
Abstract
Background and Objective
DBPR108 (prusogliptin) is a novel, orally bioavailable dipeptidyl peptidase-4 (DPP‐4) inhibitor. This study investigated the pharmacokinetics and pharmacodynamic characteristics of DBPR108 tablets in patients with type 2 diabetes.
Methods
In this randomized, parallel-group, open-label, phase I study, Chinese adults with type 2 diabetes, glycated hemoglobin of 7.0–9.5%, and body mass index of 19–35 kg/m2 were randomized 1:1:1 to once-daily DBPR108 50-, 100-, or 200-mg tablet groups. The primary endpoints included pharmacokinetic and pharmacodynamic characteristics after a single dose and multiple doses of DBPR108.
Results
In total, 30 patients were randomized with 10 patients in each group. DBPR108 was quickly absorbed with median time to reach the maximum plasma concentration of 1.5–4 h at steady state. Exposure to DBPR108 at steady-state increased dose proportionally with mean maximum steady-state plasma DBPR108 concentration during dosage intervals of 119, 256, and 567 ng/mL in the 50-, 100-, and 200-mg groups, respectively. Accumulation ratio of DBPR108 ranged from 0.85 to 1.3, and steady state was reached after four continuous daily doses. After multiple doses of DBPR108, maximum inhibitory efficacy of DPP-4 increased with higher dose levels ranging from 62.1 to 89.4%. Active glucagon-like peptide-1 levels increased after DBPR108 administration. In addition, six patients experienced treatment-emergent adverse events without leading to treatment interruption or discontinuation.
Conclusions
DBPR108 was well tolerated in Chinese patients with type 2 diabetes, and both the pharmacokinetic and pharmacodynamic profiles support once-daily dosage regimens of DBPR108 in future studies.
Trial Registration
ClinicalTrials.gov (NCT05146869); registered 23 November 2021.
Similar content being viewed by others

Explore related subjects
Discover the latest articles and news from researchers in related subjects, suggested using machine learning.Avoid common mistakes on your manuscript.
The exposure to DBPR108 (prusogliptin) increased dose proportionally and exhibited minimal accumulation after multiple doses. |
A trend of increasing dipeptidyl peptidase-4 inhibition and plasma active glucagon-like peptide-1 levels was observed with higher exposure to DBPR108. |
The study results support the use of once-daily dosage regimens of DBPR108 in future studies. |
1 Introduction
Dipeptidyl peptidase‐4 (DPP-4) inhibitors are a class of oral antidiabetic agents that inhibit the degradation of the incretins, mainly glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) [1]. GLP-1 stimulates insulin secretion, reduces glucagon concentrations, and delays gastric emptying [2]. GLP-1 and GIP are released after food intake, but DPP-4 degrades them immediately. By inhibiting DPP-4, the levels of GLP-1 and GIP increase, which induces glucose-dependent insulin release and a decrease in glucagon secretion to maintain glucose homeostasis [3]. With a low risk of hypoglycemia and high tolerability, DPP-4 inhibitors, as a treatment for type 2 diabetes, are suitable for specific patient populations, including the elderly, those with renal or hepatic impairment, and as an add-on for patients whose glycosylated hemoglobin (HbA1c) levels are above recommended targets with metformin [4,5,6].
As cardiovascular safety is an important aspect of antidiabetic treatments, regulatory agencies require large cardiovascular outcomes trials for the development of novel agents for the treatment or prevention of diabetes [7, 8]. Newer classes of antidiabetic agents, including DPP-4, GLP-1 receptor agonists, and sodium-glucose transporter 2 inhibitors, were approved with efficacy in cardiovascular outcomes trials [6]. Among newer classes of glucose-lowering drugs, DPP-4 inhibitors were the most widely prescribed agents [9, 10].
DPP-4 inhibitors are a group of heterogeneous compounds that have dissimilar chemical structures and different pharmacokinetic profiles [6, 11]. For instance, a US Food and Drug Administration safety review identified that saxagliptin and alogliptin might increase the risk of heart failure, especially in patients with heart or kidney diseases [12]. Furthermore, saxagliptin is associated with an elevated risk of hospitalization for heart failure [13]. While rare, increased risks of pancreatitis were observed in large clinical trials of saxagliptin, alogliptin, and sitagliptin [14]. In addition, unlike linagliptin, dose adjustment is required for sitagliptin, saxagliptin, and alogliptin when given to patients with renal insufficiency [6, 15, 16]. These potential adverse risks associated with existing DPP-4 inhibitors underscore the need for a safer DPP-4 inhibitor option, especially for the treatment of patients with type 2 diabetes, who present with different comorbidities.
DBPR108 (prusogliptin) is a novel, potent, orally bioavailable DPP‐4 inhibitor that displays excellent selectivity toward DPP‐4 (half-maximal inhibitory concentration [IC50]: 15 nM) over DPP‐2, DPP‐8, DPP‐9, and fibroblast activation protein (IC50: > 50 nM) [17, 18]. DBPR108 exhibits high potency and selectively inhibits DPP-4 activity in the plasma of humans, rodents, dogs, and monkeys with an IC50 of 4.1–20.4 nM. By comparison, the IC50 for sitagliptin was 12.3–64.4 nM in the same species [17]. DBPR108 in combination with metformin improved glucose tolerability in diet-induced obese mice [17]. The safety, pharmacokinetics (PK), and pharmacodynamic (PD) characteristics after single and multiple doses of DBPR108 capsules in healthy participants, as well as relative bioavailability studies of DBPR108 capsules and tablets, have been carried out (unpublished data). Nevertheless, scant information on the PK and PD profiles of DBPR108 in patients with type 2 diabetes is available in the public domain. We conducted this phase I study to investigate the PK, PD, and safety characteristics to provide a basis for dose adjustments and future studies in patients with type 2 diabetes.
2 Methods
2.1 Study Design and Participants
This randomized, parallel-group, open-label, phase I study (NCT05146869) was conducted in China. Adults (aged between 18 and 75 years) diagnosed with type 2 diabetes according to the World Health Organization 1999 criteria were enrolled. Patients had HbA1c between 7.0 and 9.5% and a body mass index within the range of 19–35 kg/m2. Key exclusion criteria included fasting plasma glucose > 13.9 mmol/L, requirement of insulin treatment based on the investigator’s judgement, acute or serious complications of diabetes, a history of serious hypoglycemia, and inadequate organ function. The full inclusion and exclusion criteria are provided in the Electronic Supplementary Material (ESM) pages 2–4.
The study consisted of screening, run-in, baseline, treatment (days 1–9), and follow-up (days 9–11) periods. Eligible patients were required to have a run-in period of at least 1 week when patients followed the diet and exercise routines recommended by the investigators. After completing the run-in phase, patients’ eligibility was confirmed in the baseline period, and they then remained in the research units throughout the treatment and follow-up periods (days 1–11).
Enrolled patients were assigned 1:1:1 to the DBPR108 50-mg, 100-mg, or 200-mg groups using the block randomization method. DBPR108 tablets were administered orally at a fasting state, once daily at the same time, on days 1 and 3–9, for a total of eight doses. Block randomization was employed with a fixed block size of six to ensure balanced group assignments throughout the study. The randomization sequence was generated by an independent statistician using SAS version 9.4, and allocation was implemented through an interactive response technology system.
The study followed the principles of the Declaration of Helsinki and requirements of Good Clinical Practices and applicable regulations. The study protocol was approved by the Ethics Committee for Clinical Trials of Beijing Anzhen Hospital, Capital Medical University, and the Ethics Committee of The First Affiliated Hospital of Bengbu Medical University. All patients provided written informed consents for their study participation.
2.2 PK Assessment
Blood samples to determine DBPR108 concentration were collected from each participant at the prespecified time points: pre-dose; 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, 24, 36, and 48 h post-dose on days 1 and 9; and pre-dose on days 7 and 8. Blood samples were collected in heparin sodium-containing tubes and were centrifuged at 1700 ± 50 g at 4 °C for 10 min. Then 50% acetic acid solution was added to the isolated plasma in a 1:100 volume ratio before storing at ≤ – 60 °C until analysis.
Plasma concentration of DBPR108 was determined using a validated liquid chromatography with tandem mass spectrometry method at Nanjing Clinical Tech Laboratories Inc. (Nanjing, China). The analytic range was 1.00–2000 ng/mL with a lower limit of quantitation (LLOQ) of 1.00 ng/mL.
2.3 PD Assessment
Blood samples for determination of DPP-4 activity and active GLP-1 level were collected at pre-dose; 1, 2, 4, 6, 8, 12, 24, 36, and 48 h post-dose on days 1 and 9; and pre-dose on days 7 and 8.
Blood samples were collected in K2EDTA-containing tubes and P800 blood collection tubes for the detection of DPP-4 activity and active GLP-1 level, respectively. Blood samples were centrifuged at 1300 ± 50 g at 2–8 °C for 10 min, and the resulting plasma samples were stored at ≤ – 60 °C until analysis.
Activity of DPP-4 was determined using the EnVision HTS Microplate Reader (PerkinElmer, MA, USA) with a LLOQ of DPP-4 activity at 21.23 μU/mL. Active GLP-1 level was determined by the MSD® V-PLEX GLP-1 Active Kit (Meso Scale Discovery, MD, USA) with a LLOQ of 0.182 pM. Bioanalyses of DPP-4 and GLP-1 were carried out by Accurant Biotech (Ningbo, China).
2.4 Safety Assessment
Safety evaluation included measurements of vital signs, physical examinations, electrocardiogram, and laboratory tests. Patients were monitored for adverse events from screening to study completion or early withdrawal.
Adverse events were coded in accordance with Medical Dictionary for Regulatory Activities Terminology version 23.0, and severity was graded as mild, moderate, or severe.
2.5 Statistical Analysis
The primary endpoints included PK and PD characteristics after a single dose and multiple doses of DBPR108. Safety was evaluated as a secondary outcome. According to the National Medical Products Administration guidelines [19, 20], and on the basis of the experience of a previous study [21], phase I studies generally include 8–12 patients in each group. This study planned to enroll ten patients in each group.
Demographic and baseline characteristics were summarized in the full analysis set (FAS), which included all randomized patients who received at least one dose of DBPR108. Safety was evaluated in the safety set (SS), which included patients who received at least one dose of DBPR108, and patients in the SS were grouped according to the actual dose level they received. The PK concentration set (PKCS) included all randomized patients who had received DBPR108 and had at least one measurable PK concentration. PK parameters were calculated from the PK parameter set (PKPS), which included patients who were randomized, had received DBPR108, and had at least one evaluable PK parameter. Patients who deviated from the protocol or used prohibited concomitant medications that would influence PK parameters were excluded from the PKPS. PD was analyzed in the PD set (PDS), which included all patients in the SS who had at least one evaluable PD assessment.
PK concentrations and plasma concentration–time profiles were summarized by treatment group in the PKCS. PK parameters including maximum plasma drug concentration (Cmax), area under the concentration–time curve from time zero to infinity (AUCinf), AUC from time zero to the time of the last measurable concentration (AUClast), AUC during a dosage interval (AUCtau), terminal elimination rate constant (λz,), time to reach the maximum plasma concentration (Tmax), apparent volume of distribution during the terminal phase (Vz/F), elimination half-life (t½), apparent total body clearance for extravascular administration (CL/F), maximum plasma drug concentration at steady state (Cmax,ss), minimum steady-state plasma drug concentration during a dosage interval (Cmin,ss), AUC from time zero to infinity at steady state (AUCinf,ss), AUC from time zero to the time of the last measurable concentration at steady state (AUClast,ss), AUC during a dosage interval at steady state (AUCtau,ss), time to reach the maximum plasma concentration at steady state (Tmax,ss), elimination half-life at steady state (t½,ss), apparent total body clearance for extravascular at steady state (CLss/F), and accumulation ration (Rac) were calculated from individual plasma concentration and actual sampling times using noncompartmental analysis in the PKPS.
PD analyses of DPP-4 inhibition and active GLP‐1 level were summarized descriptively in the PDS. DPP-4 inhibition was defined as the percentage of change in DPP-4 activity from baseline. Relationships between PK parameters and indicators of PD were analyzed using linear regression and Emax models.
PK parameters were calculated using Phoenix WinNonlin version 8.3 (Certara USA, Inc.). All other analyses were conducted using SAS version 9.4.
3 Results
3.1 Patients
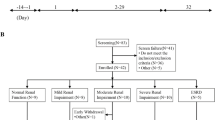
Between 30 December 2021 and 28 March 2022, 55 patients were screened in two study centers, and 30 patients were randomized into 50-, 100-, and 200-mg groups with 10 patients in each group (Fig. 1). All randomized patients received DBPR108 according to assigned dose levels and planned schedule without dose modification. All patients completed the study and were included in the FAS, PKCS, PKPS, PDS, and SS. The data cutoff date was 28 March 2022.

Study flowchart. FAS full analysis set, PDS pharmacodynamics set, PKCS pharmacokinetics concentration set, PKPS pharmacokinetics parameter set, SS safety set
Baseline characteristics were similar across the treatment groups (Table 1). In the overall population, the mean (standard deviation [SD]) age was 51.0 (9.5) years; 60% (n = 18) of patients were men; the majority of patients (n = 28 [93.3%]) were Chinese. The mean (SD) body mass index was 26.1 (2.3) kg/m2 and mean (SD) HbA1c was 8.2% (0.7%).
3.2 PK
After a single dose or multiple doses of DBPR108, the patterns of plasma concentration–time profiles were generally similar between groups (Fig. 2). DBPR108 was absorbed quickly after dosing, with a median Tmax of 1.5–4.5 h and t½ ranging from 5.0 to 12.3 h after a single dose (Table 2) and median Tmax,ss of 1.5–4.0 h and t½ ranging from 7.4 to 10.2 h after multiple doses (Table 3). Exposure to DBPR108 increased with dose.

Mean plasma concentration–time profiles of DBPR108 (PKCS) (a) single dose in linear scale, (b) single dose in semi-logarithmic scale, (c) multiple doses in linear scale, and (d) multiple doses in semi-logarithmic scale. PKCS pharmacokinetics concentration set
The plasma concentration–time profiles of multiple doses were comparable to those of a single dose (Fig. 2). DBPR108 exhibited minimal accumulation with Rac (AUC) of 0.95 (0.23), 1.02 (0.24), and 1.10 (0.29) and Rac (Cmax) of 0.85 (0.29), 1.09 (0.26), and 1.30 (0.84) in the 50-mg, 100-mg, and 200-mg groups, respectively. Steady state was achieved on day 7 (i.e., four continuous daily doses of DBPR108) (Electronic Supplementary Material Fig. 1).
Under the power model, the plasma concentration increased dose-proportionally from 50 mg to 200 mg after a single dose and multiple doses of DBPR108. The slope point estimates were near 1 while their 90% confidence intervals included 1 for Cmax and AUC (Electronic Supplementary Material Table 1).
3.3 PD
The percentage of DPP‐4 inhibition increased from baseline after a single administration of DBPR108, with greater inhibition observed at higher doses (Fig. 3a and b). After a single dose of DBPR108, the median time to Emax of DPP-4 was 2, 4, and 6 h and mean (SD) Emax,DPP-4 was 64.7% (7.2%), 72.6% (9.7%), and 88.7% (9.0%) for the 50-mg, 100-mg, and 200-mg groups, respectively. After multiple doses of DBPR108, the Emax,DPP-4 was reached between 3–4 h and Emax,DPP-4 increased with higher dose levels with a mean (SD) Emax,DPP-4 of 62.1% (7.3%) in the 50-mg group, 69.9% (12.5%) in the 100-mg group, and 89.4% (8.0%) in the 200-mg group.

Pharmacodynamic activity of DBPR108 (PDS) (a) inhibition of plasma DPP-4 activity following a single dose of DBPR108, (b) inhibition of plasma DPP-4 activity following multiple doses of DBPR108, (c) active GLP‐1 concentration-time profiles after a single dose of DBPR108, and (d) active GLP‐1 concentration-time profiles after multiple doses of DBPR108. DPP-4 dipeptidyl peptidase-4, GLP-1 glucagon-like peptide-1, PDS pharmacodynamic set
Higher concentration of active GLP‐1 was associated with increased dose levels of DBPR108 after both a single dose and multiple doses (Fig. 3c and d). After single administration of 50 mg, 100 mg, and 200 mg DBPR108, the median time to Emax,GLP-1 was 8, 12, and 12 h, and mean (SD) Emax,GLP-1 was 4.5 (1.6) pM, 4.8 (1.4) pM, and 5.5 (2.1) pM, respectively. After multiple doses of DBPR108, the Emax,GLP-1 was reached between 6–7 h with the mean (SD) Emax,GLP-1 of 5.7 (2.2) pM in the 50-mg group, 6.6 (2.3) pM in the 100-mg group, and 5.8 (3.2) pM in the 200-mg group.
3.4 PK–PD Relationship
After a single dose and multiple doses of DBPR108 from 50 mg to 200 mg, linear correlation was observed between DPP-4 inhibition and DBPR108 exposure, but no linear correlation was noted between changes in the plasma concentration of active GLP-1 and DBPR108 exposure.
The relationship between DPP-4 inhibition and DBPR108 exposure was fitted with the Emax model (Fig. 4). The predicted DPP‐4 inhibition was comparable to the observed DPP‐4 inhibition, indicating that the Emax model could accurately describe the relationship between DPP-4 inhibition and DBPR108 exposure. The Emax,DPP-4 was 120.4% and half maximal effective concentration (EC50) was 104.8 ng/mL after a single administration of DBPR108 and 106.2% and 80.8 ng/mL after multiple doses, respectively.

DPP‐4 inhibition versus DBPR108 plasma concentration (PKPS/PDS) (a) after a single dose of DBPR108 and (b) after multiple doses of DBPR108. C concentration at the receptor, DPP-4 dipeptidyl peptidase-4, Emax maximum drug effect, EC50 effective concentration at 50% of Emax, PDS pharmacodynamic set, PKCS pharmacokinetics concentration set
3.5 Safety
Among 30 enrolled patients, 20% of patients (n = 6; 1 in the 50-mg group, 4 in the 100-mg group, and 1 in the 200-mg group) experienced treatment-emergent adverse events (TEAEs). Most TEAEs were mild; only one event of hypertriglyceridemia in the 100-mg group was considered moderate in severity. TEAEs that occurred in the study were hypertriglyceridemia (n = 2 [6.7%]); hyperlipidemia, gingivitis, white blood cell count increase, neutrophil count increase, and iritis (n = 1 [3.3%] each). No severe TEAEs, serious TEAEs, or TEAEs leading to treatment interruption or discontinuation occurred in the study.
4 Discussion
This single- and multiple-dose, randomized study characterized the PK and PD of DBPR108 in Chinese patients with type 2 diabetes. DBPR108 had a favorable PK profile with relatively quick absorption, dose proportionality from 50 to 200 mg, and minimal drug accumulation. The PD results showed that the percentage of DPP-4 inhibition increased with higher DBPR108 exposure, and the plasma concentration of active GLP-1 increased after DBPR108 administration.
The PK profile of DBPR108 in patients with type 2 diabetes was in line with that in healthy subjects [22]. After a single dose of 100 mg DBPR108 in healthy subjects, the median Tmax was 4.48 (range 1–6) h with mean (SD) of Cmax 218.8 (101.8) ng/mL; AUCinf of 1447.3 (431.4) h × ng/mL; and t½ of 8.2 (8.6) h [22]. After multiple doses, DBPR108 was absorbed relatively quickly, with no obvious drug accumulation, as indicated by a t½,ss of 7.4–10.2 h and Rac range of 0.85–1.3. The steady state of DBPR108 was reached within a week (four continuous doses in a once-daily dosing schedule in this study). These characteristics support once-daily dosing of DBPR108.
The relationship between DPP-4 inhibition and DBPR108 exposure was fitted by the Emax model, with an Emax,DPP-4 of 106.19% and an EC50 of 80.8 ng/mL after multiple doses. After multiple doses, Emax,DPP-4 was 62.1% with 50 mg DBPR108, 69.9% with 100 mg DBPR108, and 89.4% with 200 mg DBPR108. The effect of the DPP-4 inhibition of DBPR108 was generally comparable with other DPP-4 inhibitors with Emax,DPP-4 of 90.4% after multiple doses of 100-mg sitagliptin once daily in Chinese patients with type 2 diabetes [23], 98.3% after multiple doses of 50-mg vildagliptin twice daily in Japanese patients with type 2 diabetes [24], 83.1% after a single dose of 5-mg saxagliptin in healthy subjects [25], 92.3% after multiple doses of 5-mg linagliptin once daily in male patients with type 2 diabetes [26], 92.7% after a single dose of 25-mg alogliptin in patients with type 2 diabetes [27], and 86.0% after 168 h post single dose of 25-mg omarigliptin in healthy Japanese subjects [28]. Observed increases in active GLP-1 level were consistent with the known mechanism of action of DBPR108. Following multiple doses of DBPR108, median time to Emax,GLP-1 was 6–7 h.
Similar to other DPP-4 inhibitors, DBPR108 was well tolerated in patients with type 2 diabetes. In the current study, no patients experienced serious TEAEs or TEAEs leading to dose modification. Furthermore, no cardiovascular adverse events were observed.
Results from this study provided an overall profile of the PK and PD effects of DBPR108 at different dose levels in Chinese patients with type 2 diabetes without missing doses or dose reductions and with completed assessments for PK and PD evaluations. Furthermore, the PK and PD profiles in Chinese patients with type 2 diabetes, as characterized in this study, inform further development of DBPR108, particularly for selecting the optimal dose regimen for this treatment. However, the study only enrolled Chinese patients. In addition, each group only included ten patients and each patient only received eight doses of DBPR108. A study with a larger sample size and longer treatment duration would provide a comprehensive PK, PD, and safety profile for DBPR108 in Chinese patients with type 2 diabetes.
In summary, single and multiple doses of DBPR108 had a favorable PK profile with a clinically meaningful effect on DPP-4 inhibition and increases of plasma active GLP-1 level. Greater exposure to DBPR108 was associated with increased DPP-4 inhibition. DBPR108 was well tolerated without any cardiovascular adverse observed in patients with type 2 diabetes. These results support the use of once-daily dose regimens of DBPR108 and the continued clinical development of DBPR108 in patients with type 2 diabetes.
References
Kasina S, Baradhi KM. Dipeptidyl peptidase IV (DPP IV) inhibitors. Treasure Island: StatPearls Publishing LLC.; 2024.
Edwards CM. The GLP-1 system as a therapeutic target. Ann Med. 2005;37:314–22. https://doi.org/10.1080/07853890510037400.
Capuano A, Sportiello L, Maiorino MI, Rossi F, Giugliano D, Esposito K. Dipeptidyl peptidase-4 inhibitors in type 2 diabetes therapy—focus on alogliptin. Drug Des Dev Ther. 2013;7:989–1001. https://doi.org/10.2147/dddt.S37647.
Davies MJ, D’Alessio DA, Fradkin J, Kernan WN, Mathieu C, Mingrone G, et al. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2018;2018(41):2669–701. https://doi.org/10.2337/dci18-0033.
Davies MJ, Aroda VR, Collins BS, Gabbay RA, Green J, Maruthur NM, et al. Management of hyperglycemia in type 2 diabetes, 2022. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2022;2022(45):2753–86. https://doi.org/10.2337/dci22-0034.
Florentin M, Kostapanos MS, Papazafiropoulou AK. Role of dipeptidyl peptidase 4 inhibitors in the new era of antidiabetic treatment. World J Diabetes. 2022;13:85–96. https://doi.org/10.4239/wjd.v13.i2.85.
US Food and Drug Administration. Guidance for industry: type 2 diabetes mellitus—evaluating the safety of new drugs for improving glycemic control. https://www.fda.gov/media/135936/download. Accessed 19 Aug 2024.
European Medicines Agency. Guideline on clinical investigation of medicinal products in the treatment or prevention of diabetes mellitus. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-medicinal-products-treatment-or-prevention-diabetes-mellitus-revision-2_en.pdf. Accessed 19 Aug 2024.
Yang A, Wu H, Lau ESH, Zhang X, Shi M, Fan B, et al. Glucose-lowering drug use, glycemic outcomes, and severe hypoglycemia: 18-year trends in 0·9 million adults with diabetes in Hong Kong (2002–2019). Lancet Reg Health West Pac. 2022;26: 100509. https://doi.org/10.1016/j.lanwpc.2022.100509.
Athanasakis K, Prodromiadou E, Papazafiropoulou A, Koutsovasilis A, Driva S, Ziori M, et al. Twenty-year trends in the prescription costs of type 2 diabetes: real world data and empirical analysis in Greece. Diabetes Res Clin Pract. 2020;162: 108095. https://doi.org/10.1016/j.diabres.2020.108095.
Kumar S, Mittal A, Mittal A. A review upon medicinal perspective and designing rationale of DPP-4 inhibitors. Bioorg Med Chem. 2021;46: 116354. https://doi.org/10.1016/j.bmc.2021.116354.
US Food and Drug Administration. FDA drug safety communication: FDA adds warnings about heart failure risk to labels of type 2 diabetes medicines containing saxagliptin and alogliptin. Accessed 19 July 2024.
Packer M. Do DPP-4 inhibitors cause heart failure events by promoting adrenergically mediated cardiotoxicity? Clues from laboratory models and clinical trials. Circ Res. 2018;122:928–32. https://doi.org/10.1161/circresaha.118.312673.
Tkáč I, Raz I. Combined analysis of three large interventional trials with gliptins indicates increased incidence of acute pancreatitis in patients with type 2 diabetes. Diabetes Care. 2017;40:284–6. https://doi.org/10.2337/dc15-1707.
Graefe-Mody U, Friedrich C, Port A, Ring A, Retlich S, Heise T, et al. Effect of renal impairment on the pharmacokinetics of the dipeptidyl peptidase-4 inhibitor linagliptin(*). Diabetes Obes Metab. 2011;13:939–46. https://doi.org/10.1111/j.1463-1326.2011.01458.x.
McGill JB, Sloan L, Newman J, Patel S, Sauce C, von Eynatten M, et al. Long-term efficacy and safety of linagliptin in patients with type 2 diabetes and severe renal impairment: a 1-year, randomized, double-blind, placebo-controlled study. Diabetes Care. 2013;36:237–44. https://doi.org/10.2337/dc12-0706.
Yeh KC, Yeh TK, Huang CY, Hu CB, Wang MH, Huang YW, et al. DBPR108, a novel dipeptidyl peptidase-4 inhibitor with antihyperglycemic activity. Life Sci. 2021;278: 119574. https://doi.org/10.1016/j.lfs.2021.119574.
Yeh TK, Tsai TY, Hsu T, Cheng JH, Chen X, Song JS, et al. (2S,4S)-1-[2-(1,1-dimethyl-3-oxo-3-pyrrolidin-1-yl-propylamino)acetyl]-4-fluoro-pyrrolidine-2-carbonitrile: a potent, selective, and orally bioavailable dipeptide-derived inhibitor of dipeptidyl peptidase IV. Bioorg Med Chem Lett. 2010;20:3596–600. https://doi.org/10.1016/j.bmcl.2010.04.124.
National Medical Products Administration. [H] GCL1-2: guidance for clinical pharmacokinetics studies of medicinal products. https://www.nmpa.gov.cn/wwwroot/gsz05106/07.pdf. Accessed 19 Aug 2024.
National Medical Products Administration. [H] Guideline for clinical trials of drugs and biological products for the treatment of diabetes mellitus. https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjyp/20120515120001975.html. Accessed 12 Feb 2025.
He YC, Liu TH, Wu LL, Qin LL. Review of the run-in period design in randomized clinical trials related to diabetes mellitus. Chin J New Drugs. 2018;27:2641–4.
Zhang H, Bian Y, Zhao W, Miao L, Zhang H, Cui J, et al. A phase I clinical study of the pharmacokinetics and safety of prusogliptin tablets in subjects with mild to moderate hepatic insufficiency and normal liver function. Curr Drug Metab. 2024;25:140–51. https://doi.org/10.2174/0113892002288120240221111336.
Zhou C, Zhou S, Wang J, Xie L, Lv Z, Zhao Y, et al. Safety, tolerability, pharmacokinetics and pharmacokinetic-pharmacodynamic modeling of cetagliptin in patients with type 2 diabetes mellitus. Front Endocrinol (Lausanne). 2024;15:1359407. https://doi.org/10.3389/fendo.2024.1359407.
Yamaguchi M, Saji T, Mita S, Kulmatycki K, He YL, Furihata K, et al. Pharmacokinetic and pharmacodynamic interaction of vildagliptin and voglibose in Japanese patients with type 2 diabetes. Int J Clin Pharmacol Ther. 2013;51:641–51. https://doi.org/10.5414/cp201902.
Upreti VV, Boulton DW, Li L, Ching A, Su H, Lacreta FP, et al. Effect of rifampicin on the pharmacokinetics and pharmacodynamics of saxagliptin, a dipeptidyl peptidase-4 inhibitor, in healthy subjects. Br J Clin Pharmacol. 2011;72:92–102. https://doi.org/10.1111/j.1365-2125.2011.03937.x.
Heise T, Graefe-Mody EU, Hüttner S, Ring A, Trommeshauser D, Dugi KA. Pharmacokinetics, pharmacodynamics and tolerability of multiple oral doses of linagliptin, a dipeptidyl peptidase-4 inhibitor in male type 2 diabetes patients. Diabetes Obes Metab. 2009;11:786–94. https://doi.org/10.1111/j.1463-1326.2009.01046.x.
Dudkowski C, Tsai M, Liu J, Zhao Z, Schmidt E, Xie J. The pharmacokinetics and pharmacodynamics of alogliptin in children, adolescents, and adults with type 2 diabetes mellitus. Eur J Clin Pharmacol. 2017;73:279–88. https://doi.org/10.1007/s00228-016-2175-1.
Tsuchiya S, Friedman E, Addy C, Wakana A, Tatosian D, Matsumoto Y, et al. Single and multiple dose pharmacokinetics and pharmacodynamics of omarigliptin, a novel, once-weekly dipeptidyl peptidase-4 inhibitor, in healthy Japanese men. J Diabetes Investig. 2017;8:84–92. https://doi.org/10.1111/jdi.12538.
Acknowledgements
The authors would like to thank the patients who participated in this study and their families and the investigators, study teams, and nurses who assisted with the study conduct. Medical writing and editorial assistance were provided by Tingyuan Yang of CSPC Pharmaceutical Group Co., Ltd. as well as Shao-Hua Chin and Lawrence Law of Parexel, funded by CSPC Zhongqi Pharmaceutical Technology (Shijiazhuang) Co., Ltd.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This study was funded by CSPC Zhongqi Pharmaceutical Technology (Shijiazhuang) Co., Ltd.
Conflict of interest
Juan Wang, Tianhao Zhang, Lingli Yao, Huanhuan Qi, Xiaofang Zhang, and Rui Jia are employees of CSPC Pharmaceutical Group Co., Ltd. The other authors declare that they have no conflicts of interest.
Data availability statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Ethics approval
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee for Clinical Trials of Beijing Anzhen Hospital, Capital Medical University, and the Ethics Committee of The First Affiliated Hospital of Bengbu Medical University.
Consent to participate
Informed consent was obtained from all patients included in the study.
Consent for publication
Not applicable.
Code availability
Not applicable.
Author contributions
Shan Jing, Xiaoli Li, and Tianhao Zhang conceptualized and designed the study; Lingli Yao, and Rui Jia provided administrative support; Juan Wang and Tianhao Zhang supplied study materials; Wenfang Liu, Kexu Yang, Yang Lin, Chunyan Lu, Jingyi Liu, and Huan Zhou performed data collection; Shan Jing, Wenfang Liu, Kexu Yang, Huanhuan Qi, and Xiaofang Zhang analyzed and interpreted the data; Shan Jing, Wenfang Liu, and Kexu Yang drafted the manuscript; all authors approved the final version; and Shan Jing and Xiaoli Li are accountable for all aspects of the work.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Liu, W., Yang, K., Lin, Y. et al. Pharmacokinetics and Pharmacodynamics of Prusogliptin (DBPR108), a Once-Daily Dipeptidyl Peptidase-4 Inhibitor, in Patients with Type 2 Diabetes. Clin Pharmacokinet 64, 703–713 (2025). https://doi.org/10.1007/s40262-025-01501-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-025-01501-8