
Abstract
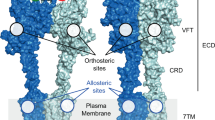
The metabotropic glutamate receptors (mGlus) are class C G protein-coupled receptors (GPCR) that form obligate dimers activated by the major excitatory neurotransmitter L-glutamate. The architecture of mGlu receptor comprises an extracellular Venus-Fly Trap domain (VFT) connected to the transmembrane domain (7TM) through a Cysteine-Rich Domain (CRD). The binding of L-glutamate in the VFTs and subsequent conformational change results in the signal being transmitted to the 7TM inducing G protein binding and activation. The mGlu receptors signal transduction can be allosterically potentiated by positive allosteric modulators (PAMs) binding to the 7TMs, which are of therapeutic interest in various neurological disorders. Here, we report the cryoEM structures of metabotropic glutamate receptor 5 (mGlu5) purified with three chemically and pharmacologically distinct PAMs. We find that the PAMs modulate the receptor equilibrium through their different binding modes, revealing how their interactions in the 7TMs impact the mGlu5 receptor conformational landscape and function. In addition, we identified a PAM-free but agonist-bound intermediate state that also reveals interactions mediated by intracellular loop 2. The activation of mGlu5 receptor is a multi-step process in which the binding of the PAMs in the 7TM modulates the equilibrium towards the active state.
Similar content being viewed by others


Introduction
The major excitatory neurotransmitter in the mammalian nervous system, L-glutamate binds to various receptors in the membrane to modulate neuronal activity1,2. Among them, the metabotropic glutamate receptors (mGlu) are class C G protein-coupled receptors. This family is composed of eight subtypes, divided into three groups based on their pharmacology, sequence similarity, and primary cellular signalling pathways. The structure of mGlu receptor is modular with an extracellular region composed of the venus-fly trap domain (VFT) that binds the agonist L-glutamate and the cysteine-rich domain (CRD), which connects the VFT to the transmembrane domain (7TM)3. Typical of all class C GPCRs, mGlu receptors also form obligate homo- and heterodimers, dimerisation being essential for their biological function. Binding of the orthosteric agonist, L-glutamate induces the closure of the VFTs that mediates a large conformational change resulting in the signal being transmitted to the 7TM and binding of intracellular signalling partners such as the G protein4,5,6,7,8,9. Surprisingly, no natural ligands that bind to the 7TM of mGlu receptors have been identified yet several small synthetic molecules can bind, acting either as positive or negative allosteric modulators, PAM and NAM, which enhance or inhibit receptor signalling, respectively2.
The different mGlu receptors are widely expressed in the human brain where they modulate glutamatergic transmission, thus contributing to many important physiological processes2. Aberrations in glutamatergic transmission result in many neurological disorders, making mGlu receptor targets of therapeutic interest2,10,11. The mGlu5 subtype is an important modulator of synaptic plasticity involved in learning and memory12. The mGlu5 positive allosteric modulation has strong potential for alleviating different symptoms of schizophrenia13. However, some of the PAMs have been shown to be associated with adverse side-effects and thus developing safe mGlu5 PAMs is an important requirement and at the same time a difficult task14,15. Understanding the effects of PAMs is a real challenge, and how they bind and impact on the dynamics of the mGlu activation mechanism is of great physiological and pharmacological interest.
Pharmacologically, mGlu5 PAMs trigger diverse signalling outcomes, resulting in reported biased signalling16,17,18 likely associated with multiple receptor conformations. Biased signalling refers to the ability of different ligands binding to the same receptor but engage with different signalling complexes, triggering differential cellular responses19. Amongst the large number of PAMs that have been developed to date, some are defined as pure PAM that only potentiate the glutamate action (VU040955116 and VU2920), but they can display a low agonist-like activity (intrinsic agonist activity) depending on the signalling pathway and cellular context17,21,22,23. Other PAMs, such as VU042446515, are called ago-PAM that display a strong intrinsic agonist activity and can induce receptor signal transduction without the need for glutamate binding to the VFTs, either by activating the 7TM domain alone or by displacing the equilibrium between the inactive and active state of the receptor.
Several studies have reported the PAM-bound conformation of class C GPCRs and how the PAM and the G protein modulate the receptor conformation4,5,6,7,9,24,25. The class C GPCR activation mechanism is asymmetric i.e., with only one G protein bound and activated at a time by the receptor dimer6. The 7TMs form an asymmetric interface in the activated state both in the presence and the absence of G protein5,6,25. In mGlu2 receptor, only one ago-PAM was found bound to the G protein-coupled 7TM5, while in other class C receptors, such as the calcium sensing receptor (CaSR), PAMs are bound to both protomers in the absence or presence of G protein, although they adopt different binding pose or mode25,26. Recent single-molecule FRET analysis proposed a multi-state process including an agonist bound intermediate-active conformation between the inactive and active state27,28. mGlu activation mechanism is a dynamic, multi-step process and the PAMs are likely to modulate this receptor state equilibrium, promoting the stabilisation of the active state. Despite the conserved domain architecture in mGlu receptors, the differences in the 7TM sequence provides an opportunity to selectively modulate the mGlu’s activity in an isoform dependent manner and may lead to the discovery and the design of new molecules with therapeutic potential and high level of selectivity. Thus, understanding the mechanism of PAM binding and the activation of the receptor is highly relevant. While structures of mGlu5 7TM with NAM bound have been reported9,29,30,31, the binding mode of PAM in the mGlu5 7TM and their impact on mGlu5 conformations has remained elusive.
Here, using cryogenic-sample electron microscopy (cryoEM), we report structures of full-length mGlu5 in detergent micelles bound to the high affinity orthosteric agonist quisqualate and to chemically distinct PAMs: the ago-PAM VU0424465 and the PAMs VU0409551 and VU29. We decipher the diversity of PAM binding mode and how they impact mGlu5 receptor conformation and function, providing insights into the effect of PAMs on the conformations and activity of mGlu5 receptor.
Results
PAMs of mGlu5 with different scalffold and pharmacological properties
To elucidate the effect of PAM binding on mGlu5 receptor conformations and the extent of the structural changes they bring about in the 7TM, we chose three PAMs, VU04424465, VU29 and VU0409551 (Fig. 1a), which were selected based on their difference in chemical scaffold, reported affinity, as well as their different intrinsic agonist activity15,17,22. In the literature, VU0424465 is described as a high affinity ago-PAM15 contrary to VU29 and VU0409551, which are less able to activate mGlu5 by themselves without agonist glutamate17,22. Here, we confirm that VU0424465 possesses a high intrinsic agonist activity in absence of the agonist quisqualate, being able to almost fully activate mGlu5 at 1 µM (83 ± 6% of the maximal activation induced by quisqualate in our assay) (Fig. 1a; Supplementary Table 1). In contrast, VU29 and VU0409551 display a low intrinsic agonist activity, being able to partially activate the receptor at 1 µM (20 ± 9% and 15 ± 1% of maximal activation induced by the agonist quisqualate for VU29 and VU0409551, respectively) (Fig. 1a; Supplementary Table 1). Increasing the VU29 and VU0409551 concentration raise the ago-PAM activity to around 50% of the maximum response, reflecting their lower potency. The allosteric effect of PAMs in mGlu receptor signal transduction is well documented32,33. How these ligands bind and modulate mGlu5 receptor function has been extensively investigated using ligand binding, single-point mutagenesis and cell-based functional assays2. However, understanding the possible PAM binding mode and how they modulate the receptor conformational landscape, at the molecular level, remains unclear. Thus, here we set out to determine the structures of mGlu5 with the above-mentioned PAMs. The structures of PAM-bound mGlu5 receptor were solved by using the thermostabilised mGlu5 receptor (mGlu5-5M) that contains five thermostabilising mutations (T742A5.42, S753A5.53, T777A6.42, I799A7.29, A813L7.43) and truncation at its C-terminus after residue A856 (mGlu5-Δ856)9,34. We have previously shown that this thermostabilised receptor is fully functional, retaining its capability of activating the G protein when expressed in HEK293 cells9,34.

a Evaluation of VU0424465, PAM VU29 and VU0409551 intrinsic agonist activity using cell-based assay. Dose-dependent potentiation of quisqualate agonist-induced response of mGlu5 by VU0424465, VU29 and VU0409551 were determined by the measurement of IP1 accumulation in HEK cells transiently transfected with mGlu5-Δ856. Data are normalised to the response measured for quisqualate alone and are mean ± SEM of three independent experiments (n = 3) performed in technical duplicates. Parameters from the dose-response curves are listed in Supplementary Table 1. Source data are provided as a Source Data file. b mGlu5 structure displays binding of the ago-PAM VU0424465 (yellow sphere) to both 7TMs and both VFTs display bound quisqualate (blue spheres). c CryoEM map (magenta) sharpened with B = −95 Å2 at 8 σ showing density for the ago-PAM VU0424465 in stick representation (3 Å carving around the atoms; carbon atoms in yellow) surrounded by key side chain residues shown (carbon atoms in blue and green). d Superposition of the two VU0424465-bound 7TMs from the same mGlu5 homodimer. VU0424465 induces a shift of TM6 along its helical axis, towards the extracellular side of the receptor. Resolved density for ICL2 displays an extended helix 3 that stretches out of the detergent micelles. e The active state is stabilised by a limited number of residues localised at the top of TM6. Molecular contact between N796 (ECL3) and D722, P724, Y730 (ECL2) observed only in one protomer. f Ligand binding site of VU0424465 in each protomer. VU0424465 make polar interaction with S805 and with the W785 carbonyl. It is stabilised by hydrophobic contact involving M802, V806, S809 in TM7; W785, Y793, F788, L789 in TM6 pushing out F788. g, h PAMs and NAMs occupy a common binding site. Panels show the superposition of the X-ray structure of thermostabilized mGlu5 7TM bound to NAM alloswitch-1 (g; Cyan, PDB code 7P2L) and M-MPEP (h; Purple, PDB code 6FFI) with 7TM of the mGlu5-5M bound to ago-PAM VU0424465 (blue and green) from this work. Figure panels (b–h) were generated using PyMOL (The PyMOL Molecular Graphics System, Schrödinger, LLC.).
Structures of the ago-PAM VU0424465-bound mGlu5 receptor
The first structure described here is the ago-PAM VU0424465-bound mGlu5 receptor at a global resolution of 3.1 Å (we define ago-PAM VU0424465 as PAM1, data set 1 as PAM1_d1; Fig. 1b–f) with no symmetry imposed. The 7TM region is of lower resolution when compared to VFT (Supplementary Figs. 1 and 2), a common feature in all the data sets described in this study (the statistics of data processing and model building of all data sets are provided in Supplementary Table 2 and the 3D FSC plots describing the anisotropy of the maps and orientation distribution plots in Supplementary Fig. 3). The density for inner TM helices is better resolved and there is unambiguous density for the PAM in both protomers and for the surrounding residues (Fig. 1c and Supplementary Fig. 2). We were also able to build the loop ICL2 in this data set (using the deepEMhancer35 maps), which has been implicated in G protein binding26,36 (Supplementary Figs. 1 and 2).
Previously, the class C GPCR structures of mGlu2 revealed that only one ago-PAM is bound in the 7TM whereas two PAMs are bound in both the 7TMs of CaSR but in different binding poses5,25. Nevertheless, despite the differences, an asymmetrical dimer interface was reported in both cases. In contrast, we observe ago-PAM bound in similar binding pose in both protomers in mGlu5 (Fig. 1c and f). VU0424465 (PAM1) shares a common binding site with the NAMs such as alloswitch-19 or M-MPEP31 (Fig. 1g and h). The fluorophenyl group is found between G6242.45, I6252.46, G6282.49, S6543.39, P6553.40, V8067.36, S8097.39 with a potential interaction with A8107.40 in one monomer only. Although the PAM is positioned in the same binding pocket as NAM, the main differences are localised at the top of TM6, where the ago-PAM structure displays a kink at P7906.55 compared to inactive NAM bound state in both 7TMs (Fig. 1g and h) but a slight shift of TM6 towards the extracellular side for only one protomer (Fig. 1d). As a consequence, the PAM that induced the TM6 shift is slightly tilted upwards (toward the vertical axis of the 7TM) and also makes an interaction with the S6583.43 and L8137.43, one of the thermostabilising mutation introduced in the receptor9 (Fig. 1f). In both monomers, the side chain of W7856.50 faces towards the 7TM and interacting with the PAM, and form a potential polar interaction with S8097.39.
The pyridinecarboxamide of VU0424465 occupies the same pose in both protomers making van der Waals interaction with M8027.32, L7445.44, and W7856.50 in TM6 (Fig. 1f). For both VU0424465 molecules, the N-pyridine atom makes polar contact to the S8057.35 and to the W7856.50 carbonyl. The dimethylpropyl group of VU0424465 molecule contacts L7445.44, F7886.53, V7896.54, Y7926.57, F7936.58 in both protomers, whereas in one monomer (monomer B) it is shifted towards TM7 and makes an additional interaction with T8017.31.
A second independent data set for VU0424465 (PAM1)-bound receptor was collected with cold FEG and Falcon 4i detector housed at the bottom of the Selectris energy filter. The batch of the receptor and grid preparation was completely independent. In this data set, two different conformations of the receptor were obtained, at overall resolutions of 3.2 Å (PAM1, conformer 1 in dataset 2 as PAM1_d2_c1) and 3.5 Å (PAM1, conformer 2 in dataset 2 as PAM1_d2_c2) respectively (Supplementary Fig. 4). The difference between these two conformations includes some variability in PAM density that reflect a differential occupancy with global rmsd of 1.5 Å and 1.6 Å for the monomers. Here, the occupancy is used to describe as the fraction of the receptor molecules with PAM bound and after averaging, produce a well-defined density, allowing to unambiguously infer the presence of a PAM bound within each protomer. In these two conformations, the VFT (0.3 Å rmsd for Cα) overlay well but the CRD and the TMD show greater deviations (0.6 Å and 0.8 Å rmsd respectively). The PAM density is observed in both protomers but the conformer 1 (Supplementary Fig. 4F) has slightly weaker density when compared to PAM1_d2_c2 (Supplementary Fig. 4F and G), and PAM1_d1 (Supplementary Fig. 2). These independent data sets provide confidence in the mode of PAM binding presented above in different biochemical preparation and illustrate the conformations of the 7TM that the receptor can adopt.
The binding modes in PAM1_d2_c1 and PAM1_d2_c2 are very similar. Notably PAM1_d2_c1 model shows that S8097.39 - W7856.50 are in a favourable distance for hydrogen bonding in only one monomer, similar to PAM1_d1. The PAM1_d2_c2 model displays the same polar contacts in monomer A, whereas the second protomer has an additional interaction between Y6593.44 and T7816.46. The small differences observed could be because of the resolution and the quality of the maps in particular at the TMD (Supplementary Figs. 1, 2 and 4). However, the structures suggest that the VU0424465 mediated molecular reorganisation of the network of interactions revolving around S8097.39, Y6593.44, W7856.50 is a feature of mGlu5 allosteric modulation.
Structures of mGlu5 receptor bound to VU29 and VU0409551
Subsequently, cryoEM reconstructions of the PAMs VU29- and VU0409551-bound mGlu5 receptor at global resolutions of ~3.2 Å (PAM2/VU29) and 3.0 Å (PAM3/VU0409551), respectively were obtained (Supplementary Figs. 5 and 6). The VU29 map had reasonably well-defined PAM density in one of the monomers that allowed model building of the PAM (Fig. 2a and Supplementary Fig. 5E) and of few bulky side-chains around the PAM binding sites are prominent. The second protomer displayed poorer density for the ligand suggesting a lower occupancy and thus the PAM was not built in this monomer, but the residual density indicates a similar binding pose (Supplementary Fig. 5E). VU29 occupies a binding site similar to VU0424465 but with some significant differences. The main difference comes from W7856.50 being pushed away from the helical bundle by the phenyl ring of VU29, similar to what is observed with CDPPB37, another mGlu5 PAM, whereas F7886.53 moves in, interacting with the VU29 (Fig. 2a). VU29’s nitrobenzamide group is localised in a similar position compared to the fluoro phenyl moiety of VU0424465, but is likely to push Y6593.44 side chain. VU29 is further stabilised by interactions with S6583.43 and S8097.39. In addition, VU29 engages with several residues from TM5 including V7405.40 and I7515.51. The 7TM has lower resolution and positions of only few side-chains can be assigned with confidence, suggesting a dynamic nature of VU29 binding and, at the moment, we cannot exclude other binding modes. However, like for the VU0424465 PAM structures, the binding mode presented here highlight a molecular reorganisation of the residue network of interaction between S8097.39 - Y6593.44, W7856.50.

a CryoEM structure of the PAM VU29-bound mGlu5 with only one PAM modelled and the binding mode of VU29 (teal blue). b–f Structure comparison of mGlu5 receptor dimer bound to ago-PAM VU0424465, PAMs VU29 and VU0409551 and VU0424465-bound to mGlu5 W785A mutant. The 3D alignment or superposition was performed with residues 25-191 and 331-461 of both lobe-1 of the PAM1_d1 data set (one protomer in green and the other in blue) (VU0424465-bound mGlu5 receptor) and those of VU29 (PAM2, yellow); VU0409551 (PAM3, light purple); the different conformations of VU0424465 including PAM1_d2_c1 (salmon) and PAM1_d2_c2 (cyan) and VU0424465-bound mGlu5 W785A mutant (PAM1_W785, olive). g PAM diversity of mGlu5 recpetor interface at TM6. Extracellular view of the 7TM dimer interface with P790 in stick representation show the difference in interface as illustrated by the movement of TM6 and P790. The panel represents the superimposition of VU042264 (blue and green), VU29 (yellow) and VU0409551 (purple). Figures were generated using PyMOL.
In the case of VU0409551, only weak density for the PAM was observed in both protomers, preventing the model building of the ligand (PAM3_c1) of this PAM. However, this data set has well resolved VFT allowing us to build few water molecules in both the protomers and the region around the agonist binding site and the 7TM is also better resolved than the VU29 map (Supplementary Figs. 6 and 7).
Structure of VU0424465-bound mGlu5-W785A mutant
In our previous study, we took advantage of high-resolution X-ray structure of the mGlu5 receptor 7TM bound to the NAM alloswitch-1 to understand the role of the residues in mGlu5 signal transduction. We observed that mutation of residue W7856.50 to alanine results in the alloswitch-1 becoming a PAM instead of NAM9, as also reported for other NAMs38, highlighting the key role of W7856.50 in ligand binding at the 7TMs and in mGlu5 signal transduction. In addition, W7856.50 in mGlu5 has been shown to interact with ligands previously18. Thus, we hypothesised that W7856.50 might play a crucial role and might adopt different positions depending on the PAM bound and prompted us to determine the structure of the thermostabilised receptor with an additional mutation, W785A (W7856.50).
We co-purified the full-length mGlu5-5M W785A mutant bound to the ago-PAM VU0424465 and the 3D reconstruction has a global resolution of ~3.4 Å (Supplementary Fig. 8). Removing the bulky W7856.50 side chain strongly impairs the PAM occupancy as assessed from the calculated map, which reveals some less defined density found in both monomers in the same region compared to other data sets (PAM1_d1, PAM1_d2_c1 and _PAM1_d2_c2). However, the ligand could not be modelled with confidence and thus the structure presented here suggests a role for W7856.50 in VU0424465 ago-PAM binding.
The diversity of PAM-bound mGlu5 receptor conformations
We have determined six 3D reconstructions for the PAM-bound mGlu5 receptor. It includes two conformations of the receptor bound to VU0424465, one bound to VU29, one to VU0409551, and one for the mGlu5-5M-W785A receptor purified with VU0424465. Surprisingly, only two of the VU0424465 structures (PAM1_d1 and PAM1_d2_c1) display an almost identical backbone superposition, from the VFTs to the 7TM when superposing the dimer (Fig. 2; rmsd 0.7 Å and 0.8 Å for chain A and B respectively, Fig. 2e). This illustrates some subtle dynamics around the 7TM-7TM interaction in the active state of the receptor. When monomers of each structure are compared (i.e., PAM1_d1; Fig. 1d, Supplementary Fig. 9), all VU0424465 structures (PAM1 models) display an asymmetrical interface, with a shift of TM6 for only one monomer and a pronounced kink at P7906.55 of TM6, both much less pronounced in the two other PAM structures and W785A mutant structure (Fig. 2b–d; Supplementary_Fig. 9).
The PAM VU0424465 strongly stabilises the TM6 asymmetric interaction. In the VU29 bound 7TM, the TM6 is clock-wise shifted and the same is observed for the VU0409551 model despite the fact that the PAM could not be modelled (Fig. 2g). A similar but less pronounced effect is observed for the W785A mutant structure. To appreciate the difference of the relative position of the 7TMs for each structure reported here and compare the different models, we measured the distance between the Cα of P7906.55 that is localised at the top of TM6 of each monomer, facing each other within the dimer interface. This distance in the VU29, VU0409551 and in the VU0424465 + W785A mutant models, significantly increase (Fig. 2g) from ~7.5 Å in the VU0424465 models (PAM1_d1, PAM1_d2-c1 and PAM1_d2-c2) up to 11 Å for the VU29 structure. This illustrates the differences of the 7TM interface promoted by different PAMs and suggest a contribution of the W7856.50 that not only affect the VU0424465 binding but also the different interfaces depending on the PAM bound.
The different maps and models presented here shows that different PAMs bind differently to the receptor 7TMs. They also illustrate the dynamics of the 7TM depending on the nature of the PAM, notably the ago-PAM VU0424465 is better stabilising the active state Acc conformation. With all these structures we can now define the signature of mGlu5 PAM bound state.
Induced conformational diversity in 7TM Allosteric modulator binding site
The structures presented here highlight previously identified residues important for PAM binding and signal transduction18,21,38,39. In order to analyse the functional impact of the PAM interactions observed in the described structures, we performed functional characterisation using mGlu5 mutants.
The W7856.50 differentially affects the activity of the PAMs (Fig. 3; Supplementary Table 3). The mutation of this residue to alanine has surprisingly very little effect on the signalling activity induced by VU0424465 (Fig. 3a), likely due to its strong affinity. In contrast, the potency of the other two PAMs VU29 (Fig. 3c and d) and VU0409551 (Fig. 3e and f) is significantly affected by W785A mutation (Supplementary Table 3). The allosteric effect of VU29 is improved in this mutant but the effect by VU0409551 is reduced. In the model proposed here, VU29 binding pushes the W7856.50 side chain out and thus removing the side-chain is likely to facilitate its efficient binding to the receptor (Figs. 2a, 3i). In other words, when mutating W7856.50 to alanine, more space is created for the PAM VU29 than for ago-PAM VU0424465 and VU0409551.

a, b VU0424465, c, d VU29, e, f VU0409551-dependent agonist-induced activity of different mutants in the PAM binding site. Single residues were mutated into alanine and functional consequences were determined by measuring the production of inositol monophosphate (IP1) in HEK293 cells transiently expressing the mGlu5-Δ856 and mutated mGlu5-Δ856 receptors. Experiments were performed in the presence of a fixed dose of the orthosteric agonist quisqualate (10 nM) and different doses of PAMs. Bar plots (a, c and e) are pEC50 average values of at least 3 independent experiments +/- SEM. Dose response curve (b, d, and f) are presented for mutants displaying statistically significant effect as compared to the WT type receptor. pEC50 value are presented in Supplementary Table 3. Each independent experiments were performed in technical duplicates (p value are indicated above each bar when required). For VU04244465, pEC50 value were determined for mGlu5-Δ856 (n = 11), for all mutants (n = 4) except for Y659A, F788A and A813L (n = 3). For VU29 pEC50, mGlu5-Δ856 (n = 10), for all mutants (n = 3) except for T781A, W785A, S805A and S809A (n = 4). In case of VU0409551, pEC50 value were determined for mGlu5-Δ856 (n = 12), for mutants T781A, W785A, F788A, A813L (n = 3), for mutants S658A, Y659A, Y792A and F793A (n = 4), and for S809A (n = 5). The Source data are provided as a Source Data file. g–i Network of polar interaction between Y659, S809 and W785 is reframed by PAM binding. mGlu5 inactive state relies on W785, Y659, S809 forming polar contact with the NAM alloswitch-1 ligand (g; PDB 7P2L). Ago-PAM VU0424465 binding destabilises polar interactions between Y659, S809 and W785 (h). VU29 pushes away Y659 and W785 but is stabilised by F788 (i). VU0424465 and VU29 stabilise different 7TM conformations. Figures were generated using PyMOL.
Another key residue in the binding site of mGlu5 is Y6593.44. Replacing this tyrosine abolishes the VU29 and VU0409551 modulation and strongly impairs the effect of VU0424465, although some activation was measured (Fig. 3; Supplementary Table 3), and it also seems to reduce the constitutive activity of the receptor. Y6593.44 is localised at the bottom of the binding site and was found in a network of polar contacts in the inactive conformation of the 7TM structure solved by X-ray crystallography9 (Fig. 3g). Here, the Y6593.44 is likely displaced towards T7816.46 and the Y6593.44 - S8097.39 interaction likely disrupted. In the protomer where the VU29 is modelled, the nitrobenzamide group pushes the Y6593.44 towards TM5 and the interaction with S809 is also disrupted. Pharmacology presented here highlights the role Y6593.44 but also of T7816.46, revealed by the reduced potency of VU0424465 and VU0409551 for the receptor in which these residues have been mutated.
A third important residue is the F7886.53 that undergoes the most dramatic movement when all structures are compared (Fig. 3g–i). The position of the F7886.53 may adopt multiple conformations depending on the nature of PAM or NAM binding. Indeed, in the structure of alloswitch-1, a photoswitchable NAM has forced the F7886.53 away from the 7TM (Fig. 3g) similar to PAM VU0424465 (Fig. 3h), whereas it moves and interacts with VU29, stabilising its binding as suggested by its reduced potency in F788A mutation (Fig. 3c, d and i). F7886.53 was shown to have a differential effect on PAM and NAM affinity18,38 that can be explained by the different rotameric position presented here, which might improve the binding of some PAMs such as VU29 or reduce it for molecule such as VU0409551, likely restricting the access to the binding pocket.
Finally, Koehl et al.7 had previously shown that the apex of ECL2 is important for orthosteric agonist induced signal transduction. Here, we observe potential interactions between N796 (ECL3) and D722, P726 and Y730 (ECL2) in the PAM1_d1 model. Such molecular contacts may help to stabilise the active conformation (Fig. 1e). This interaction is also observed in the two other VU0424465 structures (PAM1_d2_c1 and PAM1_d2_c2). However, mutating these residues has only a weak effect on the efficacy of PAM or on the orthosteric agonist quisqualate (Supplementary Figs. 10 and 11, Supplementary Table 4).
The structural analysis of ago-PAM and PAM-bound receptor presented here reveals a dynamic/flexible network of contacts between residues in the ligand binding site stabilising the mGlu5 receptor active state and ultimately to promote G protein binding. The global differences within the 7TM between the inactive state and the active state are not extensive and revolve around the residues Y6593.44, W7856.50, F7886.53 and the residue interaction network they form with T7816.46 and S8097.39. These residues have been shown to be important for the inactive state of the receptor and for transmitting receptor signalling29,39. They also clearly but differentially affect PAM’s signal transduction, and represent a hub for mGlu5 receptor activation and allosteric modulation. Overall, the Y6593.44, W7856.50 and F7886.53 appears to provide account for the complexity of mGlu5 allosteric modulation and for the diversity of PAMs binding modes.
Quisqualate-bound, Rcc conformation of mGlu5
Activation of class C GPCRs requires a number of conformational changes that first take place in the VFT upon glutamate binding and then transmitted to the 7TM3,7. In absence of glutamate or presence of orthosteric antagonist, the VFTs adopt an open (o) state and a resting (R) conformation, with respect to lobe-I orientation3,28. Binding of L-glutamate or analogues such as quisqualate induces the closure of the VFTs and the reorientation of lobe-I that brings the lobe-II in proximity. This movement is translated to the 7TM that are far apart in the open and resting state of mGlu5, but form a dimeric interface in the receptor active (A) state7,9. The multiple states that the receptors can adopt have been defined based on the conformation of the VFTs40. Thus, the antagonist-bound receptors typically adopt a resting and open conformation of the VFTs (Roo)9 and the active and closed conformation (Acc)7,9. However, the receptors can also adopt other conformations in between28,37, including an Rco4,5 conformation. Here we report an intermediate quisqualate-bound Rcc conformation of mGlu5.
Although agonists and PAMs have been included throughout the purification of the receptor in the study, we identified a subset of particles that showed distinguishable 2D classes compared to the PAM-bound active state (Acc) in the different data sets. When processing multiple PAM-bound data sets (3 different PAMs and one mutant), we identified population of particles visually identical to mGlu5 inactive state receptor (i.e., with the TM domain separated apart) but only in the PAM3 data set, the number of particles was sufficient for a medium-resolution EM reconstruction to ~4.1 Å (Fig. 4; PAM3_c2; Supplementary Fig. 12). We could calculate only low-resolution maps from particles of the VU0424465 and VU29 data sets. The medium resolution map for the PAM3 data set (Supplementary Fig. 12) has slight anisotropy when compared to other data sets (Supplementary Fig. 3).

a Two views of cryoEM map of the Rcc conformation illustrating molecular interaction between the ICL2 loops. b 3D model of the Rcc quisqualate-bound state show closed conformation of the VFTs induced by quisqualate binding and the close proximity of ICL2s. c Comparsion of the monomers of Rcc model with antagonist-bound Roo state (PDB 7FD9) and the agonist and PAM-bound Acc state (PAM1_d1; green) conformation. d CryoEM density for quisqualate (blue mesh) standard binding pose (monomer A), as observed in VU0409551-bound mGlu5 structure, and its alternate binding mode (monomer B) in intermediate state (Rcc). The sharpened map with a B-factor of -70 Å2 at σ = 11 and carve of 2 Å was used to generate the maps with Pymol. e Quisqualate (yellow stick representation) binding mode observed in VU0409551-bound mGlu5 structure (green/magenta)) and its alternate pose in intermediate Rcc state. f Intracellular view of the mGlu5 receptor structures superposition of intermediate Rcc and active Acc states. The ICL2s meet in the Rcc intermediate state during the transition between the Roo (not illustrated in the figure here) and the Acc state. ICL2 movement is illustrated by the I680 localised in ICL2. Figures were generated using PyMOL and Chimera57.
The overall map of the receptor dimer is well resolved and the 7TMs are separated apart, as previously reported in the antagonist LY341495-bound mGlu5 receptor9 (Fig. 4a–c). This structure reveals closed conformation (c) of the VFTs, with quisqualate bound, but remains stabilised in an inactive (R) state, which we propose to be in an intermediate state with the receptor in resting state and define as Rcc (Fig. 4c) representing an additional and different conformation of the receptor.
In the map, the VFT of one of the monomers is better resolved than the other and shows quisqualate bound in the classical pose as in the other structures determined7,9 (Fig. 4d and e). The ligand is stabilised by several polar interactions (Y64, S152, S173, T175, G280, D305, R310, K396) and van der Waals contact (W100, G150, S151, A174, Y223, E279, G306,). In the second VFT, the quality of the map is lower indicating flexibility and the quisqualate adopts a different pose, the carboxylic group orients towards Lobe-II, making fewer van der Waals contacts with residues Y233, E279, F277, S304, G306, and two polar interactions with D305 and S173, and suggesting a transient binding mode (Fig. 4d and e).
The weak density for one of the VFT is reflective of the receptor dynamics. Indeed, the 3D variability analysis41 (3DVA) of this subset of particles and subsequent analysis of the models by phenix.varref 42 showed that one VFT is closed (c) and in a stable conformation whereas the second VFT samples multiple states, from a closed to a more open conformation, compatible with quisqualate being weakly bound (Supplementary Movie 1 and 2). Although, we could not confidently build the Lobe-1 in this monomer due to poor density, we however can see that the helices B and C are starting to move and to undergo the transition from the open to the closed state when compared with the orthosteric antagonist LY341495-bound VFT conformation (Fig. 5a and b). This transition suggests that whilst one VFT is stabilised in the closed state (c), the second VFT is oscillating between open (o) and close (c) conformations displacing the receptor equilibrium towards a more active-like A state, initiating the motion to be translated to the CRD and to the 7TM. This Rcc conformation superpimposes well with the recently reported intermediate state 1a (as defined by the authors), in which both VFTs are closed although lobe-I is still in Resting R state37, as opposed to Rco conformation reported for the mGlu2-mGlu3 heteromer4 (described in Fig. 5c and d) or the mGlu2 homomer5.

a Superimposition of VU0424465-bound mGlu5 structure (PAM1_d1; green) and quisqualate-bound intermediate state (Rcc) (PAM3_c2; purple) and b Superimposition of the quisqualate-bound intermediate state (Rcc) (PAM3_c2; purple) and inactive state Roo (Orange; 7FD9) illustrate the close conformation of the VFTs in the intermediate Rcc state. c Superimposition of the quisqualate-bound intermediate state (Rcc) (PAM3_c2; purple) and intermediate 1a state (Cyan; 8T7H) show a similar conformation. d Superimposition of the quisqualate-bound intermediate state (Rcc) (PAM3_c2; purple) and Rco state of mGlu2/3 heterodimer (Blue; 8JD1) show the difference of open and close conformation for one VFTs. Figures were generated using PyMOL.
An interesting feature of this intermediate state is the ICL2, which extends from TM3, similar to the VU0424465-bound conformation (PAM1_d1), (Fig. 4a, b; Supplementary Fig. 1). In the Rcc conformation, the ICL2 from each protomer face each other, most likely stabilising the intermediate Rcc state (Fig. 4c and f). From the intracellular side of the receptor, the superposition between the Rcc and Acc show that ICL2s are moving away in the active Acc conformation, since TM3 and TM4 from each protomer are oriented oppositely (Fig. 4f). The ICL2 loop of mGlu5 has a unique motif KKICTKK and mutating these residues affects the efficacy of the PAM signalling with a dramatic effect in particular for the residue I680 (Supplementary Fig. 13; Supplementary Table 4). In addition, receptor constitutive activity seems to be affected in I680A mutant suggesting a role of this residue in G protein interaction and activation. Interestingly, the ICL2 loop has been proposed to be important for G protein interaction of class C GPCRs26,36 but also other class A and B receptors43,44.
Despite a modest global resolution for this state of the receptor, several key residues involved in PAM binding described above have defined density whereas no clear density could be detected for the PAM. The side chains of W7856.50 and F7886.53 are pointing towards the helical bundle (Fig. 4f). Although W7856.50 stabilises the VU04244645 in PAM1 models (Fig. 1), the presence of F7886.53 is likely to cause steric hindrance and allows us to exclude the possibility that the PAM VU0409551 is bound in the 7TM. As a consequence, we further define this conformation as a PAM-free intermediate state. This observation supports the view that the PAMs stabilise the active Acc conformation. The weaker the PAM affinity, the less the Acc state is stabilised even with agonist quisqualate bound in the VFT, allowing particles to distribute between the Rcc and the Acc state, explaining why we obtained the largest number of Rcc single particles populations in the VU0409551 data set.
Discussion
To transmit a signal across the membrane, upon ligand binding the receptors undergo structural dynamics, which trigger downstream signalling. Such structural changes in GPCRs have been well-studied as the agonist binding leads to changes in the cytoplasmic side of TM helices resulting in G protein binding4,6,24,26,45. In the GPCR family, the multi-domain containing class C GPCRs transmit the signal from the ECD to the 7TM across a distance of ~120 Å. Due to this multi-domain architecture and structural changes in the whole molecule, the receptor can sample different conformations. In fact, before the structure determination of full-length class C receptors, analysis by single molecule fluorescence and crystal structures of extracellular domain had revealed the structural changes upon agonist binding27,28,40. The agonist and their analogues bind to the VFT and its closure results in transfer of the signal to 7TM via the CRD to reach an active state7. The agonist binding in both VFTs is required to approach the active state but still only an asymmetric activation occurs, that is one G protein binds to one of the monomers and not to both at the same time5,6,24,25. Thus, the class C GPCRs can exist in multiple conformations from the resting to the active state with the VFTs open or closed. Another intriguing feature among the mGlu isoforms is the distance between the 7TM of the two monomers, which are closest in the active state highlighted by the proximity of the TM6, which occurs through the rotation of the TM helices. However, the distance between the TM domain of the monomers in the resting state differs in mGlu isoforms, closer in mGlu2 but further apart in mGlu55,7,8,9. The exact reason for such differences in the positions of the 7TM among mGlu isoforms is currently unclear. While the agonist glutamate is sufficient to activate the mGlu isoforms and stabilise the active state, the potentiation and stabilisation of the receptor is further enhanced by PAMs that bind to the 7TM. Many of the residues in the 7TM of the mGlu isoforms are conserved but still subtype selectivity can be achieved2. Thus, the structures of different mGlu isoforms with bound PAM and NAM are highly sought after.
Allosteric modulation of class C GPCRs is the subject of intense research with the potential of discovering high affinity and selective molecules for specific receptor subtype. The main therapeutic potential for mGlu5 PAMs is in the treatment of different symptoms of schizophrenia13. However, the development of a safe mGlu5 PAM with antipsychotic properties has remained a real challenge15,16. One of the main reasons is the complexity of the mGlu5 signal transduction and the adverse side effects associated with some of these PAMs in preclinical studies. The complex signalling of mGlu5 receptor has been associated to biased signalling16,17,18,21. Indeed, the VU0424465, VU29 and VU0409551 are all associated with biased signalling as observed in specific cellular context17,46. They also display different affinity that translates into different potency15,16,17,18,20. However, these molecules are selective for mGlu5. Here, we intended to understand the molecular and structural basis of mGlu5 allosteric modulations and how PAM could differentially modulate the receptor conformation and activity.
The cryoEM structures of the mGlu5 receptor show conformational diversity, particularly in the 7TMs bound to different PAMs. Despite the presence of agonist and PAM during purification, some population of the mGlu5 remains in the resting state with 7TMs splayed apart, reflecting a set of the particles, which have not bound PAM in the first place or the PAM has dissociated but with the agonist bound. In the data set with VU0409551, large population of such a resting state was observed, perhaps due to the lower potency and affinity of PAM3 (Fig. 4 and Supplementary Fig. 12)15,16,17,18. This population of the receptor was observed to exist in the closed VFT and in the inactive resting (R) state allowing us to determine the structure of an intermediate state (Rcc) conformation similar to the intermediate state 1a37 reported for mGlu5 in which both VFT are closed (Fig. 5c). Here, the L-glutamate analogue, quisqualate, is bound to both VFTs but adopts different poses: a standard pose, as previously reported7,9 and seen in all active state described here, and an alternate pose that is likely to be energetically unfavourable, suggesting significant degree of VFT dynamics oscillating between open and close state (Fig. 4d and e, Supplementary movies 1 and 2). This observation is in agreement with the Rco structure reported for the mGlu25 and mGlu2-mGlu3 heterodimer4, although the mGlu2-mGlu3 heterodimer structure clearly displays one close and one open VFTs. As a consequence, this analysis strongly suggests a sequential conformational change that starts with the binding of quisqualate, or L-glutamate, and the stable closure of one of the VFTs that will promote the closure of the second one. The structural dynamic analysis suggest that the Rcc state occurs upon stabilisation of the closed conformation of the second VFT of an Rco state before converting to the activated Acc state, where the 7TMs come together.
We note that the two ICL2s are in close proximity that consequently may contribute in stabilising the Rcc state (Figs. 4a and f), hampering the movement of the 7TM that likely requires the stable closure of the second VFT and the reorientation of Lobe-I from R to A in order for the receptor to reach the active state. The molecular interaction between ICL2s identified in this intermediate state might transiently stabilise the receptor in the Rcc state and keeps the 7TMs apart. Additionally, the ICL2 intermolecular contact likely blocks the binding of G protein, making this conformation an intermediate state that needs to be overcome before mGlu5 reaches the active state and activate G protein. In class C GPCRs, ICL2 was shown to be important for G protein binding and activation26,45, the ICL2-ICL2 position presented here might hint towards the inability of G protein to bind due to steric hindrance. In summary, ICL2s of the Rcc state may act as a checkpoint in multi-step activation process of the mGlu5 receptor before it can reach the active state. In studies involving both agonist such as quisqualate and PAMs, binding of the PAM to the 7TMs may favour the transition by altering the ICL2 conformation37 and allow the TM6 motion, interfering with the Rcc state stability, and facilitating the transition between inactive and active state as well as the cooperativity with orthosteric agonists. In a cellular context, where there are no PAMs, other factors might play a role in promoting the active state. Once both VFTs are stabilised in the closed state, the 7TMs are in close proximity and the PAM will provide further stabilisation to the active state Acc, with some diversity as illustrated here (Fig. 2b–f) that will likely affect the signalling properties of the receptor.
The high occupancy and the better density of the ago-PAM VU0424465 reported in this study correlates with higher affinity15 than for PAMs VU2920 and VU040955117,18. The receptor occupancy affects the conformation and stability of the mGlu5 active state (Fig. 2b and c), correlating with the difference in receptor signal transduction in cells i.e., a higher proportion of mGlu5 receptor in the active state will better bind and activate the G protein. An important observation is that the ago-PAM VU0424465 is bound to both protomers and stabilises the asymmetric interface characteristic of class C receptor dimer and better stabilises the active state (Figs. 1 and 2). The situation is similar to the CaSR for which each protomer has a PAM bound, although they display clear differences in binding mode but different for mGlu2 where the PAM is only bound to the 7TM coupled to G protein5 (Fig. 6a–c). We observe density in both monomers for the PAMs VU29 and VU0409551 albeit at lower occupancy (or weak density), which probably allows the receptor to explore more conformations.

a–c superposition of VU0424465-bound mGlu5 receptor (salmon) structure to PAM bound mGlu2 + Gi receptor and PAM-bound CaSR+Gi (grey). Superposition shows only one PAM bound to the G protein-coupled subunit of mGlu2, whereas CaSR displays one PAM bound to each subunit. The PAM VU0424465 is shown in yellow stick representation and the other PAMs of mGlu2 and CaSR are in magenta. The helix of Gi that interacts with the receptor is shown in green colour and the ICL2 of the receptor that interacts with G protein is highlighted in blue. d–h mGlu2 and mGlu5 NAM and PAM binds in similar allosteric binding site in the 7TMs but adopting different poses. Figures were generated using PyMOL.
We have unveiled the binding site for the ago-PAM VU0424465 and identified a binding pose for VU29 illustrating the differential contribution of TM6 residues mainly W7856.50 and F7886.53 (Fig. 3h and i). The PAM binding site is similar to the NAM binding pocket. Functional analysis confirms the importance of the mGlu5 receptor residues W7856.50, F7886.53, Y6593.44, S8097.39 and T7816.46 in PAM allosteric modulation (Fig. 3), that were also previously reported to affect the PAM binding and biased signalling at the mGlu5 receptor18,21.
The W7856.50 is central to differential stabilisation of the PAMs in the binding site, either positively for VU0424465 and VU0409551 or negatively for VU29 (Fig. 3). It was previously shown to be a key residue for mGlu5 receptor activation and biased signalling9,18,21,38, and was also proposed to contribute to the selection of the G protein-coupled subunit in mGlu receptor heteromers by diffentially stabilising the inactivate state of mGlu2, mGlu3 and mGlu4 receptor subunits4. Mutating this residue to alanine (W785A) has a direct effect on both the VU0424465 occupancy and the active state conformation that may be comparable to low affinity PAM such as VU0409551 (Fig. 2). However, despite the clear molecular interaction in the binding site, VU0424465 still induce G protein signalling in the W785A mutant. Although W7856.50 contributes differentially to the different PAM binding modes and receptor function, its role may also be essential for stabilising the mGlu5 receptor 7TMs inactive state. This is supported by the fact that some NAMs, such as alloswitch-1, that binds in a similar pose to the ago-PAM VU0424465, turns into weak PAM with mutation of W7856.50 to alanine9, destabilising the inactive state of the receptor.
The differences between the inactive and active state within the 7TM is subtle and in agreement with the absence of TM6 motion as compared to the molecular signature for the class A and B GPCR active conformations47. Comparison of the structures of NAM-bound mGlu5 show some similarity between the NAM and the PAM binding site9,29,31 (Fig. 1) but PAMs destabilise the tight polar network that involves Y6593.44, S8097.39, W7856.50 and T7816.46 through a water molecule, important for mediating NAM effect9, blocking the receptor activation. Amongst these residues, Y6593.44 is a key residue for the PAM signal transduction and for the NAM inhibition of the receptor. Interestingly, Y659A mutant has no effect on signal transduction induced by the orthosteric agonist quisqualate9, suggesting that the receptor dimer can still sample the 7TM asymmetric dimer interface, in absence of a PAM.
The isosforms of mGlu share high sequence similarity in the 7TM and yet, the ligand binding (PAM) pose is different in different isoforms (Fig. 6d–h) and how PAM selectivity is achieved is unclear. Even within mGlu5 isoform, the crystal structures with NAM and cryoEM models with PAM as described in the current study show similar binding modes, and different PAMs (VU0424465, VU29 and CDPPB37) bind in the same place but they differentially modulate the 7TM conformation as illustrated for VU29 or CDPPB37 that extend towards the membrane, pushing W7856.50 outwards (Fig. 7). In the structures of mGlu2 with G protein bound (Figs. 6a, b and 7), PAM was modelled in one of the monomers, with a different pose compared to VU0424465, the equivalent residue of W7856.50 point towards the 7TM bundle as reported here for mGlu5 with VU0424465, but adopting a different pose. The mGlu2 PAM likely pushes against W6.50 (Fig. 7) in TM6, stabilising the asymmetric interface, which both in PAM bound and PAM + G protein conformation seems to be identical5. The mGlu5 PAMs differentially stabilise the receptor active state, both by differentially interacting with the side chains (Fig. 7) in the 7TM and impacting the membrane domain interface (Fig. 2). It remains however challenging and unclear to come up with any rule in designing PAMs with desired properties for a given isoform of mGlu receptor or more generally for PAMs for class C receptors.

a Superposition of PAM-free 7TM conformation (magenta) in the intermediate Rcc and Alloswitch-1 X-ray structure (grey), b and VU0424465-bound mGlu5 (blue), c and VU29-bound mGlu5 (yellow) and d ADX55164-bound mGlu2 receptor (green; 7MTR). Figures were generated using PyMOL.
The multi-step activation process of the mGlu5 receptor perhaps includes internal check-points such as the ICL2 and the conformations of the residues in the PAM binding sites that can be modulated by small molecules and biologics and may offer opportunities to control the receptor function in the future. The strong stabilising potency of an ago-PAM such as VU0424465 is likely to trigger a much higher level of signal transduction by potentiating orthosteric L-glutamate as would a pure PAM perform, but also due to intrinsic agonist activity that triggers signal transduction in absence of L-glutamate, likely giving account for undesired side-effect. Thus, the structures reported here provide a template for exploring positive allosteric modulation to be integrated in structure-based drug discovery program for identifying PAM for mGlu5 receptor.
Methods
Expression and purification of the human full-length thermostabilized mGlu5 receptor bound to activators
The thermostabilised mGlu5-5M receptor and mGlu5-5M_W785A mutant was expressed and purified as previously reported9. Briefly, mGlu5-5M receptor and mGlu5-5M_W785A constructs were subcloned into the BacMam vector. The constructs contain peptide signal from baculovirus GP64 envelope surface glycoprotein of the Autographa californica nuclear polyhedrosis virus (AcNPV baculovirus), the flag (DYKDDDDK), 10xHis tag, and the precision protease recognition site (LEVLFQGP) at the N terminus, followed by the human mGlu5-5M or mGlu5-5M_W785A. The construct also contains H350L mutation, initially designed for binding of a nanobody and a N445A mutation to remove a glycosylation site. The mGlu5-5M_W785A includes the additional mutation W785A and the N445A glycosylation mutation was reversed.
High-titre recombinant baculovirus was obtained using the Bac-to-Bac Baculovirus expression system (Invitrogen) as described by the manufacturer. Briefly, Sf9 cells grown at 28 °C in EX-CELL 420 medium (Sigma Aldrich) were infected at a density of 3×106 cells using human mGlu5-5M and mGlu5-5M_W785A virus and P2 virus were harvested by centrifugation 48 hours post-infection. HEK293S cell line lacking N-acetylglucosaminyltransferase I (HEK293S GnTI-)48 were grown at 37 °C in humidified 5% CO2 in shaking incubator (110 rpm/minute) in Freestyle medium complemented by 10% of Foetal Bovine Serum (Sigma Aldrich). HEK293S GnTI- cells were infected at a density of 3 × 106 cells per mL with 6% of freshly produced P2 virus (60 mL per litre) and 10 mM sodium butyrate was added 24 hours post-infection. Cells expressing mGlu5-5M and mGlu5-5M_W785A were harvested 96 hours post-infection and stored at −80 °C for further purification.
For membrane preparation, frozen cells were resupended in a lysis buffer containing 25 mM HEPES (pH 7.4), 10 mM MgCl2, 20 mM KCl, 5 mM EDTA, 1 mM PMSF (Sigma Aldrich), and the cOmplete protease inhibitor cocktail tablet (Roche). The pellets were washed two times with the lysis buffer and an additional wash performed using high salt concentration (25 mM HEPES (pH 7.4), 10 mM MgCl2, 20 mM KCl, 1 M NaCl). Membrane fraction was harvested by centrifugation at 45,000 rpm ( ≈ 200,000 g) for an hour and resuspended with a lysis buffer supplemented with 40% glycerol and then stored at −80 °C. Membrane preparation from 2 litres of cell culture expression were resuspended into a final volume of 120 mL of 25 mM HEPES buffer (pH 7.4), 0.4 M NaCl, 10% Glycerol, 10 μM quisqualate (Abcam), 10 mM iodoacetamide (Sigma), and the cOmplete protease inhibitor cocktail tablets and complemented with 10 μM of either PAM such as VU04244659 or VU29 (med chem express) and VU0409551 (R&D system). The mixture was then incubated at room temperature (RT) for 1 hour and the membranes were solubilised by adding 1% (w/v) n-Dodecyl-β-D-Maltopyranoside (DDM, Anatrace), and 0.2% (w/v) cholesteryl hemisuccinate (CHS, Anatrace) for 1.5 hours at 4 °C, under shaking. Solubilised receptors were harvested by centrifugation at 45,000 rpm for 1 hour and supplemented with 10 mM imidazole (Sigma) before loading onto a Nickel-NTA (Ni2+) resin (HisTrap HP, GE Healthcare) at 0.5 mL/min, overnight at 4 °C. The resin was pre-equilibrated in 25 mM HEPES (pH 7.4), 400 mM NaCl, 10 μM quisqualate, 10 μM of PAM, 5% (v/v) glycerol, 0.2% (w/v) DDM, 0.04% (w/v) CHS, and 10 mM imidazole. The resin was washed using a step gradient of imidazole with concentrations of 10 mM, 20 mM, 40 mM and 50 mM and the receptor was eluted with 25 mM HEPES (pH 7.4), 400 mM NaCl, 10 μM quisqualate, 10 μM of PAM, 5% (v/v) glycerol, 0.05% (w/v) DDM, 0.01% (w/v) CHS, and 250 mM imidazole. The eluted receptors were incubated onto a Flag affinity resin (Anti-Flag M1, Sigma) for 1 hour at 4 °C, washed with 10 column volumes of a wash buffer (25 mM HEPES (pH 7.4), 150 mM NaCl, 10 μM quisqualate, 10 μM PAM, 0.03% (w/v) DDM, 0.006% (w/v) CHS, and 2 mM Ca2+). The receptor was eluted with 0.2 mg/mL Flag peptide in a buffer containing 25 mM HEPES pH 7.4, 0.15 M NaCl, 10 μM quisqualate, 10 μM VU0424465, 0.03% DDM (w/v), 0.006% CHS (w/v), 2 mM EDTA. The eluted fractions were then concentrated using a Amicon Ultra-4 centrifugal concentrator 100 kDa (Millipore), centrifuged for 10 minutes at 80,000 rpm to eliminate aggregates, and then loaded on a size exclusion chromatography (SEC) column (Superdex 200 Increase 10/300, GE Healthcare) pre-equilibrated with 25 mM HEPES pH 7.4, 0.15 M NaCl, 10 μM quisqualate and 10 μM PAM, 0.03% DDM (w/v) and 0.006% CHS (w/v). Eluted and purified mGlu5-5M receptor and mGlu5-5M_W785A receptor mutant bound to quisqualate and PAM were concentrated to 5-6 mg/ml using Amicon Ultra-4 centrifugal concentrator 100 kDa (Millipore) for cryoEM grid preparation and analysis.
CryoEM sample preparation, data collection and image processing
VU0424465 (PAM1) with K3 detector
The agonist quisqualate and PAM VU0424465 bound mGlu5 FL receptor in detergent micelles, at 5 mg/mL was applied to a Quantifoil® Au 0.6/1.0 300 mesh grids. Grids were rendered hydrophilic by glow-discharging for 10 seconds at 25 mA using a PELCO EasiGlow instrument. Grids were plunge-frozen using EM GP2 Plunge Freezer (Leica) with the chamber equilibrated at 95% RH and 4°C. Grids were blotted with Whatman no. 1 filter paper for 4.5-6 seconds and plunge frozen in liquid ethane. The frozen grids were stored in liquid nitrogen until further use. Data was collected on Titan Krios G3i (ThermoFisher) at the European Synchrotron Radiation Facility (ESRF, Grenoble, France) with a K3 detector in counting mode housed at the end of Bio-quantum energy filter and 20 eV slit width was used. Data was collected from one grid with a dose of ~15.2 e-/p/s, and 2.4 second exposure at nominal magnification of 105000x, corresponding to pixel size of 0.84 Å. A total of 19830 movies were used for image processing. Movie files were imported into cryosparc 3.249. Motioncorr was carried out with cryosparc in-built patch motion correction (multi) and CTF estimation was performed in-built patch CTF estimation (multi). Initial particles picking was performed using Blob picker in cryosparc and particles were initially extracted with large box size in order to have the particles centred and for subsequent analysis they were down sampled. From initial large set of particles obtained after 2D classification, ab-initio model generation was performed followed by several rounds of 2D and 3D classifications to select good particles and obtain final map using refined and reconstructed using non-uniform refinement50 (NU) with C1 symmetry. The complete workflow is described in Supplementary Fig. 1. Local resolutions were estimated with Relion51.
VU0424465 (PAM1) with Falcon4i detector
Another independent data set of the mGlu5 bound with quisqualate and PAM VU0424465 was collected with a Falcon4i detector (PAM1_d2). Quantifoil® Au 0.6/1 300 mesh grids were treated with 9:1 Ar:O2 plasma (in a Fischione 1070 chamber) for 60 s to render them hydrophilic and 3 μl of 5 mg/ml of receptor was applied to the grids. Grids were plunge-frozen using Vitrobot Mk IV (ThermoFisher) with the chamber equilibrated at 100% RH and 10°C. Grids were blotted with Whatman no. 1 filter paper for 5 seconds with a blot force of 10 without any waiting or draining time and plunge frozen in liquid ethane.
The data acquisition was carried out using the cold field emission gun (CFEG) Titan Krios G4 microscope (ThermoFisher), at the ThermoFisher NanoPort facility in Eindhoven. The CFEG microscope was equipped with a Falcon 4i detector operating in electron counting mode and was integrated at the base of a Selectris Energy Filter. The microscope featured fringe-free imaging (FFI) and aberration-free image shift (AFIS)45, enabling the acquisition of multiple images before stage movement.
Data was collected with a sampling rate of 0.727 Å per pixel, employing an electron flux of ~5 e-/Ų/s and a total fluence of 50 e-/Ų. Movies were processed using Relion 4.051. A total of 19,115 movie frames underwent motion correction using Relion’s built-in program, followed by contrast transfer function (CTF) estimation carried out with CTFFIND452. Initial particle picking was executed using Topaz with a specified threshold of -253. Reference-free two-dimensional (2D) classification was employed to identify high-quality particles. A subset of these particles was then employed to train a new Topaz model, which was subsequently utilised for repicking particles. These particles were extracted with a box size of 360 pixels and down sampled to speed up processing. From the 2D classification, a total of 396,207 particles were selected and subsequent 3D classification resulted in 221,875 particles. These selected particles were refined in 3D with C1 symmetry, resulting in a resolution of 3.5 Å after postprocessing, as determined by Fourier shell correlation (FSC) at 0.143. The map was further enhanced through one round of Bayesian Polishing and two rounds of CTF refinements following the method outlined by Zivanov et al. (2018)54, resulting in a final resolution of 2.7 Å. C2 symmetry expansion and 3D classification with focus on the TMD (using a T value of 128 and no image alignment) revealed the presence of two conformers. Particles corresponding to these two conformers were re-refined separately in Relion51.
Particles associated with these two conformers were subsequently imported into Cryosparc v4.3.049. Duplicate particles arising from symmetry expansion were eliminated before the import. Non-uniform (NU) refinement, local CTF refinement, and local refinement were carried out independently for each conformer. These refinements yielded overall resolutions of 3.2 Å (2.9 Å) and 3.5 Å (3.0 Å) for conformer 1 (PAM1_d2_c1) and conformer 2 (PAM1_d2_c2), respectively (the numbers in brackets are the resolution with tight mask and corrected FSC in cryosparc). The complete workflow is described in Supplementary Fig. 4. Local resolutions were estimated with Relion51.
VU29-bound mGlu5 receptor
The agonist quisqualate-bound mGlu5 WT in detergent micelles, at 5 mg/mL and PAM VU29 was applied to a Quantifoil® Au 0.6/1.0 300 mesh grids. Grids were rendered hydrophilic by glow-discharging for 10 seconds at 25 mA using a PELCO easiGlow instrument. Grids were plunge-frozen using EM GP2 Plunge Freezer (Leica) with the chamber equilibrated at 95% RH and 4°C. Grids were blotted with Whatman no. 1 filter paper for 4.5–6 seconds and stored in liquid nitrogen till imaging.
The data acquisition was carried out using the cold field emission gun (CFEG) Titan Krios G4 microscope (ThermoFisher), at the ThermoFisher NanoPort facility in Eindhoven equipped with a Falcon 4i detector integrated at the base of a Selectris Energy Filter. Data was collected using fringe-free imaging (FFI) and aberration-free image shift (AFIS) with a sampling rate of 0.727 Šper pixel, employing an electron flux of ~6.1 e-/Ų/s and a total fluence of 49.62 e-/Ų. The processing of this data set was done with Cryosparc49. Movie files (9469) were imported into cryosparc 3.2. Motioncorr was carried out with cryosparc in-built patch motion correction (multi) and CTF estimation was performed in-built patch CTF estimation (multi). Initial particles picking was performed using Blob picker in cryosparc and particles were initially extracted with large box size in order to have the particles centred and for subsequent analysis they were down sampled. From initial large set of particles obtained after 2D classification, ab-initio model generation was performed followed by several round of 2D and 3D classifications to select good particles and obtain final map using refined and reconstructed using non-uniform refinement50 (NU) with C1 symmetry. The complete workflow is described in Supplementary Fig. 5. Local resolutions were estimated with Relion51.
VU0409551-bound mGlu5 receptor
The agonist quisqualate-bound mGlu5 WT in detergent micelles, at 5 mg/mL and PAM VU0409551 was applied to a Quantifoil® Au 0.6/1.0 300 mesh grids. Grids were rendered hydrophilic by glow-discharging for 10 seconds at 25 mA using a PELCO easiGlow instrument. Grids were plunge-frozen using EM GP2 Plunge Freezer (Leica) with the chamber equilibrated at 95% RH and 4°C. Grids were blotted with Whatman no. 1 filter paper for 4.5–6 seconds and plunge frozen in liquid ethane. Data was collected on a Titan Krios G3i (ThermoFisher) at the European Synchrotron Radiation Facility (ESRF, Grenoble, France) with a K3 detector in counting mode with a Bio-quantum energy filter and 20 eV slit width. Data collection was performed with a dose of ~18.4 e-/p/s, an exposure of 1.9 seconds at 105000x, corresponding to pixel size of 0.84 Å. A total of 19943 movies were used for image processing. Movie files were imported into cryosparc 3.249. Motioncorr was carried out with cryosparc in-built patch motion correction (multi) and CTF estimation was performed in-built patch CTF estimation (multi). Initial particles picking was performed using Blob picker in cryosparc and particles were initially extracted with large box size in order to have the particles centered and for subsequent analysis they were down sampled for 2D classification. From initial large set of particles obtained by 2D classification, ab-initio model generation was performed, which identified two different conformations followed by several round of 2D and 3D classifications to select good particles and obtain final maps using using non-uniform refinement50 (NU) with C1 symmetry for both the active and the RCC conformation. The complete workflow is described in Supplementary Figs. 6 and 12. For the Rcc conformation, 3D variability analysis in cryosparc was performed and the resulting maps was used for analysis with Phenix.varref 42 (described below). Local resolutions were estimated with Relion51.
mGlu5 W785A
The agonist-bound mGlu5 W785A in detergent micelles, at 6 mg/mL with quisqualate and PAM VU0424465 was applied to a Quantifoil® Au 1.2/1.3 300 mesh grids. Grids were rendered hydrophilic by glow-discharging for 1 min at 25 mA using a PELCO EasiGlow instrument. Grids were plunge-frozen using Vitrobot Mk IV (ThermoFisher) with the chamber equilibrated at 100% RH and 16°C. Grids were blotted with Whatman no. 1 filter paper for 3-3.5 seconds with a blot force of 0 and plunge frozen in liquid ethane. The frozen grids were stored in liquid nitrogen until further use.
Data were collected on Titan Krios G3i (ThermoFisher) at the National CryoEM facility in Bangalore with a Falcon 3 detector in counting mode and faster data acquisition with AFIS55. Data were collected from two independent grids made on different days with a dose of ~0.54 e-/p/s and an exposure of 60 seconds at 75000x, corresponding to pixel size of 1.07 Å. Images from both data sets were manually curated to remove crystalline and thicker ice, followed by import of movie files into Relion 4.051,54. Motioncorr was carried out with relion’s in-built program and CTF estimation was performed with CTFFIND452. Based on CTF resolution and upper limit of defocus to 35000 Å, a total of 2751 and 3001 images from each batch were selected further processing (the complete workflow is described in Supplementary Fig. 8). Low-pass filtered templates from prior data sets were used for automated particle picking with Gautomatch (https://www2.mrc-lmb.cam.ac.uk/download/gautomatch-053/) and particles were extracted with 4.28 Å sampling and 2 rounds of 2D classification was performed to select good particles. These selected particles were extracted with a box size of 384 and 1.07 Å sampling and further enriched for good particles with 2D classification. The particles from batch 1 and batch 2 were individually refined in 3D with C1 symmetry resulting in 4.8 and 6.1 Å respectively after postprocessing (all resolutions described here are of FSC at 0.143). Subsequently, the particles were subjected to Bayesian Polishing54 and both B-factor weighted particles (shiny.star) were joined and refined. The combined data with 260,251 particles after 3D refinement and postprocessing resulted in a 4.3 Å map.
The combined data was subsequently imported into Cryosparc v4.3.049 and ab-initio reconstruction was performed with 5 classes and maximum resolution of 12 Å followed by heterogenous refinement. Three of the best classes or volumes were combined (that had the same resolution) and both homogenous and Non-uniform (NU) refinement50 were performed resulting in 4.3 Å and 3.6 Å maps respectively. Local and global CTF refine was then carried out on the particles with the volume from NU refinement and subsequent refinement resulted in an overall resolution of 3.4 Å. Local resolution was estimated in relion and the local resolution of the maps revealed that the VFT of the receptor had higher resolution than the CRD and the TMD as observed for the other data sets described in this study. The complete work flow is described in Supplementary Fig. 8.
For all the data sets, the anisotropy or the sphericity was estimated using the 3DFSC server56 (Supplementary Fig. 3).
Model building and refinement
Previous agonist-bound active state model (7FD8) was used for rigid body fitting into PAM1_d1 map using Chimera57 and manually inspected and rebuilt where required using Coot58. Multiple maps were used during model building in all data sets including the maps with different B-factor sharpening and the deepEMhancer maps35. For instance, the ICL2 loop in PAM1_d1 was built using the deepEMhancer map generated with mask option and for modelling water molecules, sharpened maps with automatic B-factor estimation were used. Ligand restraint files were generated using JLigand59. FoFc or difference maps were generated using Servalcat60 and used to verify the ligands and solvent.
For the subsequent data sets, the refined model from PAM1_d1 was used as template and monomers were individually fit using Chimera. This global rigid body gave good fit for the VFT and the CRD and 7TM regions were manually fit to the density with Coot first using rigid body and then with real space refinement (in the case of W785A mutant, the residue was changed to alanine). Unlike the other data sets, in the W785A mutant data set, there was also extra density at N445 for glycosylation and N445 was restored from alanine, but a sugar has not been modelled. In all the data sets, the density for ICL2 loop is ordered to varying degrees but only in PAM1_d1 and PAM3_c2, they have been modelled. For the PAM3_c2 data set, the VFT and CRD from PAM1_d1 was used and the 7TM was rigid body with 7FD9 as template along with 7P2L.
Water molecules in the VFT region of PAM1_d1 and PAM3_c1 were conservatively picked using Coot and they were manually checked using the hydrogen bonding distance and density as the criteria. Models were refined using Phenix61,62 in particular the real space refinement and Servalcat/Refmac in CCP-EM60,63,64. For the real space refinement of the model with Phenix, the map sharpened map with half the value of B-factor estimated by the relion postprocess was used and when refmac was used for refinement, the half-maps were provided. The statistics of the final models are provided in Supplementary Table 2.
For the Rcc conformation, the maps from 3D variability analysis and the model without any ligands was used as input to Phenix.varref42 to obtain the dynamic nature of the VFT (as shown in Supplementary Movies 1 and 2).
Cell culture, transfections, expression and inositol phosphate one (IP1) accumulation assay
HEK293 cells (ATCC CRL-1573) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS and were maintained at 37 °C in a humidified atmosphere with 5% CO2. Absence of mycoplasma was checked regularly to guarantee the use of mycoplasma-free cells. An alanine mutant library of the SNAP-tagged human truncated mGlu5 receptor (mGlu5-Δ856, designated as WT in the following experiments) was generated using Quick-change strategy (Agilent technologies) and verified by sequencing (Eurofins Genomics)9. Cells were transiently transfected using electroporation with the indicated mGlu5 construct. The DNA mixture for transient transfection included 0.6 µg of the indicated mGlu5 mutants, 2 µg of the glutamate transporter EAAC1 cDNA (to reduce the influence of glutamate that may remain in the assay medium as released by the cells) per 10 million of cells. Cells were seeded at the density of 100,000 cells per well into white bottom 96-well culture plates (Greiner Bio-one). Following 24 hours of transfection, expression and IP1 production were measured.
Wild-type and mutant mGlu5 receptor expression was determined by labelling the SNAP-tag receptors with BG- lumi4-Tb. In brief, cells were incubated for 1 hour at 37 °C with 100 nM of BG-Lumi4-Tb in 50 μL of glutamate-free DMEM GlutaMAX-I. Cells were then washed twice with Tag-Lite buffer, and then 100 μL of Tag-Lite buffer was added to each well (Revvity). Lumi4-Tb fluorescence was then measured with BMG Pherastar.
Before the commencement of the IP1 accumulation assay, HEK293 cells were incubated for 1.5 hours with glutamate-free DMEM GlutaMAX-I (Life Technologies), to reduce the extracellular concentration of glutamate. The IP1 accumulation assay kit (Revvity, France) was used for the direct quantitative measurement of IP1 in HEK293 cells transiently transfected with mGlu5 constructs. Cells were stimulated with various concentrations of allosteric compounds with a fixed concentration of the orthosteric ligand quisqualate (10 nM) for 30 minutes at 37 °C, 5% CO2. For the Schild experiments, cells were stimulated with various concentrations of quisqualate with a fixed concentration of the allosteric compounds. Then, cells were lysed using the lysis buffer for 15 min and half of the lysate was incubated with conjugate-lysis buffer containing the d2-labelled IP1 analogue and the terbium cryptate-labelled anti-IP1 antibody in a 384 well plate, according to the manufacturer’s instructions. After 1 hour incubation at RT, the HTRF measurement was performed after excitation at 337 nm with 50 μs delay, terbium cryptate fluorescence and tr-FRET signals were measured at 620 nm and 665 nm, respectively, using a PheraStar fluorimeter (BMG Labtech).
Data were fitted by a 3 parameters equation and statistical analyses were perfomed using one-way ANOVA followed by a Dunnett’s post-hoc test, using GraphPad Prism version 10 (San Diego, CA).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryoEM density map and model coordinates have been depositied in the Electron Microscopy Data Bank (EMDB) and the Protein Data Bank (PDB) respectively, under the accession code: EMD-37973 and PDB-8X0B (PAM1_d1, VU0424465- and quisqualate -bound mGlu5-5M, active state); EMD-37974 and PDB-8X0C (PAM1_d2_c1, VU0424465- and quisqualate -bound mGlu5-5M, active state); EMD-37975 and PDB-8X0D (PAM1_d2_c2, VU0424465- and quisqualate -bound mGlu5-5M, active state); EMD-37976 and PDB-8X0E (PAM1, VU0424465- and quisqualate-bound mGlu5-5M-W785A, active state); EMD-37977 and PDB-8X0F (PAM2, VU29- and quisqualate-bound mGlu5-5M, active state); EMD-37978 and PDB-8X0G (PAM3_c1, VU0409551- and quisqualate-bound mGlu5-5M, active state); EMD-37979 and PDB-8X0H (PAM3_c2, quisqualate-bound mGlu5-5M, intermediate state, Rcc). Source data are provided with this paper.
References
Sladeczek, F., Pin, J. P., Récasens, M., Bockaert, J. & Weiss, S. Glutamate stimulates inositol phosphate formation in striatal neurones. Nature 317, 717–719 (1985).
Gregory, K. J. & Goudet, C. International union of basic and clinical pharmacology. cxi. pharmacology, signaling, and physiology of metabotropic glutamate receptors. Pharm. Rev. 73, 521–569 (2021).
Pin, J.-P. & Bettler, B. Organization and functions of mGlu and GABAB receptor complexes. Nature 540, 60–68 (2016).
Wang, X. et al. Structural insights into dimerization and activation of the mGlu2-mGlu3 and mGlu2-mGlu4 heterodimers. Cell Res 33, 762–774 (2023).
Seven, A. B. et al. G-protein activation by a metabotropic glutamate receptor. Nature 595, 450–454 (2021).
Lin, S. et al. Structures of Gi-bound metabotropic glutamate receptors mGlu2 and mGlu4. Nature 594, 583–588 (2021).
Koehl, A. et al. Structural insights into the activation of metabotropic glutamate receptors. Nature 566, 79–84 (2019).
Du, J. et al. Structures of human mGlu2 and mGlu7 homo- and heterodimers. Nature 594, 589–593 (2021).
Nasrallah, C. et al. Agonists and allosteric modulators promote signaling from different metabotropic glutamate receptor 5 conformations. Cell Rep. 36, 109648 (2021).
Stansley, B. J. & Conn, P. J. Neuropharmacological insight from allosteric modulation of mglu receptors. Trends Pharm. Sci. 40, 240–252 (2019).
Nicoletti, F. et al. Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology 60, 1017–1041 (2011).
Lüscher, C. & Huber, K. M. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65, 445–459 (2010).
Dogra, S. & Conn, P. J. Metabotropic glutamate receptors as emerging targets for the treatment of schizophrenia. Mol. Pharm. 101, 275–285 (2022).
Parmentier-Batteur, S. et al. Mechanism based neurotoxicity of mGlu5 positive allosteric modulators–development challenges for a promising novel antipsychotic target. Neuropharmacology 82, 161–173 (2014).
Rook, J. M. et al. Unique signaling profiles of positive allosteric modulators of metabotropic glutamate receptor subtype 5 determine differences in in vivo activity. Biol. Psychiatry 73, 501–509 (2013).
Rook, J. M. et al. Biased mGlu5-positive allosteric modulators provide in vivo efficacy without potentiating mGlu5 modulation of NMDAR currents. Neuron 86, 1029–1040 (2015).
Sengmany, K. et al. Biased allosteric agonism and modulation of metabotropic glutamate receptor 5: Implications for optimizing preclinical neuroscience drug discovery. Neuropharmacology 115, 60–72 (2017).
Hellyer, S. D. et al. Probe dependence and biased potentiation of metabotropic glutamate receptor 5 is mediated by differential ligand interactions in the common allosteric binding site. Biochem Pharm. 177, 114013 (2020).
Wootten, D., Christopoulos, A., Marti-Solano, M., Babu, M. M. & Sexton, P. M. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 19, 638–653 (2018).
Chen, Y. et al. Interaction of novel positive allosteric modulators of metabotropic glutamate receptor 5 with the negative allosteric antagonist site is required for potentiation of receptor responses. Mol. Pharm. 71, 1389–1398 (2007).
Sengmany, K. et al. Differential contribution of metabotropic glutamate receptor 5 common allosteric binding site residues to biased allosteric agonism. Biochem Pharm. 177, 114011 (2020).
Gregory, K. J. et al. Investigating Metabotropic Glutamate Receptor 5 Allosteric Modulator Cooperativity, Affinity, and Agonism: Enriching Structure-Function Studies and Structure-Activity Relationships. Mol. Pharm. 82, 860–875 (2012).
Noetzel, M. J. et al. Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol. Pharm. 81, 120–133 (2012).
Shen, C. et al. Structural basis of GABAB receptor–Gi protein coupling. Nature 594, 594–598 (2021).
Gao, Y. et al. Asymmetric activation of the calcium-sensing receptor homodimer. Nature 595, 455–459 (2021).
He, F. et al. Allosteric modulation and G-protein selectivity of the Ca2+-sensing receptor. Nature 626, 1141–1148 (2024).
Olofsson, L. et al. Fine tuning of sub-millisecond conformational dynamics controls metabotropic glutamate receptors agonist efficacy. Nat. Commun. 5, 5206 (2014).
Lecat-Guillet, N. et al. Concerted conformational changes control metabotropic glutamate receptor activity. Sci. Adv. 9, eadf1378 (2023).
Doré, A. S. et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 511, 557–562 (2014).
hristopher, J. A. et al. Fragment and Structure-Based Drug Discovery for a Class C GPCR: Discovery of the mGlu5 Negative Allosteric Modulator HTL14242 (3-Chloro-5-[6-(5-fluoropyridin-2-yl)pyrimidin-4-yl]benzonitrile). J. Med. Chem. 58, 6653–6664 (2015).
Christopher, J. A. et al. Structure-based optimization strategies for g protein-coupled receptor (gpcr) allosteric modulators: a case study from analyses of new metabotropic glutamate receptor 5 (mGlu5) X-ray. Struct. J. Med. Chem. 62, 207–222 (2019).
Trinh, P. N. H., May, L. T., Leach, K. & Gregory, K. J. Biased agonism and allosteric modulation of metabotropic glutamate receptor 5. Clin. Sci. (Lond.) 132, 2323–2338 (2018).
Hellyer, S., Leach, K. & Gregory, K. J. Neurobiological insights and novel therapeutic opportunities for CNS disorders from mGlu receptor allosteric and biased modulation. Curr. Opin. Pharm. 32, 49–55 (2017).
Nasrallah, C. et al. Direct coupling of detergent purified human mGlu5 receptor to the heterotrimeric G proteins Gq and Gs. Sci. Rep. 8, 4407 (2018).
Sanchez-Garcia, R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 4, 874 (2021).
Gomeza, J. et al. The second intracellular loop of metabotropic glutamate receptor 1 cooperates with the other intracellular domains to control coupling to G-proteins. J. Biol. Chem. 271, 2199–2205 (1996).
Krishna Kumar, K. et al. Stepwise activation of a metabotropic glutamate receptor. Nature 629, 951–956 (2024).
Gregory, K. J. et al. Identification of specific ligand–receptor interactions that govern binding and cooperativity of diverse modulators to a common metabotropic glutamate receptor 5 allosteric site. ACS Chem. Neurosci. 5, 282–295 (2014).
Gregory, K. J. et al. Probing the metabotropic glutamate receptor 5 (mGlu5) positive allosteric modulator (PAM) binding pocket: discovery of point mutations that engender a ‘molecular switch’ in PAM pharmacology. Mol. Pharm. 83, 991–1006 (2013).
Vafabakhsh, R., Levitz, J. & Isacoff, E. Y. Conformational dynamics of a class C G protein-coupled receptor. Nature 524, 497–501 (2015).
Punjani, A. & Fleet, D. J. 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol. 213, 107702 (2021).
Afonine, P. V. et al. Conformational space exploration of cryo-EM structures by variability refinement. Biochim Biophys. Acta Biomembr. 1865, 184133 (2023).
Qiao, A. et al. Structural basis of Gs and Gi recognition by the human glucagon receptor. Science 367, 1346–1352 (2020).
Hilger, D. et al. Structural insights into differences in G protein activation by family A and family B GPCRs. Science 369, eaba3373 (2020).
Zuo, H. et al. Promiscuous G-protein activation by the calcium-sensing receptor. Nature 629, 481–488 (2024).
Arsova, A. et al. Positive allosteric modulators of metabotropic glutamate receptor 5 as tool compounds to study signaling bias. Mol. Pharm. 99, 328–341 (2021).
Venkatakrishnan, A. J. et al. Molecular signatures of G-protein-coupled receptors. Nature 494, 185–194 (2013).
Reeves, P. J., Callewaert, N., Contreras, R. & Khorana, H. G. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc. Natl Acad. Sci. USA 99, 13419–13424 (2002).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Scheres, S. H. W. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166 (2018).
Weis, F. & Hagen, W. J. H. Combining high throughput and high quality for cryo-electron microscopy data collection. Acta Crystallogr D. Struct. Biol. 76, 724–728 (2020).
Tan, Y. Z. et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D. Biol. Crystallogr 66, 486–501 (2010).
Lebedev, A. A. et al. JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr D. Biol. Crystallogr 68, 431–440 (2012).
Yamashita, K., Palmer, C. M., Burnley, T. & Murshudov, G. N. Cryo-EM single-particle structure refinement and map calculation using Servalcat. Acta Crystallogr D. Struct. Biol. 77, 1282–1291 (2021).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D. Struct. Biol. 75, 861–877 (2019).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D. Struct. Biol. 74, 531–544 (2018).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D. Biol. Crystallogr 67, 355–367 (2011).
Nicholls, R. A., Tykac, M., Kovalevskiy, O. & Murshudov, G. N. Current approaches for the fitting and refinement of atomic models into cryo-EM maps using CCP-EM. Acta Crystallogr. D. Struct. Biol. 74, 492–505 (2018).
Acknowledgements
We acknowledge the MRC Laboratory Molecular Biology EM and Scientific Computing facilities (Cambridge, UK), the European Synchrotron Radiation Facility for provision of beam time on CM01 and we would like to thank Eaazhisai Kandiah, Alessandro Grinzato and Gregory Effantin for assistance (Grenoble, France). We acknowledge the METi imaging facility, member of the national infrastructure France-BioImaging supported by the French National Research Agency (ANR-10-INBS-04). We acknowledge the platform of pharmacology Arpeges of the Institut de Génomique Fonctionnelle (Montpellier, France), and Eric Mauger from IT department. We acknowledge the National cryoEM Facility at Bangalore for data collection and computational support. We acknowledge Lourdes Muñoz and Carme Serra from SimChem (IQAC-CSIC) for analytical and synthetic support. L.B. was supported by Ligue Contre le Cancer. G.F. was supported by SWITCH-ON (ANR-20-CE11-0019). J.F.-I., and A.L. were supported by Ministerio de Ciencia e Innovación, Agencia Estatal de Investigación and ERDF - A way of making Europe (PID2020-120499RB-I00) and by the Catalan government (2021 SGR 00508). G.C. was supported by Medical Research Council grant MC-U105184322. K.R.V. acknowledges the DBT B-Life grant DBT/PR12422/MED/31/287/2014, and the support of the Department of Atomic Energy, Government of India, Government of India, under Project Identification No. RTI4006. KRV is part of EMBO Global Investigator Network. G.L. was supported by SWITCH-ON (ANR-20-CE11-0019) and Ligue Contre le Cancer.
Author information
Authors and Affiliations
Contributions
G.C. and L.B. contributed equally to the work and share the co-first authorship. L.B. performed expression and purification of the full-length mGlu5-5M bound to quisqualate and three different PAM samples for cryoEM analysis, with the help of G.F. L.B. and A.F. performed grid freezing. G.C. prepared cryoEM grids, screened, collected and processed cryoEM data. S.B. performed grid screening. A.K. performed data collection at the TFS cryoEM facility. F.M. performed and analysed pharmacological experiments on the mGlu5 receptor and receptor mutants under the supervision C.G. J.F.I. synthesised the VU0424465 under the supevision of A.L. K.R.V. and G.L. performed image processing and building/refinement of full-length mGlu5-5M models. K.R.V. and G.L. interpreted the data and wrote the manuscript with contributions from all the authors. G.L. designed, supervised and coordinated the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Shane Hellyer and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cannone, G., Berto, L., Malhaire, F. et al. Conformational diversity in class C GPCR positive allosteric modulation. Nat Commun 16, 619 (2025). https://doi.org/10.1038/s41467-024-55439-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-55439-9
