
Abstract
Here we report a patient with holoprosencephaly (HPE) associated with 45, XY,der(18)t(18;21)(p11.2;q21.3),-21 derived from a paternal balanced reciprocal translocation. Array comparative genomic hybridization analysis revealed 18p11.32-p11.21 and 21q11.2-q21.3 deletions. So far, nine cases of monosomy 18p with an unbalanced translocation (18;21) have been reported, four of which presented with HPE. Our case provides a detailed long-term clinical course and helps us to better understand these rare genetic events.
Similar content being viewed by others

Holoprosencephaly (HPE) is estimated to occur in 1 in 16,000 live births, typically between the 18th and 28th day post-fertilization. It is a developmental disorder of the central nervous system formation and midline facial structures, caused by impaired left–right differentiation of the fetal forebrain1. HPE is classified into alobar, semi-lobar and lobar types, with the severity of facial malformations ranging from cyclopia to normal, correlating with the degree of central nervous system abnormalities. While patients with milder forms of HPE survive in adolescence or adulthood, severely affected patients do not survive beyond early infancy. Regarding the genetic background, 25–50% are due to chromosomal structural abnormality, such as trisomy 13, del(13q), dup(13q), del(18p), del(7)(q36), dup(3)(p24-pter), del(2)(p21) and del(21)(q22.3) and 18–25% are due to pathogenic variants of single genes, such as SHH, ZIC2, SIX3, TGIF, PYCH1, CDON, GLI2, FOXH1 and NODAL. 1,2.
Chromosome 18p deletion is characterized by intellectual disability, growth retardation and craniofacial dysmorphisms. Approximately 10% of patients with this syndrome show symptoms of HPE1,2. About two-thirds of the cases are de novo, and one-third of cases are caused by a de novo unbalanced translocation3. The critical gene for HPE is 5′-TG-3′-interacting factor (TGIF) gene located at 18p11.3 1,2.
We report a patient presenting HPE with an unbalanced translocation of chromosomes 18 and 21, 45,XY,der(18)t(18;21)(p11.2;q21.3)dpat,-21 derived from a paternal balanced reciprocal translocation.
The proband was an 8-year-old boy. He was the second child of healthy nonconsanguineous parents, in their early 30s. His older sister was in good health. His parents had no spontaneous abortion. Cleft lip and palate defects were found at 23 weeks of gestational age, with no other abnormalities. He was born by vaginal delivery at 40 weeks gestation. His birth weight was 2,574 g (standard deviation (s.d.) −2.2.), his height was 46 cm (s.d. −2.1) and his occipitofrontal circumference was 31.8 cm (s.d. −1.4). The Apgar score was 8 at 5 min. Multiple malformations were found including cleft lip and palate, depressed nasal bridge, palpebral fissure upslanted, hypertelorism, widely spaced nipples, micropenis and overlapping fingers. The muscle tone was normal. On the 8th day after birth, he was diagnosed with lobar HPE by brain magnetic resonance imaging (Fig. 1a,b). He was also diagnosed with syndrome of inappropriate antidiuretic hormone and treated with antidiuretic hormone. At 5 months of age, he was diagnosed with West syndrome and treated with valproic acid and clonazepam. Inguinal hernia repair was performed at 1 year of age. At 2 years of age, Nissen fundoplication and gastrostomy were performed to address oral intake difficulties and gastroesophageal reflex. At 3 years of age, oxygen therapy was initiated for severe mixed sleep apnea, and a tracheostomy was performed at 8 years of age. Currently, he has spastic quadriplegia and is bedridden. He cannot control his head, but he moves his hands with some intention. He expresses his feelings by crying. He is being treated with valproic acid, topiramate, phenobarbital, clobazam and clonazepam. However, his seizures remain intractable, and he continues to experience partial seizures with eye deviation and secondary generalized seizures.

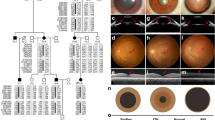
A, B Brain magnetic resonance imaging at 8 days of age. T2-weighted axial A and sagittal B images show lobar HPE. C Multicolor FISH of the patient. An unbalanced translocation at 45,XY,der(18)t(18;21),-21 was revealed. The yellow box shows -21, and the arrow indicates der(18)t(18;21). D, E Array CGH of the patient. The vertical axis represents the chromosomal position, while the horizontal axis shows the fold change in copy number variation. Array CGH revealed 45,XY,der(18)t(18;21)(p11.2;q21.3),-21, 13.8-Mb deletion at arr[GRCh37] 18p11.32p11.21(131,700_13,935,908)×1 D and 12.7-Mb deletion at arr[GRCh37] 21q11.2q21.3(14,957,733_27,609,147)×1 E.
G-banding chromosome analysis showed 45,XY,-21. However, because full monosomy 21 is lethal, a chromosomal translocation was suspected. Multicolor fluorescence in situ hybridization (FISH) analysis revealed an unbalanced translocation at 45,XY,der(18)t(18;21),-21 (Fig. 1c). Chromosome 21q was found to be translocated to chromosome 18p, and a partial deletion of chromosome 18p was suspected. To elucidate the deletion of 18p and 21q, array comparative genomic hybridization (CGH) was performed using SurePrint G3 Human CGH Microarray Kit, 4×180K after written informed consent from his parents. The test was approved by the bioethics committee for human gene analysis at Jichi Medical University.
Array CGH revealed 45,XY,der(18)t(18;21)(p11.2;q21.3),-21 with 13.8-Mb deletion at 18p11.32-p11.21 (arr[GRCh37] 18p11.32p11.21(131,700_13,935,908)×1) and 12.7-Mb deletion at 21q11.2-q21.3 (arr[GRCh37] 21q11.2q21.3(14,957,733_27,609,147)×1) (Fig. 1d,e). The Online Mendelian Inheritance in Man (OMIM) map showed that 14 morbid genes, THOC1, TYMS, SMCHD1, LPIN2, TGIF1, LAMA1, NDUFV2, APCDD1, PIEZO2, GNAL, TUBB6, AFG3L2, PSMG2 and MC2R, were deleted within the 18p11.32-p11.21, and that six morbid genes, NRIP1, USP25, TMPRSS15, MRPL39, JAM2 and APP, were deleted within the 21q11.2-q21.3. Parental multicolor FISH analysis demonstrated that his mother’s karyotype was 46,XX, and that his father’s karyotype was 46,XY,t(18;21), indicating his father’s balanced reciprocal translocation.
Chromosome 18p deletion is known to cause intellectual disability, growth retardation, craniofacial dysmorphisms and HPE1. Chromosome 21q11.2-q21.3 deletion is associated with a range of clinical phenotypes, from normal development to various degrees of speech delay, global developmental delay and other developmental disorders4,5,6. In our patient, the deleted region was approximately 12.7 Mb in size, spanning from the central region to 27.6 Mb. A patient reported by Lindstrand et al. with a 14.0-Mb deletion from the central region to 28.2 Mb presented with borderline developmental delay, aortic stenosis and inguinal hernia6. Another case reported by Errichiello et al. involved a 14.5-Mb deletion from the central region to 27.5 Mb was associated with intellectual disability, global motor impairment, dysmorphic features such as a low hairline and widely spaced nipples, amenorrhea, obesity and pituitary microadenoma5. While the clinical features of our patient are largely attributable to the del(18p), the 21q11.2-q21.3 deletion may have a subtle effect on brain development and developmental delay. In addition, inguinal hernia and widely spaced nipples may also be explained by the 21q11.2-q21.3 deletion. Our patient did not present with hypotelorism or a single maxillary central incisor but instead exhibited cleft lip and palate defects along with hypertelorism (Table 1). Although hypotelorism is observed in approximately 80% of HPE cases7, complex facial clefts with hypertelorism, as seen in our patient, are also recognized features of HPE8.
Reciprocal translocations lead to chromosomal imbalances by three types of disjunction: 3:1 segregation, adjacent 1 and adjacent 2 segregations. As chromosome 21 is acrocentric, 3:1 segregation is more likely to occur9. Previous reports of t(18;21) translocations and our case document eight instances of 3:1 tertiary monosomy10,11,12,13,14,15 and two instances of adjacent-1 segregation16,17 (Table 1). Among the eight cases with 3:1 tertiary monosomy, four presented with HPE. Their karyotypes are 45,XY,dic(18;21)(p11.1;p11.1)13, 45,XY,dic(18;21)(p11.1;p11.1)/ 46,XY,del(18)(p11.1),del(21)(p11.1)13, 45,XX,t(18;21)(q11.1;q11.1)15 and 45,XY,der(18)t(18;21)(p11.2;q21.3),-21 (our case). In addition, one case of adjacent-1 segregation, 46,XY,der(18)t(18;21)(p11.2;q22.3), was reported to have HPE17. The occurrence of HPE with 3:1 tertiary monosomy, four out of eight cases, is higher than the approximately 10% observed in overall 18p deletion syndrome. While a decrease in TGIF contributes to the development of HPE, other factors, such as disruptions in retinoic acid signaling, are also believed to be involved18. The precise mechanism underlying the higher frequency of HPE in 18p monosomy with 3:1 tertiary monosomy compared with overall 18p monosomy remains unclear and has yet to be elucidated. Among four previous cases of HPE, three cases were terminated13,15,17, and clinical course after infancy was not documented in the remaining case13. Our case highlights both the advancements and difficulties in managing HPE cases, and the long clinical course for 8 years described here will contribute to a better understanding of how to treat HPE cases in the future.
Concerning the inheritance, six instances were de novo10,12,13,14,15, three were maternally inherited11,16,17 and our case was paternally inherited (Table 1). As the father’s karyotype is expected to be 46,XY,t(18;21)(p11.2;q21.3) based on the proband’s karyotype, Stengel–Rutkowski’s method19,20 estimates the recurrence rate for this condition to be approximately 7% for paternal origin and 11% for maternal origin (Supplementary Table 1). However, precise estimation of the recurrent risk and genotype–phenotype correlation remains challenging due to limited data. Accumulating more cases is essential to establish a more accurate prediction method.
In summary, we present a boy with HPE and the karyotype 45,XY,der(18)t(18;21)(p11.2;q21.3),-21 caused by a paternal balanced reciprocal translocation. Our case helps us to better understand these rare genetic events.
HGV datbase
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3503https://doi.org/10.6084/m9.figshare.hgv.3506.
References
Dubourg, C. et al. Holoprosencephaly. Orphanet J. Rare Dis. 2, 8 (2007).
Roessler, E. & Muenke, M. The molecular genetics of holoprosencephaly. Am. J. Med. Genet. Part C 154c, 52–61 (2010).
Chen, C. P. et al. Chromosome 18p deletion syndrome presenting holoprosencephaly and premaxillary agenesis: prenatal diagnosis and aCGH characterization using uncultured amniocytes. Gene 527, 636–641 (2013).
Pavone, P. et al. A young boy with 21q21.1 microdeletion showing speech delay, spastic diplegia, and MRI abnormalities: original case report. Glob. Med. Genet. 10, 234–239 (2023).
Errichiello, E. et al. Dissection of partial 21q monosomy in different phenotypes: clinical and molecular characterization of five cases and review of the literature. Mol. Cytogenet. 9, 21 (2016).
Lindstrand, A. et al. Detailed molecular and clinical characterization of three patients with 21q deletions. Clin. Genet. 77, 145–154 (2010).
Tekendo-Ngongang, C., Muenke, M. & Kruszka, P. Holoprosencephaly Overview. In (eds Adam, M. P. et al.) GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. [Updated 2020 Mar 5]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1530/.
Castillo, M. & Mukherji, S. K. Imaging of facial anomalies. Curr. Probl. Diagn. Radio. 25, 169–188 (1995).
Jalbert, P. & Sele, B. Factors predisposing to adjacent 2 and 3:1 disjunctions: study of 161 human reciprocal translocations. J. Med. Genet. 16, 467–478 (1979).
Friedrich, U., Dalby, M., Staehelin-Jensen, T. & Bruun-Petersen, G. Chromosomal studies of children with developmental language retardation. Dev. Med. Child Neurol. 24, 645–652 (1982).
Tharapel, A. T. et al. Identification of the origin of centromeres in whole-arm translocations using fluorescent in situ hybridization with alpha-satellite DNA probes. Am. J. Med. Genet. 40, 117–120 (1991).
Artman, H. G., Morris, C. A. & Stock, A. D. 18p- syndrome and hypopituitarism. J. Med. Genet. 29, 671–672 (1992).
Wang, J. C., Nemana, L., Kou, S. Y., Habibian, R. & Hajianpour, M. J. Molecular cytogenetic characterization of 18;21 whole arm translocation associated with monosomy 18p. Am. J. Med. Genet. 71, 463–466 (1997).
Alkan, M. et al. Presumptive monosomy 21 with neuronal migration disorder re-diagnosed as de novo unbalanced translocation t(18p;21q) by fluorescence in situ hybridisation. Genet. Counsel. 13, 151–156 (2002).
Goldstein, I., Weissman, A., Brill-Zamir, R., Laevsky, I. & Drugan, A. Ethmocephaly caused by de novo translocation 18;21-prenatal diagnosis. Prenat. Diagn. 23, 788–790 (2003).
Smith, A. B., Peterson, P., Wieland, J., Moriarty, T. & DeLisi, L. E. Chromosome 18 translocation (18;21) (p11.1;p11.1) associated with psychosis in one family. Am. J. Med. Genet. 67, 560–563 (1996).
Chen, C. P. et al. Prenatal diagnosis of partial monosomy 18p(18p11.2->pter) and trisomy 21q(21q22.3->qter) with alobar holoprosencephaly and premaxillary agenesis. Prenat. Diagn. 21, 346–350 (2001).
Knepper, J. L., James, A. C. & Ming, J. E. TGIF, a gene associated with human brain defects, regulates neuronal development. Dev. Dyn. 235, 1482–1490 (2006).
Stengel-Rutkowski, S., Stene, J. & Gallano, P. Risk Estimates in Balanced Parental Reciprocal Translocations: Analysis of 1120 Pedigrees (Expansion Scientifique Française, 1988). https://books.google.co.jp/books/about/Risk_Estimates_in_Balanced_Parental_Reci.html?
Gardner, R. J. M. & Amor, D. J. Gardner and Sutherland’s Chromosome Abnormalities and Genetic Counseling (Oxford Univ. Press, 2018).
Acknowledgements
We appreciate the proband and his family to join the study. This work was supported by JSPS KAKENHI Grant Number 23K07254, AMED Grant Number 24FC1003, the Foundation for Development of the Community, and the Kawano Masanori Memorial Public Interest Incorporated Foundation for the Promotion of Pediatrics (Grant Number 35-5).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wakabayashi, H., Matsumoto, A., Komori, S. et al. Partial monosomy 18p and 21q due to a paternal reciprocal translocation leading to holoprosencephaly. Hum Genome Var 12, 10 (2025). https://doi.org/10.1038/s41439-025-00314-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-025-00314-2
