
Abstract
Azo switches are widely employed as essential components in light-responsive systems. Here, we develop an azo-fluorescent switch that is visible light-responsive and its light-responsive processes can be monitored using fluorescence imaging. Visible light irradiation promotes isomerization, accompanied by changes in fluorescence that enable the process to be monitored through fluorescence imaging. Furthermore, we document that the nanocavity size of liposome encapsulated nanoparticles containing azo changes in the isomerization process and show that this change enables construction of a light-responsive nanoplatform for optically controlled release of antimycotics. Also, natural light activation of nanoparticles of the switch loaded with an antimycotic agent causes death of Rhizoctonia solani. The results show that these nanoparticles can double the holding period in comparison to small molecule antimycotics. The strategy used to design the imaging-guided light-controlled nano-antimycotic release system can be applicable to protocols for controlled delivery of a wide variety of drugs.
Similar content being viewed by others

Introduction
Azo compounds are commonly used as molecular switches in the material sciences owing to their ability to undergo reversible isomerization between E and Z forms in response to light1,2. Owing to their non-invasive and biocompatible nature, visible light-activated azo optical switches3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21 have received great attention for potential applications in biological systems22,23,24. In particular, azo optical switches have been developed to engineer biomolecules, such as proteins and nucleic acids, so that their functions can be controlled through visible light-induced conformational changes25,26,27,28,29,30,31,32,33. Recently, azo switch-mediated optical control has been employed to explore and manipulate various cellular processes. And changes in molecular structure and the light-induced regulation of cell behavior are primarily manifested at the cellular microenvironment level34,35. It should be possible to design new types of azo switches that coordinate light control of biological processes with fluorescence imaging techniques developed for optical tracking and monitoring dynamic processes36,37,38,39,40,41,42,43,44,45,46,47,48. Thus, E-Z photoisomerization of new azo-fluorescent molecular switches, designed using this strategy, would not only function to modulate biological processes using light but also to induce fluorescence changes for signaling and assessing the light-regulated events.
The typical strategy employed to construct azo-fluorescent molecular switches involve introduction of a fluorophore directly at the ends of the azo unit or azobenzene moiety (see Fig. 1a)49,50,51,52,53,54,55,56,57,58,59. However, fluorophore-modified azo compounds are often susceptible to fluorescence quenching caused by a process known as fluorescence resonance energy transfer (FRET) between the fluorophore and the azo unit60. In addition, introduction of fluorophores can have a deleterious effect on photoisomerization efficiencies, and often elicit cytotoxicity and bio-incompatibility61,62,63. Moreover, azo compounds that have fluorophores directly linked to the azo moiety are difficult to prepare using simple synthetic methods. Direct incorporation of azoarene moieties as platforms for azo-fluorescent molecular switches might be able to circumvent some of these problems. However, it is well-known that azoarenes undergo non-radiative processes caused by rapid internal conversion that leads to dissipation of energy as heat rather than the emission of light64,65,66.

a Schematic of azo-fluorescent switches generated by direct fluorophore modification. b Schematic of D-A type azo-fluorescent switches operating via intramolecular charge-transfer. c Structure, photoisomerization of AzoPNMe2 and molecular design of AzoPJ in which TICT process is inhibited.
In contemplating alternate strategies that potentially circumvent these problems, we give attention to the design of visible light-activated azo-fluorescent molecular switches that contain the azoarene N = N double bond as a conjugating bridge separating an electron donor and electron acceptor. We reason that this approach can create a Donor-Acceptor (D-A) system (Fig. 1b).
In this work, we assess the photoswitch performance of the visible light-activated azo-fluorescent molecular switches in both in vitro and in vivo. We demonstrate that E,Z photoisomerization of this light activated-switch in the cellular microenvironment can be monitored by using fluorescence imaging. The structural changes promoted by photoisomerization of this azo-fluorescent switch is used as the foundation of a controlled release nanoplatform for the light-controlled release of drugs.
Results
Molecular design
The isomerization behavior of azo optical switches is closely related to their molecular structures. As is well known, electronically conjugated backbone having D-A feature are typically utilized for forming fluorescent molecules67, due to the presence of typical intramolecular charge transfer (ICT) processes in the system. Azo-heteroarenes are a class of visible-light-activated compounds that display excellent E-Z photoisomerization efficiencies6,7. Moreover, unsaturated heterocyclic moiety of azo-heteroarene can not only function as conjugated units, they also can serve as electron acceptors. One example of a previously described visible light-activated azo-heteroarenes possessing a typical D-A conjugated backbone is AzoPNMe2 (Fig. 1c)68. In this substance, the phenyl azo acts as the conjugating unit, while the terminal N,N-dimethylamino group and pyrazole ring serve as the respective electron donor and electron acceptor. Molecular frameworks of this type fit the design criteria of D-A type azo fluorescent switches described above. However, most studies of AzoPNMe2 have focused on exploring its photoisomerization properties, and little attention has been given to its fluorescence capabilities.
To assess its fluorescence properties, AzoPNMe2 was synthesized utilizing the reported method68. The fluorescence spectrum of this substance in DMSO at room temperature contains only a weak emission band at 475 nm (Supplementary Fig. 1). We speculate that the low fluorescence of AzoPNMe2 is associated with the occurrence of E,Z photoisomerization induced internal conversion, as well as non-radiative twisted intramolecular charge-transfer (TICT) between the N,N-dimethylamino and pyrazole groups69,70,71. Earlier observations have shown that TICT can weaken fluorescence and greatly reduce the photostability of compounds72. We reasoned that it should be possible to introduce appropriate structural constraints in AzoPNMe2 to block operation of TICT, and that this manipulation would lead to an azo-arene containing compound that fluoresces more strongly. We postulated that incorporating a structurally rigid strongly electron donating, julolidine group containing hexahydroquinolizine in place of the N,N-dimethylamino moiety in AzoPNMe2 would create the azoarene AzoPJ in which TICT is effectively restricted (Fig. 1c). Thus, unlike AzoPNMe2, AzoPJ may display efficient fluorescence.
Synthesis and characterization
To explore the proposal made above, AzoPJ was synthesized utilizing the short route shown in Fig. 2a. Reaction of julolidine with the diazonium salt generated by treatment of 4-amino-1,3,5-trimethylpyrazole with sodium nitrite led to production of the target AzoPJ in 65% yield. The structure of AzoPJ was assigned using NMR spectroscopy and mass spectrometry. To gain detailed structural information, a single crystal of AzoPJ was produced utilizing slow diffusion of hexane into a dichloromethane solution and then subjected to X-ray crystallographic analysis (Supplementary Table 1 and Supplementary Data 1). Analysis of the crystal structure (Fig. 2b) showed that the azo-moiety in AzoPJ has E-configuration, and the conjugated system including the julolidine and pyrazole groups did not present an absolute planarity. Inspection of the x-ray crystallographic data also demonstrates that AzoPJ molecules in the crystal exist in an orderly stacked arrangement that results from multiple intermolecular interactions such as C-H···π and C-H···N types (Supplementary Fig. 2). Of importance is the observation that the crystal of AzoPJ displays bright green fluorescence (excitation at 430 nm), and its fluorescence spectrum contains two distinct emission peaks at 535 and 569 nm (Supplementary Fig. 3). Moreover, the intensity of fluorescence of AzoPJ in DMSO (Fig. 2c) is 4-fold greater than that of AzoPNMe under the same conditions. These observations described above indicate that the fluorescence properties of AzoPJ are in accord with those required for an azo-arene based fluorescent switch.

a Synthetic route for preparation of AzoPJ. b Crystal structure of AzoPJ. c Normalized fluorescence spectra of AzoPNMe2 (150 μmol L−1) and AzoPJ (150 μmol L−1) in DMSO (AzoPNMe2: λex = 328 nm; AzoPJ: λex = 350 nm). d Frontier molecular orbital profiles of E-AzoPJ and Z-AzoPJ arising from TD-DFT calculations at the B3LYP/6-31 G* level with the Gaussian 09 program. Source data are provided as a Source Data file.
Time-dependent density functional theory (TD-DFT) calculations were performed to gain additional insight into the molecular structure and electronic properties of AzoPJ. Absorption maxima of the major electron transitions and the molecular orbitals involved in these transitions were calculated using this method (Fig. 2d). Analysis of the optimized structure shows that the computed absorption spectrum of E-AzoPJ should undergo two major electronic transitions, including an intense S0-S2 transition at ca. 418.64 nm with an oscillation intensity of 1.0032. Moreover, this band has a 71% contribution from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) transition. Another calculated absorption band of E-AzoPJ corresponds to a S0-S5 transition, with a maximum absorption wavelength of 283.40 nm, an oscillation intensity of 0.2464 and a 55% contribution from a HOMO-2 to LUMO transition. For its isomer Z-AzoPJ, the calculated absorption spectrum also contains two major electronic transitions, one of which corresponds to an S0-S1 transition (496.73 nm), with an oscillation intensity of 0.2180, and 61% contribution from a HOMO to LUMO transition. The other corresponds to a S0-S2 transition (371.68 nm), with an oscillation intensity of 0.3148 and a 58% contribution from HOMO-1 to LUMO transition. Inspection of calculated molecular orbital coefficients show that electron densities in the HOMO and LUMO of E-AzoPJ are distributed over almost the entire azo conjugate skeleton. However, its HOMO orbital electron density has a tendency to favor the julolidine group while the LUMO orbital electron density is uniformly distributed throughout the azo conjugated backbone. This finding indicates that E-AzoPJ will have an intramolecular charge transfer character. Furthermore, the occurrence of similar charge transfer can also be seen by analysis of electron distributions of Z-AzoPJ in HOMO and LUMO transition. The theoretical results summarized above indicate that AzoPJ is capable of acting as a fluorescent molecular switch potentially.
Photoisomerization properties of AzoPJ
Photoisomerization of a substance is an important metric for evaluating its performance as an optical switch. The photoisomerization behavior of AzoPJ in DMSO was evaluated by using UV-vis absorption spectroscopy. The absorption spectrum of AzoPJ (Fig. 3a) contains a broadband between 315–600 nm with a maximum at 440 nm, suggesting that AzoPJ exists mainly in its most stable E-form. After reaching a photostationary state created by irradiation with 440 nm light, the spectrum contains two intense absorption peaks at 376 nm and 453 nm and the color of solution is changed from yellow to orange. These observations show that a mixture of the E- and Z-isomer is formed by irradiation. Validation of the reversibility of this photochemical process was gained by showing that irradiation of the photostationary state mixture of E- and Z-AzoPJ with 535 nm light resulted in nearly full recovery of the absorption spectrum of the E-isomer. Moreover, a clear isosbestic point at 478 nm exists in the absorption spectra produced by 440 nm irradiation (Fig. 3a–c), which indicates that singlet excited state of AzoPJ undergoes exclusive visible light-induced E,Z-isomerization. The integrity of the photoisomerization process is maintained following ten alternate exposures to 440 light and 535 nm light (Fig. 3d). The 440 nm light employed to promote E-to-Z isomerization coincides with the highest intensity region of natural light radiation. Consequently, natural light can also be utilized to drive the photoisomerization reaction (Fig. 3e). The combined results demonstrate that AzoPJ has the potential of functioning as a visible light-activated optical switch.

a UV-Vis absorption spectra of AzoPJ (150 μmol L−1) in DMSO. b Absorption spectra changes of AzoPJ (E → Z) in DMSO upon irradiation with 440 nm light (50 mW cm−2). c Absorption spectral changes of a Z- and E-mixture of AzoPJ (Z → E) in DMSO upon irradiation with 535 nm light (50 mW cm−2). d Reversible photoisomerization cycles promoted by alternate irradiation of AzoPJ (150 μmol L−1) in DMSO with 440 (50 mW cm−2) and 535 nm light (50 mW cm−2). e UV-Vis absorption spectra of AzoPJ (150 μmol L−1) in DMSO after irradiation with natural light (simulated using an artificial climate box MGC-450HP-2L). f Fluorescence spectra of AzoPJ (150 μmol L−1) in DMSO upon irradiation with 440 (20 W cm−2) and 535 nm light (15 W cm−2) (λex = 350 nm). Source data are provided as a Source Data file.
Resembled most of azo compounds, AzoPNMe2 exhibited a weak fluorescence in DMSO. As described above, to improve fluorescence property, the N,N-dimethylamino group in AzoPNMe2 was replaced by the julolidine moiety so that excited state non-irradiation decay caused by C-N bond rotation (TICT process) could be inhibited. As predicted, AzoPJ in DMSO displays obvious fluorescence at 507 nm upon excitation at 350 nm (Fig. 3f). Upon irradiation with 440 nm light, the fluorescence spectrum of a solution of AzoPJ in DMSO changes in that emission at 420 nm gradually increasing while emission at 507 nm gradually decreasing (Supplementary Fig. 4). In addition, optical images of the solution show that the original green fluorescence of the solution darkens upon 440 nm irradiation, in a manner that matches changes occurring in the fluorescence spectra. At the photostationary state, the fluorescence spectrum contains two main emission peaks at 420 and 507 nm. In addition, irradiation of the mixture at 535 nm light causes emission at 420 nm to gradually weaken while the band at 507 nm gradually increases, and following a time period the fluorescence spectrum becomes nearly that of the original solution (Fig. 3f and Supplementary Fig. 4). These observations clearly show that AzoPJ has photoisomerization, absorption and fluorescence properties that could be suited to use as a fluorescent molecular switch.
Nanometer size optically controlled switch
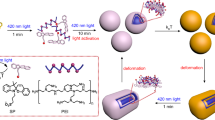
In addition to inhibiting the TICT process and increasing the luminescence, introduction of the julolidine group into AzoPJ is expected to have an impact on the space required for E-Z isomerization. This suggestion arises from a comparison of the 3.8417 × 10−22 cm3 volume of AzoPNMe2 with that of AzoPJ which is 4.3343 × 10−22 cm3. At the current time, four possible mechanisms have been proposed for photoisomerization of azo compounds mainly including simple rotation, inversion, concerted inversion and inversion-assisted rotation73. However, regardless of the pathway, azo compounds with larger-sized functional groups may require more space to support the above rotation, or inversion, etc, during E-Z isomerization (Fig. 4a). In this way, the larger size of the julolidine unit is expected to cause obvious changes in the cavity size before and after light irradiation in the resulting nano-system of AzoPJ, as described in Fig. 4b. Such switchable cavity will provide a possibility for the construction of a nano-controlled release system.

a Calculated (RHF/6-31 G** level with the Gaussian 09 program) molecular volumes of AzoPNMe2 and AzoPJ and diagrams of trajectories of the N-substituents upon N = N bon rotation. b Schematic of the volume change for AzoPJ NPs promoted by photoisomerization. Size distributions of AzoPJ NPs determined using DLS in the dark (c) and after irradiation with 440 nm light (d). TEM images of AzoPJ NPs without irradiation (e) and after irradiation with 440 nm light (f), (TEM results are collected using a Tecnai G2 F20 TEM system (FEI) at 200 kV). AzoPJ NPs: 100 μg mL−1; 440 nm light: 20 W cm−2, 6 h. e, f n = 3 independent experiments.
To assess this proposal, AzoPNMe2 and AzoPJ nanoparticles (NPs) were fabricated by mixing aqueous solutions of the matrix-encapsulation agent distearoyl phosphatidyl acetamide-methoxy polyethylene glycol-5000 (DSPE-mPEG5000) with individual THF solutions of the azo-fluorescent switches followed by solvent removal using a nitrogen stream. The nanoparticle diameters and zeta potentials of the prepared nanoparticles before and after light irradiation were determined using dynamic light-scattering (DLS). DLS analysis shows that the average hydrodynamic diameter of AzoPNMe2 NPs changes from 179.8 nm to 193.1 nm, and the zeta potential is transformed from −49.85 mV to −40.08 mV upon irradiation (Supplementary Figs. 5 and 6). Although the DLS results show that AzoPNMe2 NPs do undergo small size and potential changes upon irradiation, observation made using transmission electron microscopy (TEM) demonstrate that the morphology of AzoPNMe2 NPs is not significantly altered (Supplementary Fig. 7).
The properties of AzoPJ NPs were evaluated using the same methods. DLS results show that the average size of these NPs increases from 117.5 nm to 165.4 nm (Fig. 4c, d), and the zeta potential changes from −53.9 mV to −29.88 mV upon irradiation with 440 nm light (Supplementary Fig. 8). This observation shows that, in contrast to that of the AzoPNMe2 NPs, the size of AzoPJ NPs becomes significantly larger after 440 nm irradiation, a result that is confirmed using TEM (Fig. 4e, f). Additionally, the nanoparticles showed a trend of diminishing absorption intensity in the 365–500 nm range similar to that induced by AzoPJ photoisomerization under 440 nm light irradiation while 535 nm light irradiation was able to partially recover (Supplementary Fig. 9), probably due to the different solution environments and states in which the nanoparticles and the small-molecule AzoPJ are located74,75,76,77. Moreover, the size and zeta potential of the nanoparticles also show a trend of decreasing and then increasing when it was radiated with 440 nm and 535 nm light, respectively (Supplementary Figs. 10 and 11). The large negative zeta potentials and their trends before and after light irradiation are likely to be related to the molecular structure and property of the liposomes and the azo molecules encapsulated in it78,79,80,81,82,83. Thus, introduction of julolidine group not only improves the fluorescence intensity of the azo switch, it also creates the opportunity for the system to be utilized as a visible light controlled, nanometer scale, optical switch for fluorescence monitored release of small molecule agents. All these findings are in perfect agreement with our design.
To explore its potential for use in biological systems, AzoPJ NPs was subjected to stability and biocompatibility evaluations. For this purpose, time-dependent changes in the absorption spectrum of AzoPJ NPs were determined as a function of both time in the dark and different pH values. The findings suggest that no significant changes take place in the spectra of nanoparticles under dark conditions (Supplementary Fig. 12) and under different pH condition (Supplementary Fig. 13), suggesting that these nanoparticles have excellent stability. Further evidence was obtained by checking the changes of hydrodynamic diameter and zeta potential with time and pH (Supplementary Figs. 14 and 15). In addition, assessment of the cytotoxicity of the NPs was made using the MTT method involving incubation of B16 cells with the AzoPJ NPs for 24 h followed addition and incubation with MTT solution for another 4 h. The intensity of absorbance at 490 nm of each sample was measured using a microplate reader to yield the cell viability. The results show that B16 cells maintain high viability in the presence of AzoPJ NPs at concentrations in the range of 0–40 µg mL−1 (Supplementary Fig. 16).
Fungal imaging
Investigations in vitro as described above confirmed that AzoPJ NPs possessed an excellent optical regulatory property and good biocompatibility. To explore the fluorescence imaging capability of AzoPJ NPs in biological systems, we next conducted studies employing Rhizoctonia solani, a pathogen that causes devastating diseases to various important crops worldwide. In the effort, Rhizoctonia solani were first activated by culturing at 28 °C on Potato Dextrose Agar (PDA) medium and then incubated with different concentrations of AzoPJ NPs at 28 °C for 24 h followed by extensive washing (3x) with PBS. Confocal laser scanning microscopy (CLSM) was then conducted by monitoring fluorescence signals in the 460–580 nm region with excitation at 405 nm. As the images in Fig. 5a show, green fluorescence arises from the hyphae of the fungus with an intensity that increases as the concentration of AzoPJ NPs in the incubation mixture increases (Fig. 5b). When the AzoPJ NPs concentration used for incubation is 1 mg mL−1, the microstructure morphology of the hyphae, especially the septa on the hyphae, is clearly visible in the fluorescence image. A time-dependent fluorescence imaging study (Supplementary Fig. 17) indicates that the intensity of the fluorescence images of hyphae gradually increase with increasing incubation times (Fig. 5c), and when the incubation time is 48 h, the hyphae emit strong green fluorescence. The above results indicate that AzoPJ NPs is taken up by fungal cells and that it fluoresces in the cells.

a CLSM images of Rhizoctonia solani incubated with AzoPJ NPs at different concentrations (λex = 405 nm, λem = 460–580 nm). b Fluorescence intensity of Rhizoctonia solani incubated with different concentrations of AzoPJ NPs, the data on the bar chart indicate mean. c Fluorescence intensity of Rhizoctonia solani incubated with AzoPJ NPs (1 mg mL−1) for different times, the data on the bar chart indicate mean. d CLSM images of Rhizoctonia solani incubated with AzoPJ NPs (1 mg mL−1), before and after irradiation with 440 nm light (440 nm light: 20 W cm−2, 1 h; λex = 405 nm, λem = 460–580 nm), the data on the bar chart indicate mean. e Fluorescence intensity of Rhizoctonia solani incubated with AzoPJ NPs before and after irradiation with 440 nm light, the data on the bar chart indicate mean. a–e n = 3 independent fungal biological sample in each experiment. Statistical significance is calculated by (b, c, e) one-way ANOVA. Each graph is presented as the mean ± SD. Source data are provided as a Source Data file.
For a light-driven controlled release nano-system, it is extremely critical that AzoPJ NPs undergoes successful photoisomerization in the cell. As described above, CLSM images (Fig. 5d) of AzoPJ NPs in hyphae of Rhizoctonia solani display green emission. However, upon 440 nm irradiation the green fluorescent diminishes significantly (Fig. 5e), suggesting that AzoPJ in NPs undergoes photoisomerization in vivo. Moreover, nearly the same observations arise from fluorescence imaging studies with Staphylococcus aureus (Supplementary Figs. 18–23).
Light-controlled releasable nano-antimycotics
One of the important functions of photoswitches is their use in the construction of controlled release systems for nanodrugs84,85,86. Observations made in this investigation suggest that AzoPJ has the capability of functioning as a promising light-responsive component of a controlled release nanoplatform for use in biological systems, and that the release process can be monitored employing fluorescence imaging. In the next phase of this study, we constructed and explored the properties of a AzoPJ-based fluorescence imaging-guided nano-system for controlled release of drugs. The matrix encapsulation method described above was employed to prepare this nano-system. Accordingly, mixing an aqueous solution of DSPE-mPEG5000 with a THF solution of AzoPJ and the antimycotics flubeneteram (PEPA) followed by solvent removal using a nitrogen stream led to generation of the target AzoPJ-PEPA NPs (Fig. 6a). DLS analysis shows that the average size of AzoPJ-PEPA NPs increases by almost 100 nm from 136.1 nm to 230.7 nm upon irradiation with 440 nm light (Fig. 6b, c), the change that is confirmed using TEM (Fig. 6d, e). Also, 440 nm irradiation promotes a change in the electric potential of these NPs from −30.97 to −1.39 mV (Supplementary Fig. 24). As a control, PEPA NPs, which do not contain the azo switch, were synthesized using the same method. DLS and TEM studies show that these NPs do not undergo significant changes in size or electrical potential upon irradiation. (Supplementary Figs. 25–27). Furthermore, AzoPJ-PEPA NPs exhibit a high stability over a wide pH range (Supplementary Figs. 28 and 29).

a Schematic of formation and light-controlled release function of AzoPJ-PEPA NPs; Size distribution of AzoPJ-PEPA NPs by DLS in the dark (b) and after irradiation with 440 nm light (c). TEM images of AzoPJ-PEPA NPs without irradiation (d) and after irradiation with 440 nm light (e), (Scale bar = 200 nm, TEM results are collected using a Tecnai G2 F20 TEM system (FEI) at 200 kV). f In vitro PEPA release from AzoPJ-PEPA NPs induced by 440 nm irradiation, natural light irradiation and under dark conditions, the data on the line chart indicate mean. g Viability of B16 cells in the presence of AzoPJ-PEPA NPs (0–40 μg mL−1), the data on the bar chart indicate mean. Cells are incubated with AzoPJ-PEPA NPs (0–40 μg mL−1) for 24 h. AzoPJ-PEPA NPs: 100 μg mL−1; 440 nm light: 20 W cm−2, 6 h. d–f n = 3 independent nanoparticles solution in each experiment. g Data are collected from ten biological replicates. Statistical significance is calculated by one-way ANOVA. Each graph is presented as the mean ± SD. Source data are provided as a Source Data file.
Next, we assessed features of the light-controlled release of antimycotics by AzoPJ-PEPA NPs. First, construct a standard release curve (Supplementary Fig. 30) and the dialysis method was employed to generate data. Analysis of this curve showed that the efficiency for encapsulation of PEPA in the NPs is 80%. Release behavior of PEPA from AzoPJ-PEPA NPs in PBS buffer was then evaluated using absorption spectroscopic analysis of samples either irradiated with 440 nm light, natural light or under dark conditions as a reference. The intensity of antimycotics characteristic absorption peak around at 260 nm increased gradually not only under 440 nm light but also under natural light irradiation (Supplementary Figs. 31 and 32). However, no significant changes in the absorption spectra were detected when AzoPJ-PEPA NPs were placed in a dark condition (Supplementary Fig. 33). Meanwhile, the stability of the nano-antimycotics during PEPA release was also evaluated by DLS and zeta potential. Upon irradiation with 440 nm light, the average size of these NPs shows an increasing trend while the zeta potential is less negative with the increase of irradiation time in 0–24 h (Supplementary Fig. 34). But in dark condition, no obvious changes of DLS and zeta potential of AzoPJ-PEPA NPs are observed (Supplementary Fig. 35). Those results indicate that the nanoparticles release the antimycotic PEPA upon irradiation in the visible light region. Inspection of the release curves displayed in Fig. 6f, shows that the cumulative release of PEPA increases and reaches a plateau at ca. 40% when irradiated at 440 nm. In contrast, only 3% of PEPA is released when AzoPJ-PEPA NPs is kept in the dark for this period. In addition, the natural light radiation-driven release of antimycotics almost reached a similar efficiency of the release. Finally, an assessment carried out by determining the viability of B16 cells in the presence of AzoPJ-PEPA NPs (0–40 μg mL−1) using the MTT method showed that it has low cytotoxicity (Fig. 6g).
An investigation of the use of AzoPJ-PEPA NPs in fluorescence imaging-guided light-controlled release was carried out in Rhizoctonia solani. By viewing CLSM images in Fig. 7a, it is clearly seen that Rhizoctonia solani stained individually by AzoPJ NPs and AzoPJ-PEPA NPs display green fluorescence, and that their complete hyphae structure can be observed in the fluorescence images. And, the intensity of green fluorescence of AzoPJ-PEPA NPs stained Rhizoctonia solani significantly weakens upon 440 nm irradiation (Fig. 7b). This change in fluorescence is attributed to isomerization of the azo-switch under 440 nm light radiation. It follows that the fluorescence changes originating from the azo-switch isomerization are able to visualize the release process of antimycotics by fluorescence imaging.

a CLSM images of Rhizoctonia solani incubated individually with AzoPJ NPs, AzoPJ-PEPA NPs, or AzoPJ-PEPA NPs with 440 nm light irradiation (λex = 405 nm, λem = 460–580 nm). b Fluorescence intensities of Rhizoctonia solani incubated with AzoPJ NPs, AzoPJ-PEPA NPs, or AzoPJ-PEPA NPs with 440 nm light irradiation, the data on the bar chart indicate mean. c Expanded images of hypha treated by AzoPJ-PEPA NPs, or AzoPJ-PEPA NPs with 440 nm light irradiation, arrows designate areas of necrotic hypha. d CLSM images of Rhizoctonia solani stained with PI after being treated with PEPA, AzoPJ-PEPA NPs, or AzoPJ-PEPA NPs with 440 nm light irradiation. e Fluorescence intensities of Rhizoctonia solani stained with PI after being treated with PEPA, AzoPJ-PEPA NPs, or AzoPJ-PEPA NPs with 440 nm light irradiation, the data on the bar chart indicate mean. AzoPJ NPs: 1 mg mL−1; AzoPJ-PEPA NPs: 1 mg mL−1; PEPA: 200 mg L−1; 440 nm light: 20 W cm−2, 1 h. a–e n = 3 independent fungal biological sample in each experiment. Statistical significance is calculated by (b, e) one-way ANOVA. Each graph is presented as the mean ± SD. Source data are provided as a Source Data file.
In addition, deformation and rupture of hyphae of Rhizoctonia solani take place after staining with AzoPJ-PEPA and irradiation with 440 nm light, while almost no changes occur in hyphae treated only with AzoPJ-PEPA NPs and not subjected to irradiation (Fig. 7c). This phenomenon can be attributed to the inhibitory effect on growth of the fungus brought about by light driven released PEPA. Particularly noteworthy is the finding that deformation and rupture of hyphae of AzoPJ-PEPA treated and irradiated bacteria are synchronized with changes in fluorescence intensities. This finding suggests that light-controlled release of antimycotics in living fungal cells can be visually monitored by utilizing fluorescence imaging.
Experiments were conducted to gain solid support for the conclusion that treatment with AzoPJ-PEPA followed by 440 nm irradiation promotes damage of Rhizoctonia solani. For this purpose, the fluorescent dye propidium iodide (PI), commonly employed to characterize cell viability, was incubated with the Rhizoctonia solani that are individually treated with either PEPA, AzoPJ-PEPA NPs, or AzoPJ-PEPA NPs with 440 nm light irradiation. By analyzing fluorescence intensities of CLSM images in the range of 590–700 nm, it was found that the fungus treated with PEPA alone displays red fluorescence (Fig. 7d), suggesting that PEPA effectively damages Rhizoctonia solani. Equally red fluorescence arises from the images of fungus treated with AzoPJ-PEPA NPs and exposed to 440 nm light (Fig. 7e). In contrast, images of Rhizoctonia solani only treated with AzoPJ-PEPA NPs display only negligible red fluorescence. The combined observations clearly show that AzoPJ-PEPA NPs can be used as visible light-activated platform for controlled release of antimycotics and subsequent destruction of the cell structure and ensuing death of Rhizoctonia solani.
Antifungal activity
More detailed studies were carried out utilizing the AGAR dilution method to evaluate use of the light activated AzoPJ-PEPA NPs as an antibiotic delivery agent in Rhizoctonia solani cells87,88. We first determined the effect of different concentrations of AzoPJ-PEPA NPs on the inhibition of Rhizoctonia solani. Inspection of agar plates showed that irradiation of mixtures of AzoPJ-PEPA NPs and this fungus with either 440 nm light or natural light leads to a decrease in the growth diameter of Rhizoctonia solani in a manner that depends on the concentration of the nano-antimycotic agent (Supplementary Figs. 36 and 37 and Supplementary Tables 2 and 3). When PEPA concentration of AzoPJ-PEPA NPs in the medium is 10 mg L−1, growth of Rhizoctonia solani is 86.3% inhibited under 440 nm light irradiation condition. In addition, the antifungal effect promoted by the azo switch is also irradiation time dependent, with the average extent of inhibition reaching 80.4% after irradiation with same light for 24 h (Supplementary Fig. 38 and Supplementary Table 4). Compared to the single 440 nm light source, the natural light source is obtained by an artificial climate chamber. Due to its more stable and uniform light, the growth inhibition rate of Rhizoctonia solani can reach 89.2% upon irradiation with natural light when PEPA concentration of AzoPJ-PEPA NPs in the medium is 10 mg L−1. Furthermore, irradiation time-dependent inhibition of Rhizoctonia solani is able to reach 83.2% when natural light is used (Supplementary Fig. 39 and Supplementary Table 5).
In comparison to traditional small molecule drug treatment, protocols using nanosized drug delivery systems typically have enhanced efficacies89,90,91,92,93,94. To evaluate if this effect is also seen with light-activated AzoPJ-PEPA NPs delivery system, the mycelial growth rate method was employed by comparing times required to reach growth limits by Rhizoctonia solani treated with seven different conditions including (I) Blank, (II) Natural light irradiation, (III) AzoPJ-PEPA NPs (the dose of active ingredient PEPA: 5 mg L−1), (IV) PEPA (5 mg L−1) with natural light irradiation; (V) PEPA (7.5 mg L−1) with natural light irradiation; (VI) PEPA (10 mg L−1) with natural light irradiation; (VII) AzoPJ-PEPA NPs (the dose of active ingredient PEPA: 5 mg L−1) with natural light irradiation. To produce these sample, Rhizoctonia solani was inoculated into a blank medium containing deionized water, a medium containing the antimycotics PEPA, and a medium containing nano antimycotics AzoPJ-PEPA NPs. The light-treated groups were exposed to natural light while the light-free samples were wrapped in foil and kept in the same environment until Rhizoctonia solani reached full growth. As the images in Fig. 8a demonstrate, the growth of Rhizoctonia solani in the blank control group I reached a limit in only 3 d, and that in groups treated with natural light alone (group II) and with medium containing AzoPJ-PEPA NPs without irradiation (group III) reached full growth in 4 d. Determinations of the antifungal effects of PEPA with different concentrations in groups IV–VI showed that when the concentrations of PEPA is 5, 7.5 and 10 mg L−1, and natural light irradiation is performed, fungus growth reaches a limit within 1 to 2 weeks. For the resulting nano-system in group VII, Rhizoctonia solani in a medium containing AzoPJ-PEPA NPs (group VII) also take 2 weeks to reach the growth limit under natural light irradiation. Although AzoPJ-PEPA NPs containing 5 mg L−1 of (group VII) exhibit antifungal effects that are nearly identical to those of PEPA (10 mg L−1) alone (Fig. 8b), the dose of the active ingredient in the light treated AzoPJ-PEPA NPs is half of that used when only the active ingredient is administered. Thus, with the same efficacy, it can be seen that this light-controlled releasable nano-antimycotics was able to significantly reduce the dose of small molecule antimycotics. In addition, at the same dose level of PEPA (5 mg L−1), the group IV containing the small molecule antimycotic lasted only 7 d. The observation chronicled above clearly suggest that the light-controlled nano-system for delivery effectively increases the bioavailability and period of activity of the active antimycotic agents.

a Photographs of Rhizoctonia solani on the agar plates treated using different conditions including (I) Blank, (II) Natural light irradiation, (III) AzoPJ-PEPA NPs (the dose of active ingredient PEPA: 5 mg L−1), (IV) PEPA (5 mg L−1) with natural light irradiation; (V) PEPA (7.5 mg L−1) with natural light irradiation; (VI) PEPA (10 mg L−1) with natural light irradiation; (VII) AzoPJ-PEPA NPs (the dose of active ingredient PEPA: 5 mg L−1) with natural light irradiation. b Corresponding colony-forming unit (CFU) diagram for Rhizoctonia solani. Natural light provided using an artificial climate box (MGC-450HP-2L), the data on the bar chart indicate mean. n = 3 independent fungal biological sample in each experiment. Each graph is presented as the mean ± SD. Source data are provided as a Source Data file.
As mentioned earlier in the text, Rhizoctonia solani is a pathogen that causes a devastating disease for rice and wheat that is called sheath blight. Consequently, we explored the use of AzoPJ-PEPA NPs as a modality for treating this disease. At first, the interface affinity of AzoPJ-PEPA NPs was investigated by measuring contact angle. AzoPJ-PEPA NPs have a smaller contact angle compared to PEPA. As a result, this nano-antimycotics improve the interface affinity of PEPA on rice leaves and potentially promote drug bioavailability (Supplementary Fig. 40). Then, to explore light-controlled anti-rice blight of this agent, leaves of rice in the late tillering stage were inoculated with Rhizoctonia solani and then sprayed with different solution. Then the changes of rice leaves were recorded. The same sample protocol used in vivo agar plate assay was employed here e.g., (I) Blank, (II) Natural light irradiation, (III) AzoPJ-PEPA NPs (the dose of active ingredient PEPA: 5 mg L−1), (IV) PEPA (5 mg L−1) with natural light irradiation; (V) PEPA (7.5 mg L−1) with natural light irradiation; (VI) PEPA (10 mg L−1) with natural light irradiation; (VII) AzoPJ-PEPA NPs (the dose of active ingredient PEPA: 5 mg L−1) with natural light irradiation. Following the third day of the 7-d observation period, visible lesions appeared on the rice leaves in the blank group (Fig. 9). Leaves treated with natural light alone (group II) and those inoculated with AzoPJ-PEPA NPs and not natural light treated (III) developed lesion spots around the fourth day. Rice leaves in the groups IV and V developed lesion spots around the fourth day of culture when the concentrations of PEPA are 5 and 7.5 mg L−1. Owing to the antifungal effects of released PEPA, disease induced lesions are lower in groups IV and V than they are in groups I-III. In group VI containing leaves treated with 10 mg L−1 of PEPA, fewer visible lesion spots arise but, blight phenotype caused by Rhizoctonia solani becomes apparent on the fifth day and the near entirety of the leaf becomes blighted by day 795,96. Importantly, rice leaves sprayed with AzoPJ-PEPA NPs and then irradiated using natural light do not develop lesion spots within 7 d and only a little yellowing occurs. In addition, the magnified lesion spots photographs, additional control experiments and quantified lesion length of rice leaves (Supplementary Figs. 41 and 42 and Supplementary Table 6) further indicate that the treatment in group VII can inhibit the growth of Rhizoctonia solani. The dose of PEPA in group VII is 5 mg L−1, which is half of the active ingredient employed in Group VI. Thus, delivery of PEPA using AzoPJ-PEPA NPs is more effective against Rhizoctonia solani. Moreover, the effects of different exposure concentrations of AzoPJ-PEPA NPs (0–1 mg mL−1) on the germination rate, shoot length and root length of rice seedlings were assessed. The results show that the effects on rice seedlings are almost negligible, indicating that AzoPJ-PEPA NPs have good biocompatibility with plants (Supplementary Fig. 43). These findings show conclusively that the AzoPJ-PEPA NPs light-driven delivery system has the potential to improve the effectiveness of sheath blight disease of rice and wheat control.

Photographs of Rhizoctonia solani on rice plant leaves under different conditions: (I) Blank, (II) Natural light irradiation, (III) AzoPJ-PEPA NPs (the dose of active ingredient PEPA: 5 mg L−1), (IV) PEPA (5 mg L−1) with natural light irradiation; (V) PEPA (7.5 mg L−1) with natural light irradiation; (VI) PEPA (10 mg L−1) with natural light irradiation; (VII) AzoPJ-PEPA NPs (the dose of active ingredient PEPA: 5 mg L−1) with natural light irradiation. All samples were incubated at 28 °C. Natural light generated using an artificial climate box (MGC-450HP-2L). n = 3 independent sample in each experiment.
In the above effort, we developed a strategy to design an azo-heteroarene fluorescent molecular switch. The molecular component of the switch contains a typical D-A electronic features, a N = N double bond as a conjugated bridging chain and light activated switch center, and pyrazole and a julolidine moieties at the terminal positions. In this system, pyrazole serves as the electron acceptor and the cyclic amino group in julolidine functions as the electron donor. Moreover, the rigidity of the julolidine group leads to effective blockage of the TICT excited state quenching process and an enhancement of the fluorescence property of the azo switch. Visible light irradiation of the switch promotes E,Z-photoisomerization accompanied by a clearly visible changes in green fluorescence. It was demonstrated that azo fluorescent switch can be employed to perform switchable fluorescence imaging in living fungi and bacteria. In addition, as a result of the larger size of julolidine moiety, nanoparticles formed by liposome-encapsulation of the azo switches undergo an increase in nanocavity size upon visible light induced E,Z-isomerization. Owing to this unique property, NPs of the azo-fluorescent switch serve as the core component of a drug delivery system that undergoes light-controlled release of an antifungal agent against Rhizoctonia solani, and one that provides a fluorescence response to the release process. In terms of antifungal activity, NPs of the azo-fluorescent switch loaded with antifungal agent still exhibits higher antifungal activity in vitro, even when the active ingredient content is halved compared to the small molecule agent. And when the dose of the active ingredient was the same, the nano-antimycotics is able to double the holding period. This study has shown that the loaded NPs had a light induced control effect against rice sheath blight disease caused by Rhizoctonia solani infestation that cannot be matched by the same small molecule antimycotics. Overall, we devised a unique strategy for designing biologically functioning azo-fluorescent switches, which we believe will be applicable to construction of imaging-guided light-controlled releasable nanomedicines. More, we have reason to believe that more and more molecular imaging technologies will be applied to the field of agriculture in the future, forming a unique Agricultural Molecular Imaging (AMI) to serve precision agriculture and sustainable development.
Methods
Materials
All initial chemicals were obtained from commercial suppliers, and these materials were used without additional purification unless specified otherwise. All reactions were conducted under a nitrogen atmosphere employing standard Schlenk techniques. The progress of reactions was monitored by thin layer chromatography (TLC), and all solvents used for dehydration were deoxygenated before use.
Characterization
1H and 13C NMR were recorded on an American Varian Mercury Plus 400/600 spectrometer (400/600 MHz) with tetramethylsilane as the internal reference. Mass spectral data was collected on Agilent 6224 Accurate-Mass TOF LC/MS. UV-vis spectra were recorded via a UV-3600 visible spectrophotometer (Shimadzu), and fluorescence spectra were obtained using a Cary Eclipse fluorescent spectrophotometer and FLS1000 (Edinburgh Instruments). Dynamic light scattering (DLS) measurements were performed on the Zetasizer instrument ZEN3600 (Malvern, UK) with a 173 back-scattering angle and He-Ne laser (633 nm). Transmission electron microscopy (TEM) results were collected by a Tecnai G2 F20 TEM system (FEI) at 200 kV. TEM samples were prepared by dropping nanoparticles onto copper grids and drying them overnight. Confocal imaging was done with Leica TCS SP8 X White Light Laser Confocal Microscope. Irradiation at 415 nm, 440 nm or 535 nm was performed using light emitting diode (LED) lamp. Natural light condition was simulated via artificial climate box (MGC-450HP-2L). Contact angle values were record via Optical Contact Angle Meter (OCA20, Dataphysics).
Synthesis of AzoPJ
AzoPJ was prepared using standard diazo couplings97. In a typical procedure, 4-amino-1,3,5-trimethyl-1H-pyrazole (1.0 mmol, 1.0 equiv) was dispersed in 2 mL ice water (0–5 °C), and HCl (12 M, 0.5 mL) was added to the solution with stirring. Then, a cold aqueous solution of NaNO2 (3 mL, 1.2 mmol, 1.2 equiv) was added slowly to the above settled solution. After stirring at 0–5 °C for 45 min, the resulting diazonium salt was transferred to a solution of NaOAc (3 eq) in H2O and Julolidine (1.3 mmol, 1.3 eq) in EtOH. The mixture was allowed to stir at room temperature for 3 h and the resulting orange yellow precipitate was collected through vacuum filtration. After washing with H2O, EtOH, PE, the crude product was purified by column chromatography (eluent: ethyl acetate and PE) to afford the orange solid. (200 mg, 65% yield), the structure was confirmed by NMR and MS spectrum (Supplementary Figs. 44–46); 1H NMR (600 MHz, CDCl3): δ = 7.31 (s, 2H), 3.75 (s, 3H), 3.23 (s, 4H), 2.81 (s, 4H), 2.53 (s, 3H), 2.47 (s, 3H), 1.99 (s, 4H). 13C NMR (600 MHz, CDCl3): δ = 144.23, 143.55, 141.74, 136.35, 134.95, 121.05, 120.91, 50.00, 35.87, 27.83, 21.84, 13.71, 9.98.
Synthesis of AzoPJ NPs
AzoPJ NPs were prepared using a matrix-encapsulation method43. Briefly, DSPE-mPEG5000 (9 mg) was dissolved in deionized water (9 mL). To this solution, AzoPJ (1 mg) in THF was added resulting in mixed solution. After 30 min of ultrasonic, THF in the mixture was removed via nitrogen flow. Then, the mixture was concentrated on an Amicon Ultra centrifugal filter with a molecular-weight cut-off of 50 kDa at 1500 × g. And the nanoparticles were stored at 4 °C.
Synthesis of AzoPJ-PEPA NPs
The preparation of AzoPJ-PEPA NPs follows the same method used for AzoPJ NPs. Initially, AzoPJ (1 mg) and antibacterial agents (0.2 mg) was dissolved in THF (1 mL and 0.5 mL) respectively. The above solution was sonicated for 5 min, respectively. Then, AzoPJ solution was added to antibacterial agent solution and sonicated for 30 min to form a homogenous orange solution. Next, the mixture was quickly added to 9 mL of DSPE-mPEG5000 aqueous solution and nitrogen flow was used to remove THF. Finally, AzoPJ-PEPA NPs were obtained after centrifuging at 1500 × g with 50 kDa microsep. And the nanoparticles were stored at 4 °C.
Preparation of AzoPJ single crystals
Single crystal of AzoPJ suitable for crystallographic analysis was prepared by slowly diffusing hexane into a dichloromethane solution of AzoPJ at room temperature. The crystal was grown on a glass fiber for diffraction experiments. Intensity data were collected using a Nonius Kappa CCD diffractometer with Mo Kα radiation (0.71073 Å) at 195 K. The structures were analyzed by a combination of direct methods (SHELXS-97)98 and Fourier difference techniques and refined by full-matrix least-squares (SHELXL-97)99.
Photoisomerization of AzoPJ
UV-Vis studies on cis-trans isomerization were conducted using a UV-3600 visible spectrophotometer (Shimadzu). A stock solution of AzoPJ (1 mM) was prepared in DMSO, and test solutions were also prepared in DMSO. UV-vis spectra were recorded under ambient light conditions. These samples were exposed to a 440 nm LED lamp (50 mW cm−2 or 20 W cm−2), and UV-vis spectra were obtained to monitor the generation of the cis isomer. Subsequently, the samples were irradiated by a 535 nm LED (50 mW cm−2 or 15 W cm−2), and UV-vis spectra were recorded again. The same solutions were used to obtain fluorescence spectra of AzoPJ under identical illumination conditions.
Time dependent spectrum experiment
For the time-dependent experiment of trans-cis isomerization, AzoPJ or nanoparticles were exposed to a 440 nm LED (50 mW cm−2 or 20 W cm−2), and spectra were recorded at various time intervals. In the time-dependent experiment of cis-trans isomerization, AzoPJ was irradiated with a 440 nm LED (50 mW cm−2 or 15 W cm−2) until the trans-cis isomerization was complete. Subsequently, these samples were irradiated with a 535 nm LED (50 mW cm−2 or 15 W cm−2), and spectra were recorded at different time points. The same solutions were used to obtain time-dependent fluorescence spectra of AzoPJ under identical illumination conditions.
Spectroscopic procedures of AzoPJ under natural light
UV-Vis studies on cis-trans isomerization were conducted using a UV-3600 visible spectrophotometer (Shimadzu). UV-vis spectra were recorded under ambient light conditions. A stock solution of AzoPJ (1 mM) was prepared in DMSO, and test solutions were also prepared in DMSO. UV-vis spectra were recorded under ambient light conditions. These samples were exposed to natural light for 5 min, and UV-vis spectra were recorded to monitor the generation of the cis isomer.
Fatigue resistance
To assess fatigue resistance, AzoPJ solutions in DMSO underwent alternating exposure to 440 nm and 535 nm light (50 mW cm−2), and UV-vis spectra were recorded after each cycle, for a total of 10 cycles. A stock solution of AzoPJ (1 mM) was prepared in DMSO, and test solutions were also prepared in DMSO.
Fluorescence spectroscopic procedures of AzoPJ single crystals
Fluorescence spectra of single crystals were acquired using FLS1000 (Edinburgh Instruments) under ambient light conditions. The excitation for the fluorescence spectra measurement was conducted at 430 nm.
Fluorescence spectroscopic procedures of AzoPNMe2
Fluorescence spectra of AzoPNMe2 were measured using a Cary Eclipse fluorescent spectrophotometer. A stock solution of AzoPNMe2 (1 mM) was prepared in DMSO, and test solutions were also prepared in DMSO. Fluorescence spectra were recorded under ambient light conditions. Excitation for the fluorescence spectra measurement was conducted at 328 nm.
Spectroscopic procedures of nanoparticles
The UV-vis spectra of nanoparticles were acquired with a UV-3600 visible spectrophotometer (Shimadzu). A stock solution of nanoparticles (1 mg mL−1) was prepared in deionized water or PBS, and test solutions were also prepared in deionized water or PBS. The initial UV-vis spectra were recorded under ambient light conditions. Subsequently, the samples were exposed to a 440 nm LED (20 W cm−2), natural light and dark conditions, and the spectral changes were captured using a spectrometer. Natural light provided using an artificial climate box (MGC-450HP-2L).
A stock solution of nanoparticles (1 mg mL−1) was prepared in deionized water, and test solutions were also prepared in deionized water. UV-vis spectra were recorded under ambient light conditions. These samples were exposed to a 440 nm LED lamp (20 W cm−2), and UV-vis spectra were obtained to monitor the generation of the cis isomer. Subsequently, the samples were irradiated by a 535 nm LED (15 W cm−2), and UV-vis spectra were recorded again.
DLS characterization and zeta potential of nanoparticles
The Zetasizer instrument ZEN3600 (Malvern, UK) was utilized for DLS characterization and zeta potential measurements, employing a 173 back-scattering angle and a He-Ne laser (633 nm). A solution containing nanoparticles (0.1 mg mL−1) was prepared in deionized water and stored in darkness. Initial DLS measurements and zeta potential of the nanoparticles were obtained before exposure to 440 nm light. Subsequently, these samples underwent irradiation with a 440 nm LED lamp (20 W cm−2), and post-irradiation DLS measurements and zeta potential assessments were conducted to monitor changes in size distribution and potential.
TEM of nanoparticles
The TEM results were obtained using a Tecnai G2 F20 TEM system (FEI) operating at 200 kV. A solution containing nanoparticles (0.1 mg mL−1) was prepared in deionized water and stored in darkness. Initially, TEM images of the nanoparticles were captured before exposure to 440 nm light. Subsequently, these samples were irradiated with a 440 nm LED lamp (20 W cm−2), and TEM imaging was performed to monitor changes in morphology.
Cultures for cells
B16 cells obtained from the China Infrastructure of Cell Line were cultured in Roswell Park Memorial Institute 1640 Medium supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco). The cells were maintained in a controlled environment with 5% CO2 and 95% air at 37 °C.
Cell viability
B16 cells were plated into 96-well plates and incubated in Roswell Park Memorial Institute 1640 Medium supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco) in a 5% CO2 and 95% air environment at 37 °C. The following day, nanoparticles were introduced into the culture. After 24 h, the cell culture medium was removed, and fresh 1640 Medium without FBS was added. Live and dead cells were quantified for each condition using a microplate reader following MTT staining.
Cultivation of Rhizoctonia solani
Rhizoctonia solani sclerotia, the causative agent of Fusarium stem rot (FSR), were isolated for cultivation. This involved inoculating the pathogen onto Potato Dextrose Agar (PDA) medium. The cultures of Rhizoctonia solani were maintained at a temperature of 28 °C throughout the experiments.
Cultivation of Staphylococcus aureus
Glycerol-preserved Staphylococcus aureus were inoculated into soybean-casein digest medium and incubated overnight in incubator shakers set at 25 °C and 80 × g.
Imaging of Rhizoctonia solani
For the concentration-dependent experiment, activated Rhizoctonia solani hyphae were exposed to AzoPJ NPs (0 mg mL−1, 0.25 mg mL−1, 0.5 mg mL−1, 1 mg mL−1) for 24 h at 28 °C. After washing three times with PBS, the hyphae were fixed using the tabletting method and then imaged using a confocal laser scanning microscope. The fluorescence signal within the range of 460–580 nm was captured upon excitation at 405 nm. Each group within the study consisted of n = 3 samples.
For the time-dependent experiment, activated Rhizoctonia solani hyphae were treated with AzoPJ NPs (1 mg mL−1) for various incubation periods (0 h, 12 h, 24 h, 48 h). After washing three times with PBS, the hyphae were fixed and imaged under the same conditions as mentioned earlier. Each group within the study consisted of n = 3 samples.
Regarding fluorescence photoswitch imaging, Rhizoctonia solani hyphae were exposed to AzoPJ NPs (1 mg mL−1) for 48 h. After washing and fixing, the hyphae were irradiated with 440 nm light (20 W cm−2) for 1 h. Subsequently, fluorescence images were captured following the same procedures as described above. Each group within the study consisted of n = 3 samples.
Imaging of Staphylococcus aureus
The Staphylococcus aureus solution underwent centrifugation, and the resulting supernatant was discarded. For the concentration-dependent experiment, AzoPJ NPs (0 mg mL−1, 0.25 mg mL−1, 0.5 mg mL−1, 1 mg mL−1) were incubated with Staphylococcus aureus for 24 h. After washing three times with PBS, fluorescence signals ranging from 460–580 nm were collected upon excitation at 405 nm following the pressure slicing method. Each group within the study consisted of n = 3 samples.
In the time-dependent experiment, Staphylococcus aureus was co-incubated with AzoPJ NPs (1 mg mL−1) for durations of 0 h, 12 h, 24 h, and 48 h, respectively. Subsequently, fluorescence images were obtained using the same procedures. Each group within the study consisted of n = 3 samples.
For fluorescence photoswitch imaging, Staphylococcus aureus was introduced to a solution containing AzoPJ NPs (1 mg mL−1). Following 24 h of co-incubation, fluorescence within the 460–580 nm range was collected under excitation at 405 nm. The Staphylococcus aureus samples were then subjected to irradiation with 440 nm light (20 W cm−2) for 1 h. Subsequent images were captured following the same steps. Each group within the study consisted of n = 3 samples.
Data analysis
TD-DFT calculations at the B3LYP/6-31 G* level with the Gaussian 09 program. Calculations regarding molecular volume are at the RHF/6-31 G** level with the Gaussian 09 program. Image analysis was employed by ImageJ software. For the confocal microscope imaging, ImageJ software was employed for image analysis. The fluorescence intensity within identically sized areas was measured, and normalization of intensity was conducted for each experiment.
Concentration-dependent anti-fungal experiment
The concentration-dependent inhibition experiment was conducted using the AGAR dilution method. Initially, PDA medium was prepared, and nanoparticles were subsequently added to the PDA medium under aseptic conditions to create drug-containing plates with varying concentrations. After inoculating the fungus (6 mm in diameter), all samples were exposed to either 440 nm light (20 W cm−2) or natural light for 4 h and then placed in an incubator at 28 °C. The diameters of hyphal growth were measured daily using a vernier caliper until the fungus in the non-light group reached its growth limit. The degree of growth inhibition was calculated based on these measurements. Each group within the study consisted of n = 3 samples.
Time-dependent anti-fungal experiment
The time-dependent inhibition experiment was conducted using the AGAR dilution method. Initially, PDA medium was prepared, and nanoparticles were subsequently added to the PDA medium under aseptic conditions to create drug-containing plates with the same concentration. After inoculating the fungus, all samples were exposed to either 440 nm light (20 W cm−2) or natural light for different durations and then placed in an incubator at 28 °C. The diameters of hyphal growth were measured daily using a vernier caliper until the fungus in the control group reached its growth limit. The degree of growth inhibition was calculated based on these measurements. Each group within the study consisted of n = 3 samples.
Antimicrobial persistent effect experiment
In an antifungal assay, Rhizoctonia solani were divided into seven treatment groups: (I) Blank, (II) Natural light irradiation, (III) AzoPJ-PEPA NPs (the dose of active ingredient PEPA: 5 mg L−1), (IV) PEPA (5 mg L−1) with natural light irradiation; (V) PEPA (7.5 mg L−1) with natural light irradiation; (VI) PEPA (10 mg L−1) with natural light irradiation; (VII) AzoPJ-PEPA NPs (the dose of active ingredient PEPA: 5 mg L−1) with natural light irradiation. After the above solutions were added to PDA medium, samples requiring light were irradiated with natural light for 4 h per day, while the samples without light irradiation were placed in the same location and wrapped in tin foil. All samples were then incubated in an incubator at 28 °C for the remaining 5/6 of the time. The diameters of hyphal growth were measured daily using a vernier caliper until the fungus in the fourth group reached its growth limit. Each group within the study contained n = 3.
Anti-fungal bioassay of rice
Rice plants at the tillering stage were chosen, with their tip and base tissues removed from the same leaf position, while the middle part of the leaf was retained. The leaf tissues were then divided into seven treatment groups as follows: (I) Blank, (II) Natural light irradiation, (III) AzoPJ-PEPA NPs (the dose of active ingredient PEPA: 5 mg L−1), (IV) PEPA (5 mg L−1) with natural light irradiation; (V) PEPA (7.5 mg L−1) with natural light irradiation; (VI) PEPA (10 mg L−1) with natural light irradiation; (VII) AzoPJ-PEPA NPs (the dose of active ingredient PEPA: 5 mg L−1) with natural light irradiation. The cuticles on the center of rice leaves were punctured with a sterilized needle, and approximately 6 mm diameter mycelial cake was inoculated. These leaves were then sprayed with the corresponding preparations. Subsequently, all the inoculated rice leaves were placed in an illumination incubator set at 28 °C and 80% relative humidity, within a local environment with a photoperiod of 12 h light-dark cycle. Non-light groups were wrapped in tin foil. The changes in the leaves were recorded daily. Each group within the study contained n = 3.
Control experiments of anti-fungal bioassay of rice
Rice plants at the tillering stage were chosen, with their tip and base tissues removed from the same leaf position, while the middle part of the leaf was retained. The leaf tissues were then divided into two treatment groups as follows: (I) No treatment; (II) Blank potato dextrose agar (PDA) medium. The cuticles on the center of rice leaves were punctured with a sterilized needle, and approximately 6 mm diameter PDA was inoculated or not. Subsequently, all the inoculated rice leaves were placed in an illumination incubator set at 28 °C and 80% relative humidity, within a local environment with a photoperiod of 12 h light-dark cycle. Each group within the study contained n = 3.
Leaf wettability
The rice leaf was fixed on glass slides, and a syringe was used to deposit 5 μL of AzoPJ-PEPA NPs solution onto the rice leaf. Optical Contact Angle Meter (OCA20, Dataphysics) was used to record the droplet deposition images and contact angle values. Deionized water and PEPA solution were used as controls.
Biosafety of AzoPJ-PEPA NPs
Rice seeds were sterilized by soaking in 75% ethanol and then washed with deionized water. After soaking in ultrapure water for 48 h. 20 rice seeds were exposed to AzoPJ-PEPA NPs with different concentrations (0, 0.2, 0.4, 0.6, 0.8, 1 mg mL−1). After 48 h of seed germination, we evaluated the rice seedling growth performance. Each group within the study contained n = 3.
Encapsulation efficiency
The nanoparticles were separated from the dispersion through centrifugation at 1500 × g for 20 min. Following centrifugation, the unencapsulated drug was left in the supernatant. The supernatant was then appropriately diluted and analyzed using a UV-3600 visible spectrophotometer (Shimadzu). The entrapment efficiency was calculated as follows:
Drug release efficiency
To determine the in vitro drug release under light irradiation, the AzoPJ-PEPA nanoparticles (1 mL) were exposed to 440 nm light (20 W cm−2) or the natural light based on an artificial climate chamber containing 20 lamps (18 watts each). Following light exposure, these samples were introduced into a dialysis bag (MWCO: 14 KDa), which was then submerged into the release medium and stirred at 37 °C. Subsequently, 1 mL of the release medium was collected and analyzed using a UV-3600 visible spectrophotometer (Shimadzu). The drug release was determined based on the absorbance of PEPA and the standard absorption curves of PEPA.
Growth inhibition ratio
The growth inhibition ratio (%) was calculated as the following equation:
where \({D}_{0}\) and \({D}_{t}\) is diameters of negative control mycelial diameter and treatment mycelial diameter, respectively.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The crystal structures of AzoPJ are available free of charge from the Cambridge Crystallographic Data Center under reference number CCDC-2337626 [https://www.ccdc.cam.ac.uk/structures/Search?Ccdcid=2337626&DatabaseToSearch=Published]. Source data are provided with this paper.
References
Crespi, S., Simeth, N. A. & König, B. Heteroaryl azo dyes as molecular photoswitches. Nat. Rev. Chem. 3, 133–146 (2019).
Lancia, F., Ryabchun, A. & Katsonis, N. Life-like motion driven by artificial molecular machines. Nat. Rev. Chem. 3, 536–551 (2019).
Ahn, D., Stevens, L. M., Zhou, K. & Page, Z. A. Rapid high-resolution visible light 3D printing. ACS Cent. Sci. 6, 1555–1563 (2020).
Wang, H. et al. Visible-light-driven halogen bond donor based molecular switches: from reversible unwinding to handedness inversion in self-organized soft helical superstructures. Angew. Chem. Int. Ed. 59, 2684–2687 (2020).
Duan, H.-Y., Han, S.-T., Zhan, T.-G., Liu, L.-J. & Zhang, K.-D. Visible-light-switchable tellurium-based chalcogen bonding: photocontrolled anion binding and anion abstraction catalysis. Angew. Chem. Int. Ed. 62, e202212707 (2023).
Dang, T. et al. Thiazolylazopyrazoles as nonsymmetric bis-heteroaryl azo switches: High-yield visible-light photoisomerization and increased Z-isomer stability by o-carbonylation. Angew. Chem. Int. Ed. 62, e202301992 (2023).
Shen, X. et al. Dibenzo[b,f][1,4,5]chalcogenadiazepine photoswitches: Conversion of excitation energy into ring strain. Angew. Chem. Int. Ed. 61, e202209441 (2022).
Kerckhoffs, A. & Langton, M. J. Reversible photo-control over transmembrane anion transport using visible-light responsive supramolecular carriers. Chem. Sci. 11, 6325–6331 (2020).
Qiu, Q. et al. Visible light activated energy storage in solid-state Azo-BF2 switches. Chem. Sci. 14, 11359–11364 (2023).
Qian, H., Wang, Y.-Y., Guo, D.-S. & Aprahamian, I. Controlling the isomerization rate of an azo-BF2 switch using aggregation. J. Am. Chem. Soc. 139, 1037–1040 (2017).
Kurihara, M., Hirooka, A., Kume, S., Sugimoto, M. & Nishihara, H. Redox-conjugated reversible isomerization of ferrocenylazobenzene with a single green light. J. Am. Chem. Soc. 124, 8800–8801 (2002).
Lin, R. et al. Phenylazothiazoles as visible-light photoswitches. J. Am. Chem. Soc. 145, 9072–9080 (2023).
Yang, Y., Hughes, R. P. & Aprahamian, I. Visible light switching of a BF2‑coordinated azo compound. J. Am. Chem. Soc. 134, 15221–15224 (2012).
Siewertsen, R. et al. Highly efficient reversible Z-E photoisomerization of a bridged azobenzene with visible light through resolved S1(nπ*) absorption bands. J. Am. Chem. Soc. 131, 15594–15595 (2009).
Beharry, A. A., Sadovski, O. & Woolley, G. A. Azobenzene photoswitching without ultraviolet light. J. Am. Chem. Soc. 133, 19684–19687 (2011).
Bléger, D., Schwarz, J., Brouwer, A. M. & Hecht, S. o‑Fluoroazobenzenes as readily synthesized photoswitches offering nearly quantitative two-way isomerization with visible light. J. Am. Chem. Soc. 134, 20597–20600 (2012).
Gemen, J. et al. Disequilibrating azobenzenes by visible-light sensitization under confinement. Science 381, 1357–1363 (2023).
Meteling, H. J. et al. Versatile surface patterning with low molecular weight photoswitches. Small 18, 2203245 (2022).
Yang, Y., Hughes, R. P. & Aprahamian, I. Near-infrared light activated azo-BF2 switches. J. Am. Chem. Soc. 136, 13190–13193 (2014).
Dong, M. et al. Near-infrared photoswitching of azobenzenes under physiological conditions. J. Am. Chem. Soc. 139, 13483–13486 (2017).
Lentes, P. et al. Nitrogen bridged diazocines: photochromes switching within the near-infrared region with high quantum yields in organic solvents and in water. J. Am. Chem. Soc. 141, 13592–13600 (2019).
Samanta, S., Babalhavaeji, A., Dong, M. & Woolley, G. A. Photoswitching of ortho-substituted azonium ions by red light in whole blood. Angew. Chem. Int. Ed. 52, 14127–14130 (2013).
Halabi, E. A. & Weissleder, R. Light-deactivated fluorescent probes (FLASH-off) for multiplexed imaging. J. Am. Chem. Soc. 145, 8455–8463 (2023).
Samanta, S. et al. Photoswitching azo compounds in vivo with red light. J. Am. Chem. Soc. 135, 9777–9784 (2013).
Pfaff, P., Samarasinghe, K. T. G., Crews, C. M. & Carreira, E. M. Reversible spatiotemporal control of induced protein degradation by bistable photoPROTACs. ACS Cent. Sci. 5, 1682–1690 (2019).
Samanta, S., Qin, C., Lough, A. J. & Woolley, G. A. Bidirectional photocontrol of peptide conformation with a bridged azobenzene derivative. Angew. Chem. Int. Ed. 51, 6452–6455 (2012).
Trads, J. B. et al. Sign inversion in photopharmacology: Incorporation of cyclic azobenzenes in photoswitchable potassium channel blockers and openers. Angew. Chem. Int. Ed. 58, 15421–15428 (2019).
Steinmüller, S. A. M. et al. Visible-light photoswitchable benzimidazole azo-arenesas β-arrestin2-biased selective cannabinoid 2 receptor agonists. Angew. Chem. Int. Ed. 62, e202306176 (2023).
Aggarwal, K. et al. Visible light mediated bidirectional control over carbonic anhydrase activity in cells and in vivo using azobenzenesulfonamides. J. Am. Chem. Soc. 142, 14522–14531 (2020).
Ewert, J. et al. Photoswitchable diazocine-based estrogen receptor agonists: Stabilization of the active form inside the receptor. J. Am. Chem. Soc. 144, 15059–15071 (2022).
Kienzler, M. A. et al. A red-shifted, fast-relaxing azobenzene photoswitch for visible light control of an ionotropic glutamate receptor. J. Am. Chem. Soc. 135, 17683–17686 (2013).
Wegener, M., Hansen, M. J., Driessen, A. J. M., Szymanski, W. & Feringa, B. L. Photocontrol of antibacterial activity: Shifting from UV to red light activation. J. Am. Chem. Soc. 139, 17979–17986 (2017).
Josa-Culleré, L. & Llebaria, A. Visible-light-controlled histone deacetylase inhibitors for targeted cancer therapy. J. Med. Chem. 66, 1909–1927 (2023).
Gao, M., Kwaria, D., Norikane, Y. & Yue, Y. Visible-light-switchable azobenzenes: Molecular design, supramolecular systems, and applications. Nat. Sci. 3, e220020 (2023).
Chen, H. et al. Visible and near-infrared light activated azo dyes. Chin. Chem. Lett. 32, 2359–2368 (2021).
Huang, Y. et al. Fluorescent probes for glucolipid metabolism of bacterial cell wall. Green Chem. Eng. 4, 417–426 (2023).
Li, Z. et al. Photoswitchable diarylethenes: from molecular structures to biological applications. Coord. Chem. Rev. 497, 215451 (2023).
Ma, X. et al. The aggregates of near-infrared cyanine dyes in phototherapy. ChemMedChem 18, e202300204 (2023).
Zeng, X., Chen, W., Liu, C., Yin, J. & Yang, G.-F. Fluorescence probes for reactive sulfur species in agricultural chemistry. J. Agric. Food Chem. 69, 13700–13712 (2021).
Huang, Y., Chen, W., Chung, J., Yin, J. & Yoon, J. Recent progress in fluorescent probes for bacteria. Chem. Soc. Rev. 50, 7725–7744 (2021).
Zeng, X. et al. A protocol for activated bioorthogonal fluorescence labeling and imaging of 4-hydroxyphenylpyruvate dioxygenase in plants. Angew. Chem. Int. Ed. 62, e202312618 (2023).
Ma, X. et al. J-aggregates formed by NaCl treatment of aza-coating heptamethine cyanines and their application to monitoring salt stress of plants and promoting photothermal therapy of tumors. Angew. Chem. Int. Ed. 62, e202216109 (2023).
Ma, X. et al. Rational design and application of an indolium-derived heptamethine cyanine with record-long second near-infrared emission. CCS Chem 4, 1961–1976 (2022).
Li, D. et al. Synergistic effects of multiple rotors and hydrogen-bond interactions lead to sensitive near-infrared viscosity probes for live-cell microscopy. Sci. China Chem. 66, 2329–2338 (2023).
Liao, Y. et al. Heptamethine cyanines in bioorthogonal chemistry. Chin. Chem. Lett. 35, 109092 (2024).
Shen, S. et al. Recent progress on fluorescent probes for viruses. Chin. Chem. Lett. 35, 108360 (2024).
Zeng, X. et al. Design of an HPPD fluorescent probe and visualization of plant responses to abiotic stress. Adv. Agrochem 1, 73–84 (2022).
Huang, Y., Ma, X., Li, J., Tan, C. & Yin, J. NIR-II cyanine nanoparticles for imaging-guided tumor targeting photothermal therapy with vitamin C enhanced efficacy. Adv. Therap. 6, 2300017 (2023).
Tian, J. et al. Activatable type I photosensitizer with quenched photosensitization pre and post photodynamic therapy. Angew. Chem. Int. Ed. 62, e202307288 (2023).
Hao, B. et al. Bridging D–A type photosensitizers with the azo group to boost intersystem crossing for efficient photodynamic therapy. Chem. Sci. 13, 4139–4149 (2022).
Ouyang, G., Bialasa, D. & Würthner, F. Reversible fluorescence modulation through the photoisomerization of an azobenzene-bridged perylene bisimide cyclophane. Org. Chem. Front. 8, 1424–1430 (2021).
Wakabayashi, J., Gon, M., Tanaka, K. & Chujo, Y. Near-infrared absorptive and emissive poly(p-phenylene vinylene) derivative containing azobenzene–boron complexes. Macromolecules 53, 4524–4532 (2020).
Boucard, J. et al. Hybrid azo-fluorophore organic nanoparticles as emissive turn-on probes for cellular endocytosis. ACS Appl. Mater. Interfaces 11, 32808–32814 (2019).
Zhu, Q., Wang, S. & Chen, P. Diazocine derivatives: a family of azobenzenes for photochromism with highly enhanced turn-on fluorescence. Org. Lett. 21, 4025–4029 (2019).
Gon, M., Tanaka, K. & Chujo, Y. A highly efficient near-infrared-emissive copolymer with a N=N double-bond π-conjugated system based on a fused azobenzene–boron complex. Angew. Chem. Int. Ed. 57, 6546–6551 (2018).
Shadmehr, M., Davis, G. J., Mehari, B. T., Jensen, S. M. & Jewett, J. C. Coumarin triazabutadienes for fluorescent labeling of proteins. ChemBioChem 19, 2550–2552 (2018).
Qian, H., Shao, B. & Aprahamian, I. Visible-light fluorescence photomodulation in azo-BF2 switches. Tetrahedron 73, 4901–4904 (2017).
Yokoi, H., Hiroto, S. & Shinokubo, H. Synthesis of diazo-bridged BODIPY dimer and tetramer by oxidative coupling of β-amino-substituted BODIPYs. Org. Lett. 16, 3004–3007 (2014).
Beharry, A. A., Wong, L., Tropepe, V. & Woolley, G. A. Fluorescence imaging of azobenzene photoswitching in vivo. Angew. Chem. Int. Ed. 50, 1325–1327 (2011).
Chevalier, A., Renard, P.-Y. & Romieu, A. Azo-based fluorogenic probes for biosensing and bioimaging: Recent advances and upcoming challenges. Chem. Asian J. 12, 2008–2028 (2017).
Allard, C. et al. Confinement of dyes inside boron nitride nanotubes: photostable and shifted fluorescence down to the near infrared. Adv. Mater. 32, 2001429 (2020).
Tsou, Y.-H. Dopant-free hydrogels with intrinsic photoluminescence and biodegradable properties. Adv. Funct. Mater. 28, 1802607 (2018).
Yoshino, J., Kanoa, N. & Kawashima, T. Synthesis of the most intensely fluorescent azobenzene by utilizing the B–N interaction. Chem. Commun. 559–561 https://doi.org/10.1039/b615966d (2007).
Edwards, K. E. K., Mermut, O., Pietro, W. J. & Barrett, C. J. Optical and computational study of the trans ↔ cis reversible isomerization of the commercial bis-azo dye Bismarck Brown Y. Phys. Chem. Chem. Phys. 25, 5673–5684 (2023).
Ghosh, J., Bhaumik, S. & Bhattacharya, A. Comparison of internal conversion dynamics of azo and azoxy energetic moieties through the (S1/S0)CI conical Intersection: an ab initio multiple spawning study. Chem. Phys. 513, 221–229 (2018).
Bull, J. N., Scholz, M. S., Carrascosa, E., da Silva, G. & Bieske, E. J. Double molecular photoswitch driven by light and collisions. Phys. Rev. Lett. 120, 223002 (2018).
Chen, X., Zhang, X., Xiao, X., Wang, Z. & Zhao, J. Recent developments on understanding charge transfer in molecular electron donor-acceptor systems. Angew. Chem. Int. Ed. 62, e202216010 (2023).
Bhunia, S., Dolai, A., Bera, S. & Samanta, S. Near-complete bidirectional photoisomerization of para-dialkylamino-substituted arylazopyrazoles under violet and green or red lights. J. Org. Chem. 87, 4449–4454 (2022).
Wang, C. et al. Quantitative design of bright fluorophores and AIEgens by the accurate prediction of twisted intramolecular charge transfer (TICT). Angew. Chem. Int. Ed. 59, 10160–10172 (2020).
Miao, R. et al. A general method to develop highly environmentally sensitive fluorescent probes and AIEgens. Adv. Sci. 9, 2104609 (2022).
Shin, J. et al. Harnessing intramolecular rotation to enhance two-photon imaging of Aβ plaques through minimizing background fluorescence. Angew. Chem. Int. Ed. 58, 5648–5652 (2019).
Wang, C. et al. Twisted intramolecular charge transfer (TICT) and twists beyond TICT: from mechanisms to rational designs of bright and sensitive fluorophores. Chem. Soc. Rev. 50, 12656–12678 (2021).
Bandara, H. M. D. & Burdette, S. C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 41, 1809–1825 (2012).
Fraccia, T. P. & Martin, N. Non-enzymatic oligonucleotide ligation in coacervate protocells sustains compartment-content coupling. Nat. Commun. 14, 2606 (2023).
Feng, N. et al. Nanoparticle assembly of a photo- and pH-responsive random azobenzene copolymer. J. Colloid. Interf. Sci. 421, 15–21 (2014).
Samanta, D. et al. Reversible photoswitching of encapsulated azobenzenes in water. Proc. Natl. Acad. Sci. USA 115, 9379–9384 (2018).
Harvey, L. et al. Photo-responsive supramolecular polymer bottle-brushes: The key role of the solvent on self-assembly and responsiveness. J. Colloid. Interf. Sci. 670, 409–416 (2024).
Nunes, S. S. et al. Influence of PEG coating on the biodistribution and tumor accumulation of pH-sensitive liposomes. Drug Deliv. Transl. Res. 9, 123–130 (2019).
Guimarães, D., Cavaco-Paulo, A. & Nogueira, E. Design of liposomes as drug delivery system for therapeutic applications. Inter. J. Pharm. 601, 120571 (2021).
Smith, M. C. et al. Zeta potential: a case study of cationic, anionic, and neutral liposomes. Anal. Bioanal. Chem. 409, 5779–5787 (2017).
Garbuzenko, O., Zalipsky, S., Qazen, M. & Barenholz, Y. Electrostatics of PEGylated micelles and liposomes containing charged and neutral lipopolymers. Langmuir 21, 2560–2568 (2005).
Lin, G. et al. Robust red organic nanoparticles for in vivo fluorescence imaging of cancer cell progression in xenografted zebrafish. Adv. Funct. Mater. 27, 1701418 (2017).
Angelini, G., Scotti, L., Aceto, A. & Gasbarri, C. Silver nanoparticles as interactive media for the azobenzenes isomerization in aqueous solution: from linear to stretched kinetics. J. Mol. Liq. 284, 592–598 (2019).
Guo, X., Shao, B., Zhou, S., Aprahamian, I. & Chen, Z. Visualizing intracellular particles and precise control of drug release using an emissive hydrazone photochrome. Chem. Sci. 11, 3016–3021 (2020).
Shao, B., Fu, H. & Aprahamian, I. A molecular anion pump. Science 385, 544–549 (2024).
Tatum, L. A., Su, X. & Aprahamian, I. Simple hydrazone building blocks for complicated functional materials. Acc. Chem. Res. 47, 2141–2149 (2014).
Carpinella, M. C., Giorda, L. M., Ferrayoli, C. G. & Palacios, S. M. Antifungal effects of different organic extracts from Melia azedarach L. on phytopathogenic fungi and their isolated active components. J. Agric. Food Chem. 51, 2506–2511 (2003).
Yu, X.-D. et al. Selective antibacterial activity of patchouli alcohol against Helicobacter pylori based on inhibition of urease. Phytother. Res. 29, 67–72 (2015).
Zhao, C. et al. Silica-based nanodelivery systems loaded with matrine for the brown planthopper green control and rice growth promotion. ACS Sustainable Chem. Eng. 11, 17299–17309 (2023).
Gui, F. et al. Biosynthesis of nanocrystalline silver chloride with high antibacterial activity using bacterial extracts. Adv. Agrochem 2, 88–96 (2023).
Liu, Z., Xu, W., Kovaleva, E. G., Cheng, J. & Li, H. Recent progress in encapsulation and controlled release of pesticides based on cyclodextrin derivative carriers. Adv. Agrochem 1, 89–99 (2022).
Yao, X. et al. Design, synthesis and biological evaluation of thiazole and imidazo[1,2-a] pyridine derivatives containing a hydrazone substructure as potential agrochemicals. Adv. Agrochem 2, 154–162 (2022).
Xu, H. et al. Design, synthesis and antifungal activities of novel cis-enamides via intermediate derivatization method. Adv. Agrochem 2, 97–103 (2023).
Zhang, N., Sun, J., Yin, L., Liu, J. & Chen, C. Silver nanoparticles: from in vitro green synthesis to in vivo biological effects in plants. Adv. Agrochem 2, 313–323 (2023).
Cao, X. et al. Lanthanum silicate nanomaterials enhance sheath blight resistance in rice: mechanisms of action and soil health evaluation. ACS Nano 17, 15821–15835 (2023).
Molla, K. A. et al. Understanding sheath blight resistance in rice: the road behind and the road ahead. Plant Biotechnol. J. 18, 895–915 (2020).
Weston, C. E., Richardson, R. D., Haycock, P. R., White, A. J. P. & Fuchter, M. J. Arylazopyrazoles: azoheteroarene photoswitches offering quantitative isomerization and long thermal half-lives. J. Am. Chem. Soc. 136, 11878–11881 (2014).
Sheldrick, G. M. SHELXS-97, a program for crystal structure solution (Göttingen, 1997).
Sheldrick, G. M. SHELXL-97, a program for crystal structure refinement (Göttingen, 1997).
Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFA1207400), National Natural Science Foundation of China (22274061), the 111 Project (B17019) and Fundamental Research Funds for the Central Universities (CCNU24JCPT030).
Author information
Authors and Affiliations
Contributions
J.Y. and G.-F.Y. conceived and designed the study, supervised the project, analyzed the data, and revised the manuscript. S.H.L. and W.X. gave some suggestions for the study. Y.H. performed synthesis experiments, spectral experiments, fluorescence imaging experiments, antifungal experiments and analyzed all the data and wrote the manuscript. X.Z. and X.M. performed fluorescence imaging experiments and revised the manuscript. Z.L. performed antifungal experiments and revised the manuscript. J.S. performed biological compatibility experiments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, Y., Zeng, X., Ma, X. et al. A visible light-activated azo-fluorescent switch for imaging-guided and light-controlled release of antimycotics. Nat Commun 15, 8670 (2024). https://doi.org/10.1038/s41467-024-52855-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52855-9
