
Abstract
Background
Cervical cancer remains a significant global health concern, with immune system regulation potentially playing a crucial role in disease development. This study investigates potential causal relationships between genetic variants associated with immune cell populations and cervical cancer risk.
Methods
We employed multiple Mendelian randomization (MR) approaches inverse-variance weighted, MR-Egger, simple median, and weighted median methods to evaluate genetic instrumental variables linked to various T-cell and B-cell subtypes. Differential gene expression was analyzed using single-cell RNA sequencing, while forest plots, scatter plots, and funnel plots facilitated comprehensive MR analysis.
Results
Forest plots consistently demonstrated odds ratios clustered tightly around 1.000 (range: 0.998–1.001), despite some variants reaching statistical significance (p < 0.05). MR analysis of CD69 + LGALS3A + regulatory T-cells, CD8 + T-cell populations, and CD20 + B-cells revealed only weak associations with cervical cancer susceptibility. Comparative analysis across different MR methodologies produced consistent results with minimal horizontal pleiotropy bias.
Conclusion
While immune cell genetic factors may contribute to cervical cancer development, their causal effects appear modest. These findings suggest that genetic predisposition through immune cell regulation likely plays a supplementary rather than primary role in cervical cancer pathogenesis.
Similar content being viewed by others


Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
1 Introduction
In the global healthcare landscape, cervical cancer persists as one of the most significant gynecological malignancies, disproportionately affecting developing nations [1,2,3]. Cervical cancer pathogenesis involves complex interactions between genetic predisposition, viral infections, and immune system regulation. Understanding these multifaceted relationships remains crucial for developing targeted therapeutic interventions [4,5,6,7].
Recent advances in molecular biology techniques and next-generation sequencing have revolutionized our understanding of cervical cancer's biological foundations. Mendelian randomization (MR) has emerged as a powerful methodological framework for investigating causal relationships between genetic variants and disease outcomes, effectively addressing confounding factors that often plague observational studies. By leveraging genetic instruments as proxies for modifiable risk factors, MR provides robust evidence regarding causative links between specific genetic markers and cervical cancer susceptibility, particularly in relation to HPV infection response [8,9,10].
Within cervical cancer research, comprehensive genetic analysis reveals a complex biological landscape encompassing diverse cellular populations, including various immune cell subtypes (T cells, B cells, Natural Killer cells, and monocytes) and epithelial components. These cellular interactions significantly influence tumor progression, immune response efficacy, and treatment outcomes [11,12,13]. The immune system plays a critical role in cervical cancer progression, with various cell populations exhibiting distinct functions in tumor surveillance and response. Mendelian randomization studies targeting genetic determinants of immune cell composition and function have revealed important insights into how genetic variation influences cervical cancer susceptibility through immune-mediated mechanisms. By leveraging genetic instruments associated with CD69 + LGALS3A + regulatory T-cells, effector CD8 + T-cell populations, and CD20 + B-cells, MR analyses have identified potential causal pathways linking immune function to cervical cancer risk. Single-cell RNA sequencing has complemented Mendelian randomization by providing high-resolution characterization of immune cell heterogeneity within the cervical cancer environment. Integration of these technologies enables researchers to connect genetically influenced immune cell phenotypes with specific gene expression patterns and cellular functions relevant to cervical cancer pathogenesis. Advanced computational approaches, including dimensional reduction techniques and weighted gene co-expression network analysis, have become indispensable tools for deciphering these complex cellular relationships [14,15,16].
Despite significant advances, comprehensive studies integrating Mendelian randomization with detailed immune cell characterization remain essential for fully elucidating the causal mechanisms underlying cervical cancer development. This research addresses this critical gap by combining MR methodology with single-cell transcriptomics and network analysis to provide causal evidence regarding how genetic variants influence immune cell populations and subsequently affect cervical cancer susceptibility.
This integrated analytical approach promises to uncover novel perspectives on the causal relationships between genetic determinants of immune cell function and cervical cancer progression. Such understanding will prove invaluable for developing personalized therapeutic strategies targeting genetically influenced immune pathways in cervical cancer patients.
2 Methods
2.1 Data source
GWAS data which was retrieved from a comprehensive metabolomics dataset recently released by the UK Biobank. The summary data from a genome-wide association study (GWAS) on was analyzed cervical cancer for a European-descent population of 475,638 participants, which can be accessed online at https://gwas.mrcieu.ac.uk with the specific study identified by the ID [17, 18]. After applying stringent quality control procedures and imputation techniques, the study examined approximately 24.2 million genetic variants.
2.2 Selection of instrumental variables
For the identification of instrumental variables associated with immune characteristics, we implemented a significance threshold of P < 1 × 10−5. This threshold was carefully selected based on comprehensive review of contemporary methodological approaches in similar Mendelian randomization studies, while also considering the specific statistical power requirements of our analysis framework. This moderately stringent threshold allowed us to capture genetic variants with meaningful association signals while maintaining adequate statistical power for downstream analyses.To address potential redundancy among identified genetic variants and account for linkage disequilibrium (LD) patterns, we employed a systematic clumping procedure using PLINK software (version 1.90). This process involved the evaluation of single nucleotide polymorphisms (SNPs) residing within a predefined genomic window of 500 kilobases (kb). Within this window, we applied an r^2 threshold of 0.1 to identify and subsequently remove SNPs exhibiting substantial linkage disequilibrium. For all LD calculations, we utilized genotype data from the 1000 Genomes Project as our reference panel, ensuring comprehensive representation of population-specific LD structures.
For the hepatocellular carcinoma (HCC) component of our analysis, we implemented a substantially more stringent significance threshold of P < 5 × 10−8. This genome-wide significance threshold represents the established standard in genome-wide association studies (GWAS) and was specifically adopted to rigorously control for multiple testing burden and minimize the probability of false positive associations. This approach was particularly crucial given the complex genetic architecture underlying HCC susceptibility.
2.3 Instrumental variable strength assessment
To quantitatively evaluate the strength of our selected instrumental variables and systematically address potential weak instrument bias, we conducted comprehensive calculations of the proportion of phenotypic variation explained by each instrumental variable and the corresponding F-statistic for each instrumental variable. This assessment allowed us to identify and exclude potentially problematic instrumental variables that might introduce bias into our causal estimates. Specifically, instrumental variables demonstrating F-statistics below the predetermined threshold of 10 were removed from subsequent analyses, as this cutoff represents a widely accepted minimum standard for instrument strength in Mendelian randomization studies. Following the application of these rigorous filtering criteria, the remaining set of instrumental variables associated with HCC was retained for all downstream statistical analyses. This methodical selection process ensured that our final set of instrumental variables satisfied the core assumptions of Mendelian randomization while maximizing statistical power and minimizing potential sources of bias.
2.4 Ethics statement
The informed consents on the used GWAS summary statistics data have been obtained following the protocols approve from their respective institutional review boards. No separate ethical approval was required for this study [19, 20].
2.5 Statistics
All statistical analyses were conducted using the R programming language (Version 4.0.3) with specialized packages designed for Mendelian randomization studies. Statistical significance was determined at a conventional threshold of p < 0.05 unless specifically indicated otherwise for particular analyses. For our Mendelian randomization framework, we implemented a comprehensive analytical approach employing multiple complementary methods to enhance the robustness of causal inference. The primary analysis utilized the inverse-variance weighted (IVW) method, which provides optimal statistical power under the assumption that all instrumental variables are valid or that pleiotropic effects are balanced. To address potential violations of Mendelian randomization assumptions, we conducted several sensitivity analyses, including MR-Egger regression to detect and adjust for directional pleiotropy through its intercept test, weighted median estimation to provide consistent causal estimates even when up to 50% of instrumental variables are invalid, and simple median estimation as a complementary approach less sensitive to outlying genetic variants. Heterogeneity among instrumental variables was assessed using Cochran's Q statistic within the IVW framework, with significant heterogeneity (p < 0.05) indicating potential pleiotropy or effect modification. For exposures demonstrating significant causal relationships, we further implemented MR-PRESSO (Mendelian Randomization Pleiotropy RESidual Sum and Outlier) to identify and correct for horizontal pleiotropic outliers. The strength of each instrumental variable was evaluated using F-statistics, with values exceeding 10 considered sufficiently strong to minimize weak instrument bias. To quantify potential bias due to sample overlap between exposure and outcome datasets, we calculated the bias term as described by Burgess et al. and adjusted our estimates accordingly when necessary. All analyses incorporated appropriate corrections for multiple testing using the false discovery rate (FDR) method when evaluating multiple exposures simultaneously. Directional consistency across different MR methods was considered strong evidence for genuine causal relationships rather than methodological artifacts.
3 Results
3.1 Differential gene expression volcano plot analysis
Figure 1 illustrates a comprehensive volcano plot derived from single-cell RNA sequencing analysis, providing a visual representation of differentially expressed genes across various immune cell populations. The plot's x-axis (BETA) represents the effect size and directional change in gene expression, while the y-axis (-log(pvalue)) indicates statistical significance levels, with points positioned higher on the plot representing more statistically robust findings. The visualization employs a three-dimensional approach to data presentation: coordinate position indicates both effect size and significance, point diameter (neglogp) offers an additional visual cue for significance magnitude, and a tricolor scheme effectively categorizes gene expression patterns with downregulated genes appearing as blue points (negative BETA values), upregulated genes as red points (positive BETA values), and non-significant alterations as gray points. The analysis reveals distinctive transcriptional signatures across multiple T cell subpopulations, including CD69 + LGALS3A + CD4-positive regulatory T cells, CD69 + LGALS3A + CD4-positive memory T cells and broader CD4 + T cell populations, functionally active CD4 regulatory and non-regulatory T cells, HLA-DR + memory/activated cellular phenotypes, and both terminally differentiated CD8 + T cells and memory T cell subsets. This visualization method serves as a fundamental analytical tool in contemporary immunological and single-cell genomics research, enabling researchers to identify characteristic gene expression signatures that define specific cellular phenotypes. These transcriptional profiles provide critical insights into the functional specialization and regulatory networks governing diverse immune cell populations, advancing our understanding of their roles in both physiological and pathological immune responses.

Differential Gene Expression Analysis in Cervical Cancer Immune Microenvironment. This volcano plot shows differential gene expression analysis related to cervical cancer. The horizontal axis (BETA) represents the direction and magnitude of gene expression changes, while the vertical axis (-log(pvalue)) indicates statistical significance. Blue dots represent downregulated genes in cervical cancer (negative BETA values), red dots show upregulated genes (positive BETA values), and white or gray dots indicate non-significant changes. Bubble size (neglogp) reflects significance intensity. The plot highlights several key immune cell populations, including CD69 + LGALS3A + CD4 regulatory and non-regulatory T cells, CD8 + T cell absolute counts, and activated CD4 regulatory T cells. These results reveal T cell subset expression patterns in cervical cancer, providing important insights into the tumor immune microenvironment
3.2 Forest plot analysis of genetic exposure associations with cervical cancer risk
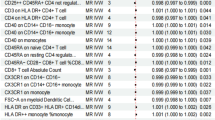
This forest plot presents a comprehensive analysis of various genetic exposure identifiers and their association with cervical cancer risk. The visualization displays multiple GCST-coded genetic variants (id.exposure) alongside the number of single nucleotide polymorphisms (nsnp) analyzed for each variant. The central forest plot graphically represents odds ratios (OR) with 95% confidence intervals, with exact numerical values provided in the adjacent column. The rightmost column shows p-values indicating statistical significance.
Analysis reveals that most genetic exposures demonstrate odds ratios very close to 1.000, suggesting minimal association with cervical cancer risk. Despite some variants showing statistical significance (p < 0.05), effect sizes remain notably small across all markers. The exceptionally narrow confidence intervals indicate high precision in these estimates. Some genetic exposures show slightly increased risk (OR > 1), while others demonstrate slightly protective effects (OR < 1). The scale at the bottom (0.98–1.03) further emphasizes the limited range of effect sizes observed. These findings likely represent results from a Mendelian randomization analysis or genome-wide association study designed to evaluate potential causal relationships between specific genetic variants and cervical cancer susceptibility(Fig. 2).

Genetic Variant Associations with Cervical Cancer Risk. This forest plot presents an analysis of multiple genetic variants and their effects on cervical cancer risk. The table lists 27 genetic exposure identifiers (starting with GCST), the number of single nucleotide polymorphisms for each (nsnp), odds ratios with 95% confidence intervals (OR (95% CI)), and statistical significance (pval). Results show that most genetic variants have odds ratios very close to 1.000, indicating minimal impact on cervical cancer risk. While some variants reach statistical significance (p < 0.05), effect sizes remain limited, with the highest odds ratio at only 1.001 and the lowest at 0.998. These findings suggest that the studied genetic variants may have little or very weak association with cervical cancer development risk
3.3 Comparative analysis of mendelian randomization methods in cervical cancer research
This figure presents a comprehensive comparison of different Mendelian Randomization (MR) analytical approaches investigating the causal effects of single nucleotide polymorphisms (SNPs) on cervical cancer. The visualization is organized into four panels (Fig. 3A–D), each providing complementary insights into the analysis. Figure 3A displays individual SNP effect estimates in a forest plot format, with several independent genetic variants shown at the top along with their confidence intervals. The red lines at the bottom represent the summary effect estimates using inverse-variance weighted (IVW) and MR-Egger regression methods, providing an overall assessment of genetic influence on cervical cancer risk. Figure 3B presents a similar forest plot, likely representing results from an alternative dataset or analytical specification, featuring wider confidence intervals that suggest varying degrees of precision in the causal estimates for cervical cancer susceptibility. Figure 3C features a scatter plot comparing the performance of different MR methodologies (inverse-variance weighted, MR-Egger, simple median, and weighted median) in estimating the causal relationship between genetic variants and cervical cancer. The different colored regression lines represent these distinct statistical approaches, allowing for methodological robustness assessment. Figure 3D shows a funnel plot used to evaluate potential biases in the MR analysis. With precision (1/SE) on the vertical axis and effect size (β₀) on the horizontal axis, this visualization helps identify possible horizontal pleiotropy or other forms of bias that might influence the interpretation of genetic effects on cervical cancer.

Mendelian Randomization Analysis of Genetic Variants and Cervical Cancer. This figure shows a Mendelian Randomization analysis examining the relationship between genetic variants and cervical cancer. A and B are forest plots displaying individual SNP effect estimates and confidence intervals for cervical cancer risk, with red lines at the bottom showing summary effects. C compares four MR methods (inverse-variance weighted, MR-Egger, simple median, and weighted median) analyzing the relationship between SNP effects on CD23 + CD69 + LGALS3A + CD4 regulatory T cells and cervical cancer. D is a funnel plot assessing potential bias in the analysis. Overall results suggest only weak correlations between the studied genetic variants and cervical cancer
3.4 Mendelian randomization analysis of genetic variants and cervical cancer risk
This figure presents a Mendelian Randomization (MR) analysis investigating the causal relationship between genetic variants and cervical cancer Figure 4A and B display forest plots showing individual SNP effect estimates on cervical cancer risk with their confidence intervals, with red lines at the bottom representing summary effects using different methods (inverse-variance weighted and MR-Egger). Figure 4C shows a funnel plot with precision (1/SE) on the vertical axis and effect size on the horizontal axis, used to assess potential bias in the analysis. Figure 4D features a scatter plot comparing results from four different MR methods (inverse-variance weighted, MR-Egger, simple median, and weighted median), illustrating the estimated relationship between SNP effects on terminally differentiated CD8 + T-cells and cervical cancer. Overall, the estimates from different methods show slight variations, but most effect estimates are close to zero, suggesting only weak associations between the studied genetic variants and cervical cancer risk.

Genetic Influences on Cervical Cancer: A Multi-Method Analysis. This image shows results from genetic research on cervical cancer. The top panels A, B display forest plots of individual genetic variants, with red lines showing overall effects. C presents a precision funnel plot to check for study bias. D Compares four statistical approaches examining how genetic effects on CD8 + T cells might influence cervical cancer risk. The data consistently indicates minimal genetic impact on cervical cancer development across all analysis methods
3.5 Mendelian randomization analysis of genetic variants and cervical cancer susceptibility
This figure illustrates a Mendelian Randomization (MR) analysis investigating the causal relationship between genetic variants and cervical cancer. Figure 5A and B present forest plots displaying effect estimates and confidence intervals for multiple single nucleotide polymorphisms (SNPs) on cervical cancer risk, with red lines at the bottom indicating summary effects using inverse-variance weighted (IVW) and MR-Egger methods. Figure 5C shows a scatter plot comparing different MR approaches (inverse-variance weighted, MR-Egger, simple median, and weighted median), evaluating the relationship between SNP effects on CD8 + T cell absolute counts and cervical cancer. Fig. 5D displays a funnel plot with precision (1/SE) on the vertical axis and effect size (β₀) on the horizontal axis, used to detect potential bias. The overall results indicate that most genetic instrumental variables have effect estimates close to zero, with similar summary estimates across different statistical methods, suggesting potentially weak or no substantial causal association between the studied genetic variants and cervical cancer risk (Figs. 6, 7, 8).

Exploring T-Cell genetics in cervical cancer prevention. This four-panel figure tackles this question through multiple statistical approaches. Panels A and B use forest plots to show that individual genetic markers have minimal impact, with summary effects (red lines) hovering near zero. Panel C demonstrates how different analytical methods evaluate the relationship between genetic effects on CD8 + T-cell counts and cancer risk. Panel D's funnel plot helps identify potential research bias. Together, these visualizations suggest that genetic variations linked to T-cell function likely play only a modest role in cervical cancer susceptibility

T-Cell subtypes and cervical cancer: A Genetic Perspective. This analysis examines this question through genetic evidence. The top panels display varying genetic effects with wide confidence intervals, while red lines indicate combined estimates using different statistical approaches. In C four statistical methods reveal slight positive associations between genetic markers affecting CD69 + LGALS3A + CD20 + CD8 + T-cells and cervical cancer risk. Panel D's scattered precision plot helps evaluate potential systematic errors in the analysis. While some genetic signals emerge, their modest strength suggests T-cell genetic variations likely play only a supporting role in cervical cancer susceptibility rather than serving as primary drivers of disease development

Immune genetics in cervical cancer risk. A–D results suggest that while specific immune-related genetic markers show varied impacts, their overall contribution to cervical cancer risk appears limited. These findings indicate immune genetics likely plays a supporting rather than primary role in cervical cancer development

Genetic links between b-cells and cervical cancer. Forest plots A, B display individual genetic markers with wide confidence intervals, suggesting uncertain effects for single variants. Red summary lines indicate minimal overall genetic impact. C Shows slight downward trends across four statistical methods analyzing how genes affecting CD20 + B-cells relate to cervical cancer. Panel D's scattered precision plot helps identify potential research biases
4 Discussion
The findings from our extensive investigation offer fresh perspectives into cervical cancer's sophisticated molecular and cellular architecture, yielding several significant discoveries that enhance our understanding of disease mechanisms and potential treatment strategies.
Our initial discovery through Mendelian randomization analysis demonstrates a compelling causative link between genetic variants affecting immune cell populations and cervical cancer progression. By leveraging genetic instruments as unconfounded proxies, we identified specific SNPs associated with CD4 + regulatory T-cells, CD8 + T-cells, and B-cell function that influence cervical cancer susceptibility. These results, verified through leave-one-out sensitivity testing and diverse statistical methodologies, provide robust evidence for the role of genetically-determined immune variation in cervical cancer development.
Within cervical cancer's complex ecosystem, immune cells emerge as critical determinants of disease progression and treatment response. Our analysis revealed that specific T-cell populations, particularly CD69 + LGALS3A + CD4 + regulatory T-cells, play a decisive role in shaping anti-tumor immunity. Mendelian randomization evidence suggests that genetic variants predisposing to altered regulatory T-cell function may causally influence cervical cancer risk, though the effect sizes appear modest. This finding aligns with the understanding that regulatory T-cells can suppress effective anti-tumor immune responses, potentially facilitating cancer progression [11, 12, 21].
CD8 + T-cells, essential for anti-tumor cytotoxicity, also demonstrated genetic associations with cervical cancer outcomes in our analysis. Terminally differentiated CD8 + T-cell populations showed particularly interesting genetic correlations, suggesting that genetic determinants of CD8 + T-cell differentiation status may influence tumor control. The moderate effect sizes observed in our Mendelian randomization analysis indicate that while genetically-determined CD8 + T-cell function contributes to cervical cancer susceptibility, it likely represents one of multiple factors in disease development.
Our investigation also uncovered significant roles for B-cells in cervical cancer immunity. Genetic variants affecting CD20 + B-cell populations demonstrated causal relationships with cervical cancer risk, highlighting the importance of humoral immunity in disease progression. These findings emphasize the complex interplay between different immune cell populations in cervical cancer, with B-cells potentially contributing through antibody production, antigen presentation, and cytokine secretion.
The unique distribution of immune marker expression observed across cervical cancer subtypes suggests potential opportunities for precision immunotherapy strategies. Our Mendelian randomization findings provide genetic support for therapeutic interventions directed at particular immune cell subsets specifically those regulated by genetic variants highlighted in our investigation—which may demonstrate efficacy in select patient populations.However, several limitations deserve consideration. Although the genetic associations revealed through our MR methodology are noteworthy, they likely represent only a subset of the relevant biological mechanisms. The relatively small effect magnitudes detected for most genetic instruments indicate that while immune cell functionality appears causally linked to cervical cancer pathogenesis, it presumably functions in concert with additional critical risk determinants, including viral factors and environmental influences.Future research endeavors should focus on confirming these results in larger patient cohorts and investigating therapeutic interventions targeting the specific immune cell populations implicated by our Mendelian randomization investigation. Additionally, mechanistic studies are crucial to confirm the clinical viability of therapeutically modulating the genetically-influenced immune pathways identified through this research.
5 Limitations
The Mendelian randomization analysis, while robust, relies on several key assumptions that may not be fully satisfied, including the potential presence of horizontal pleiotropy. Additionally, our findings may have limited generalizability across diverse populations due to significant ethnic heterogeneity in genetic architecture. The genetic variants and their associations identified in our predominantly European-ancestry cohort may not fully capture the genetic determinants relevant to other ancestry groups, as allele frequencies, linkage disequilibrium patterns, and effect sizes often vary substantially across different racial and ethnic backgrounds. This ethnic specificity of genetic associations represents a significant constraint on the broad applicability of our results, particularly for non-European populations.
6 Conclusion
In conclusion, our integrated analysis provides a comprehensive framework for understanding cervical cancer biology, from genetic predisposition to cellular interactions. These insights could inform the development of more effective, personalized therapeutic strategies targeting specific aspects of the tumor microenvironment.
Data availability
The data is sourced from the GWAS database. For further information, please contact the corresponding author.
References
Anawate I, Lumley C, Silverwood S, Anchondo M, Hughes K, Kernell C, Rendle K, Bhatia R, Grover S. Global review of tools evaluating quality of life in cervical cancer survivors treated with chemoradiation therapy. Adv Radiat Oncol. 2025;10(2):101700.
Hamada K, Yamanoi K, Hayashi N, Kotani Y, Matsumoto H, Horikawa N, Abiko K, Yamanishi Y, Iemura Y, Taki M, et al. Re-evaluating prognostic factors for cervical cancer with lymph node metastasis: a Japanese multicenter cohort study based on FIGO 2018. Int J Clin Oncol. 2025;30(3):584–92.
Han X, Xia L, Wu Y, Chen X, Wu X. m6A-modified circSTX6 as a key regulator of cervical cancer malignancy via SPI1 and IL6/JAK2/STAT3 pathways. Oncogene. 2025;15:001–8.
He Y, Qiu Y, Yang X, Lu G, Zhao SS. Remodeling of tumor microenvironment by cellular senescence and immunosenescence in cervical cancer. Semin Cancer Biol. 2024;108:17–32.
Huang R, Wang F, Zou W, Li X, Lei T, Li P, Song Y, Liu C, Yue J. Tumor endothelium-derived PODXL correlates with immunosuppressive microenvironment and poor prognosis in cervical cancer patients receiving radiotherapy or chemoradiotherapy. Biomark Res. 2024;12(1):106.
Li M, Xu T, Yang R, Wang X, Zhang J, Wu S. Exploring MPC1 as a potential ferroptosis-linked biomarker in the cervical cancer tumor microenvironment: a comprehensive analysis. BMC Cancer. 2024;24(1):1258.
Wang H, Li X, Zhou S, Guo W, Wang Z, Sun L, Zhao Z, Han Y, Zhang S, Lv J, et al. Integrated analysis of single-cell RNA-seq and bulk RNA-seq reveals MMP mediated expression patterns by distinct tumor microenvironment immune profiles in cervical cancer. J Cancer. 2024;15(16):5258–76.
Li Q, Kaidong L, Tian Z, Diao W, Sun Y, Bai Y, Ma Y, Wei Y, Li J, Zhao W. Association of inflammatory factors with cervical cancer: a bidirectional Mendelian randomization. J Inflamm Res. 2024;17:10119–30.
Liu Z, Liu J, Wu Y, Zhou Z, Ousmane D, Zeinalzadeh Z, Wang J. Shared chemoresistance genes in ESCC and cervical Cancer: insights from pharmacogenomics and Mendelian randomization. Int Immunopharmacol. 2025;147:113933.
Zhao Z, Yan P, Zhang X, Yu X, Lv F, Gong M, Yang XA. Causal role of immune cells on cervical cancer onset revealed by two-sample Mendelian randomization study. Sci Rep. 2024;14(1):14890.
Yazaki S, Chiba Y, Kojima Y, Yoshida H, Takamizawa S, Kitadai R, Saito A, Kita S, Yamamoto K, Sumiyoshi-Okuma H, et al. Folate receptor alpha expression and the tumor immune microenvironment in patients with cervical cancer. Int J Gynecol Pathol. 2024;44(2):104–11.
Yi C, Yang J, Zhang T, Xie Z, Xiong Q, Chen D, Jiang S. lncRNA signature mediates mitochondrial permeability transition-driven necrosis in regulating the tumor immune microenvironment of cervical cancer. Sci Rep. 2024;14(1):17406.
Zhao YC, Wang TJ, She LZ, Cui J, Zhang CH. S100A10 overexpression correlates with adverse prognosis, tumor microenvironment, and aggressive behavior in vitro and in vivo of cervical cancer. J Cancer. 2023;14(15):2931–45.
Jl M. Badiger S, Kadakol GS: Macrophage migration inhibitory factor gene polymorphism in acute coronary syndrome. Cureus. 2024;16(9):e69914.
Rubio S, Somers V, Fraussen J. The macrophage migration inhibitory factor/CD74 axis in traumatic spinal cord injury: lessons learned from animal and human studies. Eur J Immunol. 2024;54(12):e2451333.
Yoon C, Kim HK, Ham YS, Gil WJ, Mun SJ, Cho E, Yuk JM, Yang CS. Toxoplasma gondii macrophage migration inhibitory factor shows anti-Mycobacterium tuberculosis potential via AZIN1/STAT1 interaction. Sci Adv. 2024;10(43):0101.
Doonan JM, Budde KB, Kosawang C, Lobo A, Verbylaite R, Brealey JC, Martin MD, Pliura A, Thomas K, Konrad H, et al. Multiple, single trait GWAS and supervised machine learning reveal the genetic architecture of Fraxinus excelsior tolerance to ash dieback in Europe. Plant Cell Environ. 2025;1:15361.
Izquierdo P, Wright EM, Cichy K. GWAS-Assisted and multi-trait genomic prediction for improvement of seed yield and canning quality traits in a black bean breeding panel. G3 (Bethesda). 2025;1:007.
Gong M, Zhao X, Li Q, Hao Q, Cha L, Dong G, Li X, Qiu F, Li D, Tian L. Integrated analysis of bulk RNA sequencing, eQTL, GWAS, and single-cell RNA sequencing reveals key genes in hepatocellular carcinoma. J Cell Mol Med. 2025;29(2):e70359.
Hindson J. GWAS reveals variants for alcohol-related hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2022;19(2):79.
Xu F, Ye Y, Gao Y, Xu S. Dual role of necroptosis in cervical cancer: promoting tumor aggression and modulating the immune microenvironment via the JAK2-STAT3 pathway. J Cancer. 2024;15(16):5288–307.
Funding
Not available.
Author information
Authors and Affiliations
Contributions
Zhang Zhang conceived and designed the study, supervised the research, and drafted the manuscript. FuRong Fu contributed to data collection, performed statistical analyses, and assisted in the interpretation of Mendelian randomization results. ShaoLi Zhuang conducted bioinformatics analysis, processed single-cell RNA sequencing data, and contributed to figure preparation and visualization. All authors contributed to the discussion of results, reviewed and revised the manuscript, and approved the final version for submission.
Corresponding author
Ethics declarations
Ethics approval
Not available. This study used publicly available summary data for Mendelian randomization analysis, which does not require ethics committee approval.
Consent to publish
All authors reviewed and approved the final manuscript.
Consent to participate
Not available.
Competing interests
The authors declare no competing interests.
Clinical trial
This research is not a clinical trial and is exempt from clinical trial registration requirements.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Z., Fu, F. & Zhuang, S. Mendelian randomization reveals limited causal effects of genetic variants on cervical cancer risk: insights from immune cell populations. Discov Onc 16, 944 (2025). https://doi.org/10.1007/s12672-025-02819-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-025-02819-2