapy showed the best survival outcomes compared to other groups. A 9-gene signature was derived from the model’s importance values for subtype prediction and included TGM2, AOC3, TBXA2R, RGS3, DLC1, MMP19, ACVRL1, TCF21, and TIMP3. This signature outperformed 14 published signatures and clinical variables at survival prediction with the highest time-dependent AUC (tdAUC) and concordance index (C-index). Four machine learning models were developed using this signature, achieving tdAUC values of 0.712 and 0.684 and C-index values of 0.682 and 0.625 in the independent testing sets. An online tool for predicting survival probabilities for LUSC patients up to 10 years post-treatment is available at https://hznuduan.shinyapps.io/LCSP/.
Conclusion
We identified two LUSC subtypes by unsupervised clustering and developed an online tool for prognosis prediction using supervised machine learning models.
Similar content being viewed by others

Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
1 Introduction
Lung cancer is a significant health threat worldwide, with approximately 1.8 million fatalities and 2.2 million new cases annually [1]. LUSC constitutes about 30% of all lung cancer cases and ranks second after lung adenocarcinoma [2]. The prognosis for LUSC patients is generally not satisfying because many cases are diagnosed at an advanced or metastatic stage. Although recent advancements in molecular targeted therapies and immunotherapies have improved survival time for some patients with LUSC [3], the treatment efficacy varies due to the tumor's heterogeneity. There is an urgent need to identify the subtypes of LUSC that may contribute to developing personalized medicine for LUSC.
Genomic data has been widely used to identify molecular subtypes in LUSC. For instance, one study utilized DNA methylation data to discover seven LUSC subtypes [4]. Similarly, another study divided LUSC samples into four immune subtypes based on the expression of immune-related genes [5]. Additionally, a research group has used somatic mutations and copy number alteration data to classify LUSC into molecular subtypes [6]. In another study, two LUSC subtypes with different lactate metabolism levels were identified [7]. However, several important criteria must be met for subtype identification to be robust and clinically significant. First, the identified subtypes should be validated using independent testing datasets to ensure reproducibility. Second, these subtypes should be linked with clinical outcomes and therapeutic responses, which enhances their potential impact on clinical decision-making. Finally, developing a prediction model for subtype assignment is essential for translating these findings into a clinical setting. In our study, we have comprehensively addressed these criteria. We identified novel LUSC subtypes, validated them across multiple datasets, and developed tools for precise subtype prediction in clinical practice.
The predictive prognostic models usually used clinical and gene expression data in public databases, such as TCGA. For instance, one study established a reliable 13-gene immune-related signature that can stratify and predict the prognosis of LUSC patients [8]. A two-gene risk model, developed by multivariate Cox analysis, correlated with survival and metastasis in LUSC [9]. Our study aims to use the strengths of multiple advanced algorithms, rather than relying on a single method, to develop more robust and accurate predictive models for LUSC. Besides, providing online tools after constructing machine learning models is necessary for the clinical practice of models.
In this study, we aimed to identify novel molecular subtypes by unsupervised clustering of mRNA expression data. We developed robust machine learning models incorporating genetic and clinical factors to predict patient survival. We created an online tool for personalized survival prediction based on these models to facilitate clinical application.
2 Method
2.1 Included datasets
Datasets were retrieved from the Gene Expression Omnibus (GEO) database using the following keywords:"Pulmonary"or"Lung,""Cancer"or"Tumor"or"Carcinoma"or"Neoplasm,""Squamous cell,"and"Prognosis"or"Survival"or"Prognostic". The search was restricted to studies characterized as either"Expression profiling by array"or"Expression profiling by high throughput sequencing", with the entry type limited to"Series". Only datasets containing between 40 and 1000 samples were included. After downloading the expression files, datasets were selected according to the following inclusion criteria: (1) the study must focus on solid squamous cell carcinoma (SCC) bulk samples; (2) the sample size for SCC must exceed 50; (3) essential data, including overall survival (OS) status, OS time, and TNM staging information, must be available and complete; (4) the expression profiles must contain 12,000 or more genes; (5) the dataset must not be duplicated; (6) the dataset must have been published after 2010. Additionally, the TCGA-LUSC, independent of the GEO database, was included in this study. As all datasets used in this analysis had been previously published, no institutional ethics approval was required. Expression data were obtained on March 10, 2024, and no identifiable participant information was accessed during or after data collection.
2.2 RNA-seq data processing
For the TCGA-LUSC dataset, level 3 RNA-Seq of mRNA expression data in transcripts per million (TPM) for LUSC patients was obtained using the TCGAbiolinks package [10]. Clinical information for TCGA-LUSC was sourced from the TCGA Pan-Cancer Clinical Data Resource (TCGA-CDR) [11]. Records of pharmaceutical treatments, such as chemotherapy, were retrieved through TCGAbiolinks. For GEO datasets, GSE30219 [12] and GSE73403, expression data (series matrix) and clinical information were obtained using the GEOquery package [13]. Only the maximum values were retained for genes with multiple probes, and expression data were log2-transformed.
2.3 Identification of patient subtypes in the training cohort
2.3.1 Unsupervised clustering
The training set (TCGA-LUSC) data was used to identify subtypes. To reduce gene number for clustering analysis, a series of filtering criteria were applied in the training set: (1) genes had to be present in the GSE30219, GSE73403, and TCGA-LUSC datasets; (2) only protein-coding genes were included; (3) genes required a relevance score greater than 1 with lung cancer in the GeneCards database since a higher score indicates stronger relevance to lung cancer [14]; (4) each gene needed a coefficient of variation (CV) exceeding 1; and (5) significant genes were identified through univariate Cox analysis, retaining only those with p-values below 0.05. These filtering steps effectively minimized the gene numbers and reduced clustering complexity. Finally, unsupervised clustering was performed on the training set.
The optimal number of subtypes in the training set was initially determined using the Silhouette method, Calinski-Harabasz (CH) index, and McClain index. The Silhouette method assesses how similar an object is to its subtype compared to others, with the optimal number of subtypes maximizing the average silhouette width. The CH index evaluates the ratio between-cluster variance and within-cluster variance, where a higher value indicates more distinct clusters. The McClain index compares the average within-cluster distance to the average between-cluster distance, with lower values indicating better clustering. After selecting the most suitable number of subtypes, the k-means method was employed to assign each sample to a specific subtype. Then, we plotted the survival curves of identified subtypes in the training dataset.
2.3.2 Validation of subtype survival in independent datasets
To validate the survival differences of the identified subtypes in independent testing datasets, we developed a workflow named “Subtype Survival Validation Workflow (SSVW)”. This workflow included the following steps: (1) Based on the subtype information and expression data in the training dataset, we constructed a random forest model for subtype prediction using five-fold cross-validation to optimize model accuracy and robustness. The trained model was saved for subsequent analysis. The importance values of variables were evaluated by mean decrease in accuracy. Moreover, the top 9 genes with the highest importance values were used for the 9-gene signature for further analysis. There are some reasons for choosing 9 genes as the optimal number. Based on our experience and preliminary analyses, using 9 genes achieves a good balance between model complexity and predictive accuracy. Including more genes did not yield significant improvements, while reducing the number of genes led to a decline in performance. Many published studies in cancer survival prediction have successfully adopted 9-gene signatures [15,16,17,18,19,20,21]; (2) The trained model was then applied to predict subtypes in independent testing datasets (GSE30219 and GSE73403). Survival curves were subsequently generated for the predicted subtypes in these independent datasets. Consistency between the survival patterns observed in the training dataset and those in the independent testing datasets would serve as strong evidence for the robustness of the identified subtypes. It is noteworthy that while the gender ratio in TCGA-LUSC is approximately 3:1 (male to female), the GSE30219 and GSE73403 datasets exhibited significant gender imbalance. Specifically, the combined testing set (GSE30219 and GSE73403) initially comprised 9 female and 121 male patients. To address this imbalance, we implemented a balanced sampling approach. In each of 10 iterations, we randomly selected 27 male patients from the available pool of 121 and combined them with all 9 female patients to create a balanced dataset. The survival outcomes of the predicted subtypes were then compared within these balanced datasets to ensure robust and unbiased validation.
2.3.3 Estimation of cell infiltrations
Subsequently, we assessed the relative abundances of cell infiltrations using the EPIC algorithm [22] and gene expression data. This algorithm estimates the proportions of various cell types, such as immune cells. The predicted values for the cell types were normalized using the Z-score normalization method, and p-values were calculated using the t-test.
2.3.4 Association of subtypes with clinical variables
We analyzed the association between subtypes and various clinical variables, including age, gender, and TNM staging by comparing their distributions across subtypes. Next, we evaluated the impact of chemotherapy on survival within each subtype, comparing survival rates for patients who received chemotherapy versus those who did not. The chemotherapy treatments were specified to cisplatin, carboplatin, paclitaxel, docetaxel, vinorelbine, etoposide, pemetrexed, gemcitabine, taxol, taxotere, topotecan, adriamycin, and doxorubicin. This analysis provides a reference for potential therapeutic strategies on whether one should receive chemotherapy based on subtype characteristics.
2.4 Comparison of the 9-gene signature with published gene signatures of LUSC
To validate the significance of our gene signature, we compared it with previously published signatures identified through a PubMed search using the keywords"LUSC"and"Signature."Several studies have developed gene signatures with different focuses, including immune-related genes (GeneSg1) [23], CD8 + T cell co-expressed genes (GeneSg2) [24], DNA damage repair genes (GeneSg3) [25], pyroptosis-related genes (GeneSg4) [26], cancer stemness-based immune-related genes (GeneSg5) [27], chemokine signaling-related genes (GeneSg6) [28], and immune-related genes (GeneSg7) [29]. Other signatures include those based on tumor progression, immune infiltration, and stemness index (GeneSg8) [30], immune-related genes (GeneSg9) [31], ferroptosis-related genes (GeneSg10) [32], innate immune-related genes (GeneSg11) [33], immune-related genes (GeneSg12) [34], DNA methylation genes (GeneSg13) [35], and ferroptosis-associated genes (GeneSg14) [36].
We extracted gene signatures from each selected article and applied a consistent methodology to construct and validate our signature using independent testing sets. Subsequently, we calculated the concordance index (C-index) and the time dependent area under the curve (tdAUC) values to compare the performance of our signature with those of the published signatures. The comparison process involved the following steps: (1) In each of the 10 iterations, we randomly selected 50% of the samples from the testing dataset; (2) Using these subsets, we employed a Cox proportional hazards model to validate and compare the predictive performance of the published signatures against our 9-gene signature.
2.5 Comparison of the 9-gene signature with clinical variables
In the independent testing datasets, GSE30219 and GSE73403, we assessed the predictive performance of the 9-gene signature alongside clinical variables such as age, gender, TNM staging (T, N, and M classifications), and the AJCC Stage (Stage I-IV). The predictive ability was evaluated using tdAUC and the C-index to comprehensively compare their performance in survival prediction. The comparison process involved the following steps: (1) In each of the 10 iterations, we randomly selected 50% of the samples from the testing dataset; (2) Using these subsets, we applied a Cox proportional hazards model to validate and compare the predictive performance of the clinical variables against the 9-gene signature. In this process, the values for the M stage were consistently M0 in both the GSE30219 and GSE73403 datasets. Therefore, the tdAUC and C-index values of M0 were 0.5 and excluded in the plot.
2.6 Machine learning for predicting the survival of LUSC
In this study, we selected 3 basic survival machine learning models for survival analysis: Random Survival Forest (RSF), CoxBoost (CXB), and Gradient Boosting Machine (GBM). The RSF employs an ensemble of decision trees and could effectively handle high-dimensional data. CXB is a boosting algorithm for Cox proportional hazards models. The GBM builds a predictive model by sequentially combining the predictions of multiple weak learners (decision trees). An ensemble (ESM) model was created by averaging the prediction outputs of the RSF, CXB, and GBM models.
Clinical variables, including age, gender, TNM staging (T, N, and M classifications), and the AJCC Stage, were integrated with the 9-gene signature to enhance survival outcome prediction. A grid parameter search was conducted for these models with the following parameter settings: ntree values of 100, 200, and 500 for RSF; penalty values of 50, 100, and 200 for CXB; and ntree values of 50, 100, and 200 for GBM. The TCGA-LUSC dataset was randomly divided into training (80%) and internal validation sets (20%). Models were trained on the training set, and the internal validation set was selected to calculate the C-index for model evaluation. The optimal parameter for each model was identified through the highest C-index. The model with the optimal parameters was then validated using two independent testing datasets, GSE73403 and GSE30219. For each iteration, 50% of samples from each testing dataset were randomly selected, and the model’s predictive performance was evaluated in the selected subsets of the testing dataset. This process was repeated with 10 iterations. Then, we calculated the average C-index and tdAUC values.
2.7 Statistical analysis
In the survival analysis of subtypes, the Kaplan–Meier method was employed to calculate p-values. A chi-square test was utilized for categorical variables to analyze the distribution of clinical variables between the C1 and C2 subtypes. A t-test was conducted for numerical variables between the C1 and C2 subtypes. The hazard ratio for genes was calculated using a univariate Cox proportional hazards model. A survival machine learning model has also been developed into a Shiny app within R. The app is hosted on the Shiny server and can be accessed through a web browser.
3 Results
3.1 Gene expression dataset acquisition
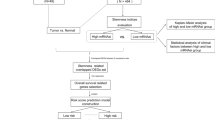
The workflow of this study is illustrated in Fig. 1A. 62 datasets were initially retrieved from the GEO database. After the selection process (Fig. 1B), two datasets (GSE30219 and GSE73403) from GEO and one LUSC dataset from TCGA were chosen for further analysis. The demographic data of these datasets are summarized in SupTable 1. Samples from the TCGA-LUSC (n = 472) were used as training sets, and GSE30219 (n = 61) and GSE73403 (n = 69) were used as independent testing sets. The average patient ages were 63.5, 59.5, and 67 years, respectively, with a predominance of male patients. Both GSE30219 and GSE73403 contained only non-metastatic (M0) cases, whereas TCGA included a few metastatic (M1) cases (n = 6). Regarding tumor staging, GSE30219 had more T1-stage patients, while GSE73403 and TCGA were primarily composed of T2 and T3-stage cases. Additionally, the median overall survival (OS) times were 6.4, 2.6, and 2.7 years, respectively, indicating differences in patient survival outcomes across datasets.

Workflow of the study (A) and the selection process of datasets (B)
3.2 Classification and validation of patient clusters
3.2.1 Clustering of LUSC patients in the training set
Based on the gene filtering criteria outlined in Sect. 2.3.1, we identified 261 genes for unsupervised clustering to uncover potential molecular subtypes among LUSC patients. We employed k-means clustering combined with three evaluation metrics: the silhouette method, the Calinski-Harabasz (CH) index, and the McClain index to determine the optimal number of clusters. The Silhouette method indicated that two subtypes were optimal (Fig. 2A). Similarly, the CH index (Fig. 2B) and the McClain index (Fig. 2C) confirmed that two subtypes were the most suitable. Following this evaluation, we selected two subtypes as the most appropriate solution. The survival curve analysis revealed that the C1 subtype exhibited significantly poorer survival than the C2 subtype (Fig. 2D).

Discovery of Subtypes in the Training Set (TCGA-LUSC). A The average silhouette width for varying numbers of subtypes. A higher average silhouette width indicates better-defined and more cohesive clusters in clustering analysis. B The calinski-Harabasz (CH) index plot illustrates the ratio of between-cluster variance to within-cluster variance. A higher CH index indicates a better-defined clustering structure. C The McClain index plot displays the average within-cluster distance compared to the average between-cluster distance. A lower McClain index suggests improved cluster separation. D The Kaplan–Meier survival plot illustrates overall survival (OS) differences among subtypes within the training cohort
3.2.2 Validation of subtype survival in independent testing sets
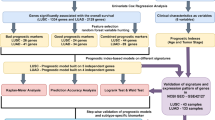
To ensure the robustness of the identified subtypes, we validated them in independent testing sets (GSE30219 and GSE73403). We built a subtype prediction model with the Random Forest classifier using the subtype information and gene expression profiles from the training set. The features of this model included the 261 genes identified in Sect. 3.2.1. The classifier was optimized through fivefold cross-validation in the training set, selecting an optimal mtry of 2 based on achieving the highest accuracy (0.97) during cross-validation. The top nine genes with the highest importance values in the Random Forest model, TGM2, AOC3, TBXA2R, RGS3, DLC1, MMP19, ACVRL1, TCF21, and TIMP3, were retained for further analysis (Fig. 3A). Partial dependency plots (PDPs) for these nine genes are shown in SupFigure 1 A–G. These plots showed that as the expression of genes increases, there is a noticeable upward trend in the probability for subtype C1. We also calculated the hazard ratio (HR) of genes in overall survival by univariate Cox analysis, and the results showed that all 9 genes are significantly associated with worse survival since all HR > 1 (SupTable 2).

Validation of subtypes in independent testing sets. A The top nine genes with the highest importance values in the Random Forest model for predicting subtypes. B Kaplan–Meier survival plot demonstrating overall survival (OS) differences among subtypes in the GSE30219 dataset. C Kaplan–Meier survival plot illustrating OS differences among subtypes in the GSE73403 dataset. D Kaplan–Meier survival plot illustrating OS differences across subtypes in the combined set of GSE30219 and GSE73403. E Distribution of tumor microenvironment (TME) cell types in the TCGA-LUSC dataset. F The impact of chemotherapy on survival outcomes within each subtype. C1_Chem represents the C1 subtype with chemotherapy. C1_No represents the C1 subtype without chemotherapy. C2_Chem represents the C2 subtype with chemotherapy. C2_No represents the C2 subtype without chemotherapy
The trained model was subsequently employed to predict subtypes within the testing sets. In the independent testing set GSE30219, survival analysis (Fig. 3B) revealed that LUSC patients classified as subtype C2 exhibited a significantly better prognosis compared to those in subtype C1 (log-rank p < 0.001, HR = 0.39, 95% CI 0.21–0.71). In contrast, for LUAD patients from the same dataset (GSE30219), no significant difference in survival outcomes (SupFigure 2) was observed between subtypes C1 and C2 (log-rank p = 0.41, HR = 0.76, 95% CI 0.41–1.42). Analysis of another independent testing set, GSE73403 (Fig. 3C), showed a trend towards better survival in subtype C2, although this difference did not reach statistical significance (log-rank p = 0.075, HR = 0.50, 95% CI 0.23–1.07). Notably, when combining data from both testing sets (GSE30219 and GSE73403, Fig. 3D), subtype C2 demonstrated significantly better survival outcomes compared to subtype C1 (log-rank p < 0.001, HR = 0.41, 95% CI 0.26–0.67).
Then, we further evaluated whether gender imbalance might affect our predictions by constructing gender-balanced independent testing sets. Specifically, in the combined testing set (which initially includes 9 female and 121 male patients), we performed a sensitivity analysis where, in each iteration, we randomly selected 27 male patients from the 121 available and combined them with all 9 female patients to form a balanced dataset. We repeated this process over 10 iterations. The results were as follows: in 2 out of the 10 iterations, subtype C1 showed a significantly worse survival outcome than C2 (HR > 1 and p-value < 0.05), and in 9 out of 10 iterations, C1 consistently exhibited worse survival outcomes (HR > 1) compared to C2 (SupTable 3).
3.3 Association of tumor microenvironment (TME), clinical variables, and chemotherapy with subtypes
We employed the EPIC algorithm to evaluate the association between TME cell types and the identified subtypes. In the training set, the C1 subtype was characterized by a higher abundance of B cells, cancer-associated fibroblasts (CAFs), endothelial cells, and macrophages (Fig. 3E), whereas CD8 + T cells were more prevalent in the C2 subtype. Regarding clinical variables, we observed that the C1 subtype was associated with significantly shorter survival time and older age (SupTable 4). Interestingly, there were no significant differences between the subtypes regarding TNM staging or the use of chemotherapy. We further examined the impact of chemotherapy on survival within each subtype. C2 patients who received chemotherapy demonstrated the best survival outcomes (Fig. 3F).
3.4 Comparison of the 9-gene signature with published signatures
We searched gene signatures from published studies for LUSC to enable a fair comparison with our 9-gene signature regarding survival prediction. The genes included in these signatures are shown in the supTable 5. We calculated the C-index and tdAUC values to assess each signature's performance using independent test sets (GSE30219 and GSE73403). In the GSE30219 dataset, our 9-gene signature demonstrated the highest performance with a tdAUC of 0.819 and a C-index of 0.763 (Fig. 4A, B). In the GSE73403 dataset, the 9-gene signature achieved the third-highest tdAUC at 0.794 (Fig. 4C) and ranked first in C-index with a value of 0.778 (Fig. 4D).

Comparison of our 9-gene signature for survival prediction with published signatures for LUSC. A The time-dependent area under the curve (tdAUC) values of signatures for predicting survival in the GSE30219 dataset. B C-index values of signatures for predicting survival in the GSE30219 dataset. C tdAUC values of signatures for predicting survival in the GSE73403 dataset. D C-index values of signatures for predicting survival in the GSE73403 dataset. GeneSg: gene signature
3.5 Comparison of the 9-gene signature with clinical variables
We compared the predictive performance of our 9-gene signature against clinical variables using independent testing sets from the GSE30219 and GSE73403 datasets. We calculated the C-index and tdAUC values for each signature to evaluate performance. In the GSE30219 dataset, our 9-gene signature demonstrated the highest tdAUC (Fig. 5A) and C-index values (Fig. 5B). Similarly, in the GSE73403 dataset, the 9-gene signature surpassed clinical variables, achieving the highest tdAUC (Fig. 5C) and C-index values (Fig. 5D).

Comparison of our 9-gene signature for survival prediction with clinical variables. A The tdAUC and B C-index values of the signatures for predicting survival in the GSE30219 dataset. C The tdAUC and D C-index values of the signatures for predicting survival in the GSE73403 dataset. The values for the M stage were consistently M0 in both the GSE30219 and GSE73403 datasets. Therefore, the tdAUC and C-index values were 0.5 and excluded in the plot
3.6 Machine learning architecture for predicting survival
We developed survival prediction models based on five clinical variables (age, gender, and TNM staging) and nine genes (TGM2, AOC3, TBXA2R, RGS3, DLC1, MMP19, ACVRL1, TCF21, and TIMP3). Four different machine learning models were evaluated during the training phase. Following hyperparameter tuning, the optimal parameters were identified as follows: for the RSF model, ntree = 500; for the CXB model, penalty = 50; and for the GBM model, ntree = 100. An ensemble model (ESM) was subsequently constructed by averaging the prediction outputs of the RSF, CXB, and GBM models.
We calculated each model’s C-index and tdAUC values in the GSE30219 (Fig. 6A, B) and GSE73403 (Fig. 6C, D) datasets. Among the models, the GBM model achieved the highest tdAUC in the GSE30219 dataset with a value of 0.712 and the highest C-index with 0.682. In the GSE73403 dataset, the GBM model again achieved the highest tdAUC with a value of 0.684 and demonstrated the highest C-index with a value of 0.625. The importance values of variables are shown in SupFigure 3.

Machine learning models for predicting survival in the testing set. Four distinct prediction models were trained on the training set (TCGA-LUSC) and validated using tdAUC (A) and C-index (B) in the GSE30219 dataset, as well as tdAUC (C) and C-index (D) in the GSE73403 dataset. The models consist of three individual machine learning algorithms: Random Survival Forest (RSF), CoxBoost (CXB), and Gradient Boosting Machine (GBM), in addition to an ensemble model (ESM) that combines the predictions of the three by averaging their outputs
3.7 Online tool for LUSC survival prediction models
The GBM model has been developed into an online tool (Fig. 7A, B), accessible at https://hznuduan.shinyapps.io/LCSP/. This tool comprises two main components: the first visualizes the distribution of the 9-gene signature expression across two subtypes (Fig. 7A); the second component allows users to manually input values for clinical and gene variables to predict survival probabilities ranging from 0 to 10 years (Fig. 7B).

Online Tool for Survival Prediction Using the GBM Model. A Visualization of the expression distribution of a 9-gene signature across two identified subtypes. B An interface that enables users to input clinical and gene expression variables to predict survival probabilities over 0 to 10 years. The online tool is accessible at https://hznuduan.shinyapps.io/LCSP/
4 Discussion
Over the past decade, genomic data have increasingly been integrated into clinical practice to enhance the management of lung cancer. However, few studies have investigated the subtypes of lung squamous cell carcinoma (LUSC) using multiple datasets. This study identified two distinct LUSC subtypes in TCGA and subsequently validated these subtypes using two independent testing datasets. Our gene signature outperformed previously published signatures, particularly in AUC and C-index values, demonstrating its robustness and potential clinical utility. Utilizing machine learning algorithms, we developed models and online tools for predicting survival outcomes.
The GSE30219 and GSE73403 datasets differ in several aspects, including sample size, patient demographics, treatment regimens, sample processing protocols, data acquisition platforms, normalization methods, and clinical features. These variations can influence the overall prognostic performance of gene signatures and the survival analyses of the identified subtypes. For instance, in the GSE30219 dataset (Fig. 3B), the C1 subtype exhibited significantly worse survival compared to C2 (p-value < 0.001). In contrast, although the C1 subtype in the GSE73403 dataset (Fig. 3C) also appeared to have poorer survival, the difference between the two subtypes was not statistically significant (p-value = 0.069). Notably, our 9-gene signature consistently outperformed published signatures across both datasets (Fig. 4A–D). These results indicate that dataset-specific characteristics may partially account for the observed differences in survival outcomes and signature performance and demonstrate the importance of multi-cohort validation for robust prognostic models.
Molecular subtypes are classifications of tumors based on their molecular characteristics, such as gene expression profiles. By categorizing tumors into these subtypes, researchers and clinicians can more accurately reflect the biological features of a tumor, thereby supporting more personalized diagnostic and treatment strategies. Among the two subtypes examined, we observed a higher abundance of cancer-associated fibroblasts (CAFs), endothelial cells, and macrophages in the C1 subtype, while CD8 + T cells were more prevalent in the C2 subtype. CAFs are known to contribute to tumorigenesis across various cancer types by promoting tumor proliferation, invasion, and metastasis [37]. This finding suggests that CAFs in the C1 subtype may negatively influence patient prognosis.
Several studies have investigated the subtypes of LUSC. One study identified four subtypes based on genes associated with immune biological processes using the TCGA-LUSC database [5]. Another study classified LUSC into three subtypes utilizing lactate metabolism-related genes from the same database [7]. A study conducted a consensus clustering analysis on the gene expression matrix of TCGA-LUSC patients and identified distinct subtypes [38]. Our study presents several advantages: (1) Instead of relying on a single cohort, we validated the subtypes identified from the TCGA dataset using two independent datasets (GSE30219 and GSE73403), enhancing the identified subtypes'robustness. (2) We developed machine learning models to predict subtype classifications, which are available on GitHub (https://github.com/bioCancerhznu/LUSCsubtype) to support potential applications in clinical practice.
Our 9-gene signature demonstrated strong performance for two primary reasons: (1) our approach incorporates genes that represent a broader spectrum of biological processes rather than being confined to a specific pathway, thereby providing a more comprehensive prediction of survival. (2) Our feature selection process employed rigorous filtering criteria, selecting genes with a relevance score exceeding 1 in the GeneCards database and retaining those with significant p-values (< 0.05) from univariate Cox analysis in the training set. A relevance score guarantees that published studies, including experimental research, validate the association of these genes with LUSC. This methodology ensured that the selected genes have a robust, statistically significant association with survival outcomes.
Previous studies have reported predictive models for LUSC patients. For example, one study utilized 13 immune-related genes to stratify and predict the prognosis of LUSC patients [8], while another study focused on two genes to predict survival outcomes [9]. In comparison, our study applied two strategies to enhance the predictive models. First, we used multiple advanced algorithms to develop more robust and accurate predictive models for LUSC rather than relying on a single method. Second, we created an online tool that displays survival rates from 0 to 10 years post-treatment. Making these predictive models accessible through an online tool is crucial for their practical application in clinical settings.
Our study found that all nine genes in the 9-gene signature were associated with poorer survival outcomes (SupTable 2). A review of the related literature revealed that five of these genes have previously been identified as contributors to worse survival. For instance, a prior study demonstrated that TGM2 could influence cancer cell proliferation, apoptosis, and invasion, with high TGM2 expression in LUSC associated with a poorer prognosis [39]. AOC3 was shown to accelerate lung metastasis in osteosarcoma by recruiting tumor-associated neutrophils and promoting tumor vascularization [40]. The oncogenic role of TBXA2R in lung cancer has been well established, with findings indicating that it enhances cell proliferation, migration, and invasion [41]. In LUSC, high expression levels of RGS3 are a risk factor for overall survival [42]. Additionally, overexpression of MMP19 has been shown to promote the migration of lung cell lines [43]. Conversely, previous research has characterized four genes in our study as tumor suppressors. The expression of DLC1 is often lost in lung cancer, and ectopic DLC1 expression significantly reduces proliferation and tumorigenicity [44]. ACVRL1 is a tumor suppressor by inhibiting lung cancer cell growth, migration, and survival [45]. TCF21 is a newly identified tumor suppressor gene that reverses epithelial–mesenchymal transition [46]. TIMP3 has been found to reduce cell numbers and induce apoptosis in lung cancer cell lines [47].
However, this study has limitations that must be acknowledged. Firstly, our research was retrospective and involved a relatively small sample size. To address the limitations of retrospective design and sample size, we propose a multicenter prospective validation study enrolling more than 100 LUSC patients. The 9-gene signature will be systematically evaluated using standardized qRT-PCR protocols alongside RNA-seq verification in subset samples. Time-dependent ROC and multivariate Cox analyses will assess its prognostic independence from clinical covariates. Additionally, several clinical parameters, such as surgery type, were not included in the survival prediction model due to the lack of available information in the GEO and TCGA datasets. Finally, the specific mechanisms underlying these prognostic genes in the pathogenesis and progression of LUSC remain unclear and necessitate further in-depth investigation in future research.
5 Conclusions
In our study, we identified and validated two distinct subtypes of LUSC and developed machine learning models to predict survival outcomes. Our prognostic prediction model strongly correlated with LUSC prognosis and outperformed existing prognostic signatures. Furthermore, we have made the machine learning models accessible online, allowing for the prediction of survival rates in LUSC patients.
Data availability
The code for data download and analysis can be found at https://github.com/bioCancerhznu/LUSCsubtype. All human data in this study come from publicly available databases, including TCGA and GEO. Researchers could get these data through TCGAbiolinks and GEOquery packages.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49. https://doi.org/10.3322/caac.21660.
Kim Y, Hammerman PS, Kim J, Yoon J, Lee Y, Sun J-M, et al. Integrative and comparative genomic analysis of lung squamous cell carcinomas in East Asian patients. J Clin Oncol. 2014;32:121–8. https://doi.org/10.1200/JCO.2013.50.8556.
Wu J, Xu C, Guan X, Ni D, Yang X, Yang Z, et al. Comprehensive analysis of tumor microenvironment and identification of an immune signature to predict the prognosis and immunotherapeutic response in lung squamous cell carcinoma. Ann Transl Med. 2021;9:569. https://doi.org/10.21037/atm-21-463.
Li X-S, Nie K-C, Zheng Z-H, Zhou R-S, Huang Y-S, Ye Z-J, et al. Molecular subtypes based on DNA methylation predict prognosis in lung squamous cell carcinoma. BMC Cancer. 2021;21:96. https://doi.org/10.1186/s12885-021-07807-7.
Yin L, Zhang W, Pu D, Zhai X, Lin Y, Wu Q, et al. Identification of immune subtypes of lung squamous cell carcinoma by integrative genome-scale analysis. Front Oncol. 2022. https://doi.org/10.3389/fonc.2021.778549.
Qian J, Chen R, Zhao R, Han Y, Yu Y. Comprehensive molecular characterizations of Chinese patients with different subtypes of lung squamous cell carcinoma. Front Oncol. 2020. https://doi.org/10.3389/fonc.2020.607130.
Wang C, Lu T, Xu R, Luo S, Zhao J, Zhang L. Multi-omics analysis to identify lung squamous carcinoma lactate metabolism-related subtypes and establish related index to predict prognosis and guide immunotherapy. Comput Struct Biotechnol J. 2022;20:4756–70. https://doi.org/10.1016/j.csbj.2022.08.067.
Yang Q, Gong H, Liu J, Ye M, Zou W, Li H. A 13-gene signature to predict the prognosis and immunotherapy responses of lung squamous cell carcinoma. Sci Rep. 2022;12:13646. https://doi.org/10.1038/s41598-022-17735-6.
Zhang X, Xiao J, Fu X, Qin G, Yu M, Chen G, et al. Construction of a two-gene immunogenomic-related prognostic signature in lung squamous cell carcinoma. Front Mol Biosci. 2022. https://doi.org/10.3389/fmolb.2022.867494.
Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44: e71. https://doi.org/10.1093/nar/gkv1507.
Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173:400-416.e11. https://doi.org/10.1016/j.cell.2018.02.052.
Rousseaux S, Debernardi A, Jacquiau B, Vitte A-L, Vesin A, Nagy-Mignotte H, et al. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci Transl Med. 2013;5:186ra66. https://doi.org/10.1126/scitranslmed.3005723.
Davis S, Meltzer PS. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics. 2007;23:1846–7. https://doi.org/10.1093/bioinformatics/btm254.
Safran M, Rosen N, Twik M, BarShir R, Stein TI, Dahary D, et al. The GeneCards Suite. In: Abugessaisa I, Kasukawa T, editors., et al., Practical guide to life science databases. Singapore: Springer Nature; 2021.
Brunner G, Reitz M, Heinecke A, Lippold A, Berking C, Suter L, et al. A nine-gene signature predicting clinical outcome in cutaneous melanoma. J Cancer Res Clin Oncol. 2013;139:249–58. https://doi.org/10.1007/s00432-012-1322-z.
Zhao J, Wang Y, Gao J, Wang Y, Zhong X, Wu X, et al. A nine-gene signature to improve prognosis prediction of colon carcinoma. Cell Cycle. 2021;20:1021–32. https://doi.org/10.1080/15384101.2021.1919827.
Park IJ, Yu YS, Mustafa B, Park JY, Seo YB, Kim G-D, et al. A nine-gene signature for predicting the response to preoperative chemoradiotherapy in patients with locally advanced rectal cancer. Cancers. 2020;12:800. https://doi.org/10.3390/cancers12040800.
Bao Z-S, Li M-Y, Wang J-Y, Zhang C-B, Wang H-J, Yan W, et al. Prognostic value of a nine-gene signature in glioma patients based on mRNA expression profiling. CNS Neurosci Ther. 2014;20:112–8. https://doi.org/10.1111/cns.12171.
Xin C, Lai Y, Ji L, Wang Y, Li S, Hao L, et al. A novel 9-gene signature for the prediction of postoperative recurrence in stage II/III colorectal cancer. Front Genet. 2023. https://doi.org/10.3389/fgene.2022.1097234.
Yang F, Zhou L, Yang H, Wang Y. Nine-gene signature and nomogram for predicting survival in patients with head and neck squamous cell carcinoma. Front Genet. 2022. https://doi.org/10.3389/fgene.2022.927614.
Wu M, Li X, Zhang T, Liu Z, Zhao Y. Identification of a nine-gene signature and establishment of a prognostic nomogram predicting overall survival of pancreatic cancer. Front Oncol. 2019;9:996. https://doi.org/10.3389/fonc.2019.00996.
Racle J, Gfeller D. EPIC: a tool to estimate the proportions of different cell types from bulk gene expression data. Methods Mol Biol. 2020;2120:233–48. https://doi.org/10.1007/978-1-0716-0327-7_17.
Zhou H, Zhang H, Shi M, Wang J, Huang Z, Shi J. A robust signature associated with patient prognosis and tumor immune microenvironment based on immune-related genes in lung squamous cell carcinoma. Int Immunopharmacol. 2020;88: 106856. https://doi.org/10.1016/j.intimp.2020.106856.
Chen L, Weng Y, Cui X, Li Q, Peng M, Song Q. Comprehensive analyses of a CD8+ T cell infiltration related gene signature with regard to the prediction of prognosis and immunotherapy response in lung squamous cell carcinoma. BMC Bioinform. 2023;24:238. https://doi.org/10.1186/s12859-023-05302-3.
Wang X, Huang Z, Li L, Wang G, Dong L, Li Q, et al. DNA damage repair gene signature model for predicting prognosis and chemotherapy outcomes in lung squamous cell carcinoma. BMC Cancer. 2022;22:866. https://doi.org/10.1186/s12885-022-09954-x.
Li X, He J. A novel pyroptosis-related gene signature for early-stage lung squamous cell carcinoma. IJGM. 2021;14:6439–53. https://doi.org/10.2147/IJGM.S331975.
Li N, Li Y, Zheng P, Zhan X. Cancer stemness-based prognostic immune-related gene signatures in lung adenocarcinoma and lung squamous cell carcinoma. Front Endocrinol. 2021;12: 755805. https://doi.org/10.3389/fendo.2021.755805.
Lai J, Yang S, Chu S, Xu T, Huang J. Determination of a prediction model for therapeutic response and prognosis based on chemokine signaling-related genes in stage I–III lung squamous cell carcinoma. Front Genet. 2022;13: 921837. https://doi.org/10.3389/fgene.2022.921837.
Li N, Wang J, Zhan X. Identification of immune-related gene signatures in lung adenocarcinoma and lung squamous cell carcinoma. Front Immunol. 2021;12: 752643. https://doi.org/10.3389/fimmu.2021.752643.
Wu R, Ma R, Duan X, Zhang J, Li K, Yu L, et al. Identification of specific prognostic markers for lung squamous cell carcinoma based on tumor progression, immune infiltration, and stem index. Front Immunol. 2023;14:1236444. https://doi.org/10.3389/fimmu.2023.1236444.
Li R, Liu X, Zhou X-J, Chen X, Li J-P, Yin Y-H, et al. Identification of a prognostic model based on immune-related genes of lung squamous cell carcinoma. Front Oncol. 2020;10:1588. https://doi.org/10.3389/fonc.2020.01588.
Wang Q, Chen Y, Gao W, Feng H, Zhang B, Wang H, et al. Identification and validation of a four-gene ferroptosis signature for predicting overall survival of lung squamous cell carcinoma. Front Oncol. 2022;12: 933925. https://doi.org/10.3389/fonc.2022.933925.
Li L, Yu X, Ma G, Ji Z, Bao S, He X, et al. Identification of an innate immune-related prognostic signature in early-stage lung squamous cell carcinoma. IJGM. 2021;14:9007–22. https://doi.org/10.2147/IJGM.S341175.
Liu Z, Wan Y, Qiu Y, Qi X, Yang M, Huang J, et al. Development and validation of a novel immune-related prognostic model in lung squamous cell carcinoma. Int J Med Sci. 2020;17:1393–405. https://doi.org/10.7150/ijms.47301.
Li G, Wu L, Yu J, Zhai S, Deng H, Wang Q. Identification and validation of three-gene signature in lung squamous cell carcinoma by integrated transcriptome and methylation analysis. Journal of Oncology. 2022;2022:1–13. https://doi.org/10.1155/2022/9688040.
Lu Y, Yang H, Cao Y, Wang Y, Wu M, He B, et al. A survival model for prognostic prediction based on ferroptosis-associated genes and the association with immune infiltration in lung squamous cell carcinoma. PLoS ONE. 2023;18: e0282888. https://doi.org/10.1371/journal.pone.0282888.
Wong KY, Cheung AH, Chen B, Chan WN, Yu J, Lo KW, et al. Cancer-associated fibroblasts in nonsmall cell lung cancer: from molecular mechanisms to clinical implications. Int J Cancer. 2022;151:1195–215. https://doi.org/10.1002/ijc.34127.
Wang H, Yang F, Wang Y, Sun L. Identification of prognostic immune subtypes of lung squamous cell carcinoma by unsupervised consistent clustering. Medicine. 2023;102: e35123. https://doi.org/10.1097/MD.0000000000035123.
Chang W, Gao W, Liu D, Luo B, Li H, Zhong L, et al. The upregulation of TGM2 is associated with poor prognosis and the shaping of the inflammatory tumor microenvironment in lung squamous cell carcinoma. Am J Cancer Res. 2024;14:2823. https://doi.org/10.62347/OBES4130.
Qi L, Gao T, Bai C, Guo Z, Zhou L, Yang X, et al. AOC3 accelerates lung metastasis of osteosarcoma by recruiting tumor-associated neutrophils, neutrophil extracellular trap formation and tumor vascularization. Heliyon. 2024. https://doi.org/10.1016/j.heliyon.2024.e37070.
Orr K, Buckley NE, Haddock P, James C, Parent J-L, McQuaid S, et al. Thromboxane A2 receptor (TBXA2R) is a potent survival factor for triple negative breast cancers (TNBCs). Oncotarget. 2016;7:55458. https://doi.org/10.18632/oncotarget.10969.
Jiang M, Li X, Quan X, Li X, Zhou B. MiR-92a family: a novel diagnostic biomarker and potential therapeutic target in human cancers. Front Mol Biosci. 2019. https://doi.org/10.3389/fmolb.2019.00098.
Yu G, Herazo-Maya JD, Nukui T, Romkes M, Parwani A, Juan-Guardela BM, et al. Matrix metalloproteinase-19 promotes metastatic behavior in vitro and is associated with increased mortality in non-small cell lung cancer. Am J Respir Crit Care Med. 2014;190:780. https://doi.org/10.1164/rccm.201310-1903OC.
Healy KD, Hodgson L, Kim T-Y, Shutes A, Maddileti S, Juliano RL, et al. DLC-1 Suppresses non-small cell lung cancer growth and invasion by RhoGAP-dependent and independent mechanisms. Mol Carcinog. 2008;47:326. https://doi.org/10.1002/mc.20389.
Fan J, Xia X, Fan Z. Hsa_circ_0129047 regulates the miR-375/ACVRL1 axis to attenuate the progression of lung adenocarcinoma. J Clin Lab Anal. 2022;36: e24591. https://doi.org/10.1002/jcla.24591.
Liu H, He R, Yang X, Huang B, Liu H. Mechanism of TCF21 downregulation leading to immunosuppression of tumor-associated macrophages in non-small cell lung cancer. Pharmaceutics. 2023;15:2295. https://doi.org/10.3390/pharmaceutics15092295.
Finan KM, Hodge G, Reynolds AM, Hodge S, Holmes MD, Baker AH, Reynolds PN. 742. Over-expression of TIMP-3 causes apoptosis in lung cancer cells. Mol Ther. 2003;7:287. https://doi.org/10.1016/S1525-0016(16)41184-6.
Funding
This research was supported by the Zhejiang Provincial Nature Foundation Fund Project (Grant No: LY16H030017) awarded to Yuefang Ye, the Zhejiang Provincial Health Department Project (Grant No: 2022KY971) awarded to Yuefang Ye, and the Natural Science Foundation of Anhui Province (Grant No: 2208085MH247) awarded to Qi Huo. No additional external funding was received for this study.
Author information
Authors and Affiliations
Contributions
Guangliang Duan, and Huiping Dai conceptualized the study. Guangliang Duan, Huiping Dai, Qi Huo, and Wei Ni conducted the formal analysis. Guangliang Duan, Fei Ding, and Yuefang Ye developed the methodology. Guangliang Duan and Tingting Tang prepared the figures. Guangliang Duan and Huiping Dai wrote the main manuscript text. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
12672_2025_2560_MOESM1_ESM.pdf
Supplementary Material 1. SupFigure 1. Partial Dependency Plotsof TGM2, AOC3, TBXA2R, RGS3, DLC1, MMP19, ACVRL1, TCF21, and TIMP3for Predicting Subtype C1. The x-axis in each plot represents the expression levels of a specific gene. At the same time, the y-axis displays the model's average predicted response for Subtype C1 as a function of that gene's expression, with all other genes held constant. Each plot illustrates the marginal effect of each gene on the likelihood of Subtype C1, demonstrating how varying expression levels influence the model’s predictions. Positive trends on the y-axis indicate that higher gene expression increases the prediction probability for Subtype C1, while negative trends suggest a decreased association
12672_2025_2560_MOESM2_ESM.pdf
Supplementary Material 2. SupFigure 2. Survival Outcomes among Molecular Subtypes in LUAD Patients from the GSE30219 Cohort
12672_2025_2560_MOESM4_ESM.docx
Supplementary Material 4. SupTable 1. Demographic Data for the Training Setand the Independent Testing Sets. SupTable 2. Hazard Ratiosand Confidence Intervalsfor nine genes in Overall Survival Based on Univariate Cox Regression Analysis. This table presents the Hazard Ratiosand 95% Confidence Intervalsfor each of the nine genes, calculated through univariate Cox regression to assess their association with overall survival. All nine genes have HR values greater than 1, indicating a significant correlation with poorer survival outcomes. Increased expression levels of these genes are associated with an elevated risk of mortality. SupTable 3. Survival Outcomes of Predicted Subtypes in the Combined Balanced Datasets. For each of the 10 iterations, a balanced dataset was created by randomly selecting 27 male patients from a pool of 121 and combining them with all 9 female patients. Hazard ratiosand corresponding p-values are reported for each iteration. An HR < 1 indicates that subtype C2 is associated with better survival outcomes than subtype C1. SupTable 4. Distribution of Clinical Variables in C1 and C2 Subtypes from the TCGA-LUSC. SupTable 5. Genes in the Published Signatures
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Duan, G., Huo, Q., Ni, W. et al. Integrative machine learning model for subtype identification and prognostic prediction in lung squamous cell carcinoma. Discov Onc 16, 886 (2025). https://doi.org/10.1007/s12672-025-02560-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-025-02560-w