
Abstract
Brome Mosaic Virus (BMV) utilizes a tRNA-like structure (TLS) within its 3’ untranslated region to mimic host tRNA functions, aiding aminoacylation and viral replication. This study explores the structural dynamics of BMV TLS interacting with tyrosyl-tRNA synthetase (TyrRS) during aminoacylation. Using cryo-EM, we capture multiple states of the TLS-TyrRS complex, including unbound TLS, pre-1a, post-1a, and catalysis states, with resolutions of 4.6 Å, 3.5 Å, 3.7 Å, and 3.85 Å, respectively. These structural comparisons indicate dynamic changes in both TLS and TyrRS. Upon binding, TLS undergoes dynamic rearrangements, particularly with helices B3 and E pivoting, mediated by the unpaired A36 residue, ensuring effective recognition by TyrRS. The dynamic changes also include a more compact arrangement in the catalytic center of TyrRS and the insertion of 3’ CCA end into the enzyme’s active site, facilitating two-steps aminoacylation. Enzymatic assays further demonstrated the functional importance of TLS-TyrRS interactions, with mutations in key residues significantly impacting aminoacylation efficiency. Furthermore, Electrophoretic Mobility Shift Assay (EMSA) demonstrated that BMV TLS binds elongation factors EF1α and EF2, suggesting a multifaceted strategy to exploit host translational machinery. These findings not only enhance our knowledge of virus-host interactions but also offer potential targets for antiviral drug development.
Similar content being viewed by others

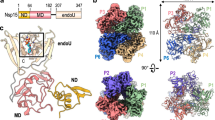

Introduction
The Brome Mosaic Virus (BMV) is a well-characterized member of the Bromoviridae family, notable for its tripartite, positive-sense RNA genome1. Each RNA segment encodes essential proteins required for the virus’s replication and host infection processes2. Common among these segments is the presence of a tRNA-like structure (TLS) at the 3′ untranslated region3,4, which mimics the physical and functional properties of cellular tRNAs5. These TLS structures exhibit various biological functions, including the regulation of RNA replication, enhancement of translation, RNA recombination, and viral assembly6,7,8,9. The mechanism by which TLS mimics tRNA involves its secondary and tertiary structures, which are similar to those of tRNA10. TLS typically possesses receptor arms and anticodon loops that resemble tRNA, and it may affect the translation process by forming analogous secondary and tertiary structures11. For instance, TLS in certain viral RNAs can bind to host aminoacyl-tRNA synthetases (aaRS)12, thereby simulating the aminoacylation process, which may aid in the translation and replication of viral RNA. Furthermore, TLS may enhance translation efficiency through interactions with the ribosome or other translation factors, similar to the role of tRNA in protein synthesis13.
Aminoacylation is a fundamental biochemical process by which tRNA molecules are charged with their respective amino acids14, enabling protein synthesis via translation15. This process, central to the life of all cellular organisms, involves the precise addition of an amino acid to the 3′ end of a tRNA molecule by aaRS16. In detail, the tRNA aminoacylation process includes two steps17: (1) amino acid activation by reacting with ATP to form an aminoacyl-adenylate intermediate (aminoacyl-AMP) and releases pyrophosphate (PPi). (2) Transfer of the amino acid: the aminoacyl group from the aminoacyl-AMP is transferred to the 2′ or 3′ hydroxyl group of the ribose at the tRNA’s 3′ end. This reaction releases AMP, resulting in a charged tRNA (aminoacyl-tRNA)18. The aminoacylated tRNA then associates with elongation factors and enters the ribosome to participate in protein translation19,20. BMV TLS undergoes a similar aminoacylation process12, but the precise molecular interactions and conformational changes remain largely unexplored. Understanding the theoretical underpinnings of aminoacylation in RNA-like structures, particularly those mimicking tRNA such as the BMV TLS, is essential for grasping how certain viruses can manipulate host cell machinery to facilitate their own replication and protein synthesis21. Structurally, BMV TLS can adopt conformations that allow it to be recognized by host aaRSs, specifically tyrosyl-tRNA synthetase (TyrRS), to charge the viral RNA with an amino acid, mimicking a tRNA function22.
Recent studies, particularly the landmark paper by Bonilla et al.23, have significantly advanced our understanding of the structural dynamics of viral TLS. Bonilla and colleagues utilized cryo-electron microscopy (cryo-EM) to obtain several conformations, both in isolation and when complexed with TyrRS, revealing the intricate conformational changes that BMV TLS undergoes to hijack the host’s TyrRS. While Bonilla et al.’s study provided groundbreaking insights, several limitations remain. The resolutions of the captured conformations were insufficient to detail specific molecular interactions within the BMV TLS-TyrRS complex, leaving questions about the precise nature of these interactions. Additionally, their study primarily focused on static snapshots of the TLS-TyrRS interactions, without fully elucidating the dynamic process of aminoacylation over time. Moreover, their work did not explore the potential impact of TLS on host cellular functions beyond the immediate interaction with TyrRS.
Given these gaps, our study aimed to build upon the foundational work of Bonilla et al. by providing a more detailed and dynamic analysis of BMV TLS during aminoacylation. We employed cryo-EM to capture not only the static conformations but also the transitional states of BMV TLS as it interacts with TyrRS. Four structures including unbound TLS (169-nt), pre-1a state (before amino acid activation), post-1a state (after amino acid activation), and the catalysis state (ready for transfer of the amino acid) were determined at 4.6, 3.5, 3.7, and 3.85 Å resolution, respectively, were characterized. These resolutions enabled us to determine atomically detailed structures, filling a critical gap in current understanding—how BMV TLS dynamically interacts with TyrRS during aminoacylation. Additionally, we investigated the functional consequences of these interactions on the efficiency of aminoacylation and subsequent steps in protein synthesis. By addressing these aspects, our research seeks to provide a more comprehensive understanding of the molecular mechanisms underpinning BMV TLS functions and its exploitation of host machinery, thereby offering potential avenues for antiviral intervention.
Results
Cryo-EM analysis of BMV TLS-TyrRS undergoing aminoacylation
To verify the catalytic capability of TyrRS with TLS, we first prepared BMV TLS through in vitro transcription (IVT) reactions to achieve high conformational homogeneity (Fig. 1a). We then conducted in vitro aminoacylation assays, demonstrating the enzymatic function of TyrRS towards BMV TLS (Fig. 1b). Various specimens were prepared to capture different conformations of TLS-TyrRS engaged in this biochemical process (Fig. 1c, d). Unbound TLS and TLS-TyrRS in the pre-1a state were obtained from the TLS-TyrRS complex preparation without adding ATP and Tyr, capturing them before the first-step aminoacylation. The post-1a state of TLS-TyrRS was acquired by preparing TLS-TyrRS complex in the presence of ATP and Tyr; however, the 2′ and 3′ hydroxyl groups at the TLS 3′ end were circularized (2′–3′ cP), preventing progression to the second step. To capture the catalytic state during the second-step aminoacylation, the assay was conducted with ATP and Tyr, where CaCl2 was added to slow down the reaction, allowing us to capture the active state (Supplementary Figs. 1–4).

a Native PAGE demonstrating the high conformational homogeneity of BMV TLS. More than 3 times this experiment was repeated independently with similar results. b In vitro aminoacylation assay comparing the activity of TyrRS with TLS and tRNA (both at the same final concentration). Data are shown as the mean ± SD (n = 3 independent experiments). Source data are provided as a Source Data file. This assay includes three controls: “TLS (no TyrRS)” indicates a reaction system without the addition of TyrRS; “TyrRS (no TLS)” indicates a reaction system without BMV TLS; and “TLS (2′–3′ cP)” refers to TLS where the 3′ terminal adenine is circularized into a 2′–3′ adenosine cyclic phosphate, which prevents aminoacylation at the 2′-OH group of the ribose. c Schematic illustration of the aminoacylation process catalyzed by TyrRS. The differently colored cylinders represent various TLS helices; gray lines between cylinders indicate the connections between helices; Magenta and cyan blocks represent the two subunits of TyrRS; the yellow star represents YMP; and the green arrow indicates the movement of TLS Helix A. d Cryo-EM maps showing various conformations of TLS-TyrRS engaged in aminoacylation. The maps correspond to the states illustrated in (c), from left to right.
A36-mediated dynamic change is crucial for TyrRS recognition
The BMV TLS comprises three groups of stacked helical domains, each exhibiting substantial functional diversity and complexity (Fig. 2a). The first domain includes a pseudoknot structure formed by Helices C, B1, and B2, with the apical loops of Helices B2 and C oriented in opposite directions. The second domain features a four-way junction connecting Helices A, D, C, and B1. In this configuration, Helix A forms a pseudoknot and functions as the acceptor stem, terminating in the conserved CCA end typical of tRNAs. The 2′-OH group on the terminal adenine’s ribose ring in Helix A can be aminoacylated with tyrosine (Tyr) by Class Ic TyrRS24. Helix A is linked to B2 via a wobble loop. The third domain consists of the dynamic Helices E and B3, which are positioned nearly perpendicular to the acceptor stem25. Helices E and B3 are connected to the TLS core structure via the unpaired A36, with Helix B3 functioning as the analog to the tRNA anticodon stem loop.

a Schematic representation of the unbound TLS structure. The left panel shows the cryo-EM map and atomic model of the unbound TLS, while the right panel illustrates its secondary structure. b Superimposed model and map of TLS-TyrRS in the pre-1a state. c Domain organization of a single TyrRS subunit. d Comparison of the unbound TLS structure (left, transparent with colors) and the bound TLS structure (right, solid colors), demonstrating conformational changes upon binding. e Overlay of the B1, B3, and E helices in the two states of TLS, along with a magnified view of the detailed alignment at residue A36 (transparent colors for unbound TLS, solid colors for bound TLS). Helix B1 remains almost unchanged, while A36, located at the junction of helices B1 and B3, shows an 8-Å displacement in the phosphate backbone. A cartoon schematic (right) illustrates how A36 mediates the dynamic rearrangement of helices E and B3. f In vitro aminoacylation assay to confirm the importance of A36. Data are shown as the mean ± SD (n = 3 independent experiments). Source data are provided as a Source Data file.
In the pre-1a state, TyrRS exists as a homodimer with aminoacylation activity, featuring an N-terminal catalytic domain composed of 11 alpha-helices surrounding four antiparallel beta-sheets in a Rossman fold arrangement. The C-terminal domain, composed of five alpha-helices and two beta-sheets, serves as the anticodon recognition region, connected by a loop of 38 amino acids (Fig. 2b, c). Comparisons between bound and unbound TLS revealed significant conformational rearrangements: in the unbound state, Helices E and B3 are approximately perpendicular to the acceptor arm, whereas in the bound state, they are parallel (Fig. 2d). Structural alignment indicates a ~90° rotation in Helices E and B3 between the two states, with Helix B1, closely following B3, remaining almost entirely superimposed and rigid (Fig. 2e). At the junction between B3 and B1, the unpaired A36 causes an 8-Å displacement in the phosphate backbone (Fig. 2e). Based on structural alignment and the distinct spatial position of A36, it was hypothesized that the A36 pivot point plays a crucial role in the dynamic changes of Helices E and B3. Enzymatic assays showed that the A36C mutant, forcing the A36 pivot to form a base pair (G129:C36), retained only 64% of the wild-type activity. In contrast, mutating the adjacent C128:G37 base pair to A128:G37 mismatch increased enzyme activity to ~119% of wild-type (Fig. 2f). This suggests that during the recognition process of TyrRS, TLS finely regulates the conformational changes of Helices E and B3 through the dynamic nature of unpaired pivot A36 to ensure efficient mutual recognition between TLS and TyrRS. The extensive conformational rearrangements necessary for TLS to bind TyrRS highlight a dynamic and complex interaction not present in tRNA-TyrRS recognition26, deepening our understanding of the dynamism and complexity of plant RNA virus TLS.
Detailed interactions between TLS and TyrRS
In the pre-1a state, structural analysis indicates that TLS binds to the positively charged regions of TyrRS, forming three sets of contact interfaces involving the acceptor stem, the B3 domain, and the wobble loop of TLS with the two subunits of TyrRS (Fig. 3a). Specifically, the catalytic domain of subunit 1 recognizes the acceptor arm of TLS, while the anticodon-binding domain of subunit 2 interacts with the B3 domain. Both catalytic domains of subunit 1 and subunit 2 collectively recognize the wobble loop (Fig. 3b–d). The interfacial area between TLS and TyrRS is larger than that between tRNATyr and TyrRS, which may explain the higher affinity and tyrosylation activity of TyrRS for TLS than tRNATyr (Fig. 1b and Supplementary Fig. 5a–c).

a Superimposed model and map of the pre-1a state, showing three sets of interaction interfaces. Black boxes indicate the regions will be shown in (b–d). b Detailed interactions between the TLS’s B3 domain and the anticodon-binding domain of TyrRS subunit 2. c Detailed interactions between the TLS’s A domain and the catalytic domain of TyrRS subunit 1. Black dashed lines indicate hydrogen bonds. d Detailed interactions between the TLS’s wobble loop and the TyrRS’s catalytic domains. e Validation of key residues for TyrRS’s enzymatic activity with TLS. “3xRA” represents the combined mutations of R171A, R174A, and R217A. Data are shown as the mean ± SD (n = 3 independent experiments). Source data are provided as a Source Data file. (f) Validation of key residues for TyrRS’s binding affinity with TLS. Data are shown as the mean ± SD (n = 3 independent experiments). Source data are provided as a Source Data file.
Further examination revealed that the anticodon-binding domain of subunit 2 primarily interacts with the B3 domain in a sequence-nonspecific manner, differing from the recognition patterns reported in TyrRS-tRNATyr interactions26,27. Notably, basic amino acid K277 forms a hydrogen bond with the phosphate backbone of C21 (cytosine 21), and C281 (cysteine 281) forms a hydrogen bond with the N3 atom of the second base, A24, of the 4-nt apical loop (5′UACA3′). The first base, U23, of the apical loop does not fully flip out; instead, its sugar ring faces the protein side, allowing K341 to form a hydrogen bond with the O2 atom of U23’s sugar ring (Fig. 3b). Additionally, A24 and C25 flip outward and are positioned within a hydrophobic patch formed by Y280, P282, P283, and P337. Single mutations of K277 and K341 to alanine didn’t significantly affect enzyme activity, but the double mutation reduced activity to 65% of the wild type, and combined mutations also led to a noticeable reduction in affinity (Fig. 3e, f). These results demonstrated that K277 and K341 in the anticodon-binding domain collectively played a role in TLS binding, and combined mutations significantly weakened binding affinity and greatly reduced enzyme activity.
Although the B3 domain of TLS structurally mimics a tRNA anticodon stem-loop for recognizing TyrRS’s anticodon-binding domain, its sequence and structure differ from the tRNA anticodon stem-loop, which potentially leads to the non-specific binding pattern of TLS with TyrRS. Typically, the anticodon stem-loop of tRNATyr consists of a 5-bp stem and a 7-nt loop28, whereas the BMV Tyr-charging TLS B3 domain has an 8-bp stem and a 4-nt loop. This arrangement gives the B3 domain a more complex and stable helical structure, maintaining overall rigidity despite positional changes. Furthermore, the tRNATyr anticodon loop contains a conserved Tyr anticodon sequence (GUA)29. In contrast, the 4-nt apical loop of BMV TLS has a conserved 5′UACA3′ sequence, which does not correspond to a Tyr anticodon but instead to the anticodons of cysteine and valine. This phenomenon is also common in the TLS family, as exemplified by the histidine-charging TLS of Tobacco Mosaic Virus (TMV)30. The specific reasons why plant virus TLS anticodon stem-loop mimic domains contain non-self-anticodons remain unknown, necessitating further in-depth research in molecular biology, virology, and botany.
Moreover, subunit 1’s catalytic domain plays a critical role in recognizing the acceptor arm of TLS. Residues R171 and R174 of subunit 1 bind to the phosphate backbone of TLS’s acceptor arm at U51 and U52, and R217 binds to U51’s phosphate backbone (Fig. 3c). Single mutations at these sites (R171A, R174A, and R217A) resulted in almost complete loss of enzyme activity, indicating these sites are crucial for correct localization of TLS on TyrRS (Fig. 3e). Affinity tests showed some variation among these sites, with R217 having a more pronounced impact on affinity (Fig. 3f). In the pre-1a state, although the pseudoknot-like acceptor arm of TLS contacts TyrRS’s catalytic domain, the 3′ CCA end extends outward, away from the enzymatic center, placing it in a pre-catalytic state. Additionally, besides the detailed interactions between TyrRS and TLS’s anticodon stem-loop and acceptor arm, TyrRS also contacts TLS’s wobble loop, an 8-nt single loop at the center of TLS connecting the acceptor arm and B2 domain, which does not exist in classic tRNATyr structures31. At the junction between TLS’s acceptor arm and wobble loop, interactions occur with TyrRS’s catalytic domains. Subunit 1’s N168 forms two hydrogen bonds with TLS’s phosphate backbone, firmly anchoring the junction. Subunit 2’s R181 specifically interacts with U158 (Fig. 3d). Mutations at these sites retained 81% and 14% enzyme activity, respectively (Fig. 3e, f). Furthermore, we conducted EMSA assays with various TyrRS mutants and compared their affinities for TLS and tRNA (Supplementary Fig. 5d) to identify specific sites distinguishing between TLS and endogenous tRNA. Mutations at R217, N168, and R181 were notably impactful; both R217A and R181A mutations drastically reduced TLS affinity with minimal effect on tRNA, while N168A produced little change in TLS binding but slightly reduced tRNA affinity. Further in vitro enzymatic assays revealed that R217A and R181A, while reducing tRNA activity by 40-50%, led to near-complete activity loss in TLS (Supplementary Fig. 5e). Structural analysis localized R217 and R181 at TLS’s pseudoknot-like acceptor arm and wobble loop, respectively (Fig. 3c, d). The acceptor arm and the wobble loop are critical structural features distinguishing TLS from tRNA, which may also contribute to the differences in their recognition mechanisms. Taken together, our affinity, activity, and structural data indicate that R217 and R181 are possible residues distinguishing viral from endogenous substrates, providing a potential way to interfere with viral invasion.
Overall, TyrRS binds to TLS’s tRNA-like acceptor arm and anticodon stem loop, with an additional interaction interface between TyrRS and TLS’s loop. This enables TLS to mimic the key functional domains of tRNATyr binding to TyrRS32, while also featuring interaction modes.
Substrate recognition and aminoacylation dynamics in TyrRS
When comparing subunit 1 and subunit 2 of TyrRS in the pre-1a state, an additional density was observed in the enzymatic center of subunit 2, which perfectly accommodates ATP, while ATP binding in the subunit 1 was not evident. Interaction analysis revealed that multiple hydrogen bonds form between TyrRS and ATP, including OE1E68:O2BATP and OGS70:O1GATP (Fig. 4a). Notably, ATP was not externally added during the in vitro preparation of the pre-1a state sample, indicating that the ATP found in subunit 2 is endogenous to TyrRS.

a ATP-binding pocket in the subunit 2 of TyrRS in the pre-1a state, showing detailed interactions between ATP and key residues. Black dashed lines represent hydrogen bonds. b YMP-binding pocket in the subunit 1 of TyrRS in the post-1a state (YMP1). Detailed interactions between YMP1 and key residues are shown, with black dashed lines representing hydrogen bonds. c YMP-binding pocket in the subunit 2 of TyrRS in the post-1a state (YMP2). d Comparison between the YMP-binding pockets in the post-1a state. The black arrow indicates that YMP1 is positioned closer to the TLS acceptor arm (forest green) compared to YMP2. e Comparison of the subunit 2’s catalytic domain between the pre-1a state (transparent colors) and post-1a state (solid colors), colored by C-alpha RMSD in ChimeraX. ATP from the pre-1a state is shown in gray, and YMP2 from the post-1a state is shown in black. The black dashed boxes highlight the regions with the most significant structural changes, specifically α-helix 105–113 and loop 180–190. Red arrows indicate the direction of movement, with the catalytic domain of subunit 2 in the post-1a state shifting closer to the active site.
To investigate how ATP and Tyr participate in the aminoacylation of BMV TLS by TyrRS, ATP and Tyr were added to the pre-1a state sample, thereby preparing the BMV TLS-TyrRS-ATP-Tyr complex (post-1a state). Unlike the pre-1a state, densities corresponding to Tyr-AMP (YMP) were observed in both subunit 1 (YMP1) and subunit 2 (YMP2) in the post-1a state (Fig. 4b, c). Despite the numerous hydrogen bonds formed by TyrRS with YMP1 and YMP2, distinct interaction residues lead to a variation in the YMP orientation, positioning YMP1 in closer proximity to the acceptor arm (Fig. 4d). The distinct recognition modes of the two YMP pockets suggest the varying functional states of the homodimer subunits and reconfirm previous findings that TyrRS naturally forms an asymmetric homodimer33,34,35.
Further comparison of subunit 2 of TyrRS in the pre-1a state and post-1a state revealed dynamic changes in the catalytic domain. Specifically, Loop 180–190 and Helix 105–113 are positioned closer to the catalytic center in the post-1a state, resulting in a more compact enzymatic pocket (Fig. 4e). This indicates that the catalytic center of TyrRS dynamically changes during substrate recognition, becoming more compact to achieve precise and efficient substrate recognition. Next, we also investigated whether the substrate recognition and aminoacylation dynamics depend on binding with TLS or tRNA. By aligning the TyrRS structures of Plasmodium falciparum (pf) and Escherichia coli, we uncovered key insights into the conserved and distinct domains of TyrRS across species. Sequence alignments showed that while the catalytic domain is highly conserved, PHAVU TyrRS (in our study) aligns more closely overall with PfTyrRS, while its anticodon recognition domain differs significantly from that of E. coli TyrRS. Structural comparison revealed that YMP within TyrRS subunit 1, which binds TLS, adopts a distinct orientation-tilting closer to the TLS receptor arm compared to YMP in PfTyrRS-YMP and E. coli TyrRS-YSA (Supplementary Fig. 6). This conformational shift suggests that as TyrRS engages TLS, the substrate YMP tilts toward the RNA arm and TLS becomes closer to the YMP pocket, thereby illustrating a dynamic recognition mechanism between aminoacyl synthetases and viral RNA.
Structural transition in TLS during aminoacylation by TyrRS
By comparing the pre-1a state with the catalytic state, we observed significant positional changes of BMV TLS relative to TyrRS. From the top view, TLS rotates approximately 30° around TyrRS (Fig. 5a, b). In the pre-1a state, although there is some contact between the pseudoknot-like acceptor arm of TLS and TyrRS, the single-stranded 3′ CCA end of the acceptor arm points outward, away from the catalytic center. In contrast, in the catalytic state, the entire acceptor arm of BMV TLS faces towards TyrRS, with the 3′ CCA deeply embedded into the catalytic center of TyrRS. The phosphodiester backbone at the end of the acceptor arm differs by as much as ~29 Å in length between the two states (Fig. 5c). Unfortunately, due to the insufficient resolution possibly derived from the on-going catalytic process (Supplementary Fig. 4e), no further changes were observed in the catalytic center.

a Superimposed models and maps of the catalysis state. b Comparison between the pre-1a state (gray) and the catalysis state (colored). TLS rotates approximately 30° around TyrRS, with the black arrow indicating the direction of rotation. c Close-up structural comparison between the pre-1a state (gray) and the catalysis state (colored). The black dashed line represents the distance between the phosphate backbones of residue A169 at the 3′ end of the TLS acceptor arm in both states, measuring approximately 29 Å. d Top 15 of GO Enrichment. Black asterisks highlight translation and translational elongation Biological Processes (GOTERM-BP). Biotin-antisense TLS is control group. Data are shown as the mean (n = 2 independent experiments). Source data are provided as a Source Data file. e Binding affinity of BMV TLS-EF1α and BMV TLS-EF2. Binding curves were generated from EMSA experiments (n = 3 independent experiments). Data are shown as the mean ± SD. Source data are provided as a Source Data file.
These structural comparisons throughout the aminoacylation process indicate dynamic changes in both TyrRS and BMV TLS (Fig. 6 and Supplementary Fig. 7). Initially, TLS undergoes conformational rearrangements, with the B3 and E helices shifting from a vertical to a horizontal orientation to bind with the TyrRS anticodon-binding domain, forming three interaction interfaces with TyrRS. Subsequently, to function effectively, the 3′ end of the acceptor arm moves from being distant from the TyrRS catalytic center to extending into it. Concurrently, TyrRS, having bound the substrates ATP and Tyr and catalyzed them into YMP, sees its catalytic center contract inward. As the TLS acceptor arm penetrates deeper, the catalytic center contracts further. These findings provide a deeper understanding of the aminoacylation process of BMV TLS by TyrRS, highlighting the dynamic interplay and structural adaptability required for efficient enzymatic function. The sequence of these conformational changes underscores the intricate coordination between the enzyme and its substrate, emphasizing the importance of structural flexibility in achieving precise biochemical interactions.

Upon binding with TyrRS, BMV TLS undergoes significant conformational rearrangements, which allow it to be recognized by TyrRS to charge with Tyr, mimicking a tRNA function. The Tyr-charging TLS then binds with EF1α or EF2 to manipulate the host’s translational machinery to its advantage. Created in BioRender. N′N′, OY. (2025) https://BioRender.com/q47d213.
BMV TLS binding to elongation factors
Elongation factors are essential for the translation process in cells. Previous studies have indicated that plant virus TLSs bind with elongation factor 1α (EF1α) to exert their functions36,37,38. In this study, we aimed to investigate the interactions between BMV TLS and host cellular components, particularly focusing on the elongation factors. Understanding these interactions could elucidate the mechanisms through which viruses exploit host cellular machinery to facilitate their own protein synthesis, thereby enhancing viral replication and pathogenesis.
To explore these interactions, we first performed Biotinylated RNA pull-down coupled with mass spectrometry, which enables the identification of TLS-binding proteins. Using in vitro synthesized BMV TLS and protein extracts derived from Nicotiana tabacum L.’s leaves, mass spectrometry results demonstrated the enrichment of ribosome-associated proteins (Fig. 5d), especially elongation factors EF1α and EF2 (Supplementary Fig. 9). We then validated these findings using electrophoretic mobility shift assay (EMSA). The EMSA results showed a clear shift in the mobility of TLS when incubated with EF1α, indicating the formation of stable complexes between TLS and EF1α (Kd = 2.60 ± 0.05 μM). Similarly, the binding interaction between TLS and EF2 (Kd = 4.22 ± 0.20 μM) was evident from the mobility shifts observed in the EMSA (Fig. 5e and Supplementary Fig. 8).
As previously reported, EF1α primarily delivers aminoacyl-tRNAs to the ribosome, while EF2 is responsible for translocating the ribosome along the mRNA strand, facilitating the elongation phase of protein synthesis39,40. The ability of BMV TLS to bind both elongation factors implies a multifaceted strategy by BMV to harness host translational machinery. This dual interaction may not only disrupt the normal function of these critical factors but could also reroute these components to favor viral protein synthesis. Understanding such interactions is crucial for elucidating the molecular basis of how BMV manipulates host cell processes to its advantage.
Discussion
Our study has provided critical insights into the structural dynamics of the BMV TLS when interacting with TyrRS. The comparative analysis revealed significant conformational changes between the unbound, pre-aminoacylation, and post-aminoacylation states, depending on the state of interaction with TyrRS. Detailedly, the unbound TLS adopts a relaxed or extended conformation that is markedly different from when it is engaged with TyrRS. This conformation may be necessary for the TLS to be recognized and bound by the enzyme. Upon binding with TyrRS, the TLS often undergoes significant conformational rearrangements, in contrast to tRNA’s interaction with TyrRS, where the overall structure of tRNATyr remains largely stable, preserving its characteristic L-shape. Particularly, the dynamic pivoting of helices B3 and E, mediated by the unpaired A36 residue, is crucial for effective interaction. These rearrangements ensure that the acceptor arm of TLS correctly aligns with the catalytic center of TyrRS. Our findings confirm the hypothesis that the A36 pivot plays a pivotal role in regulating these conformational changes, thereby ensuring efficient recognition and binding by TyrRS. The conformational changes can also include a more compact arrangement in the catalytic center of TyrRS, bringing specific reactive groups into proximity with the enzyme’s active site, facilitating the first-step aminoacylation of TLS. This detailed structural understanding addresses the limitations of previous studies, such as the work by Bonilla et al.23, which indicate the TLS-TyrRS interactions but lack details (Supplementary Fig. 9). Notably, in the pre-1a state, we only clearly detected ATP in subunit 2; subunit 1 did not exhibit distinct ATP signals, which plausibly due to its flexible positioning and limited resolution, rather than a complete absence of ATP. This deduction is derived from existing homologous structures and biochemical studies showing that both subunits of TyrRS indeed associate with ATP41,42,43 and the fact that Tyr-AMP is clearly visible in both subunits of the post-1a state. Besides, in our study, the resolution of TLS-TyrRS in the catalytic state limits the detailed analysis of precise molecular interactions and conformational changes during the second step of aminoacylation. Achieving its higher-resolution structure could further refine our understanding of the molecular mechanisms at play.
The structural flexibility of BMV TLS is a key factor in its ability to mimic host tRNA and hijack the host’s translational machinery. The ability of TLS to undergo substantial conformational changes allows it to effectively interact with TyrRS and possibly other host factors. During translation, elongation factors are crucial for the extension phase. Following aminoacylation, TLS may bind to these elongation factors, thereby influencing the host’s translation process. The interactions between BMV TLS and elongation factors EF1α and EF2, as demonstrated by our EMSA results, suggest a multifaceted strategy where the virus not only hijacks aminoacyl-tRNA synthetase activity but also the elongation phase of protein synthesis (Fig. 6). This dual manipulation likely enhances the efficiency of viral replication and protein synthesis, providing the virus with a robust mechanism to ensure its propagation within the host. Additionally, for some unknown reason, it is also possible that TLS might enter the ribosome and prematurely release proteins at Tyr codons, thereby allowing only non-Tyr or low-Tyr-containing proteins to be synthesized. In the future, it will be intriguing to explore the structural basis of TLS-elongation factor and TLS-ribosome interactions through high-resolution structural studies and the precise implications of these interactions on the efficiency of translation of both viral and host proteins. Such studies could provide deeper insights into the viral manipulation of host machinery.
BMV TLS binds the TyrRS dimer asymmetrically, with the TLS acceptor arm recognizing the catalytic domain of subunit 1 and the B3 domain binding the anticodon-binding domain of subunit 2. This interaction closely resembles the canonical recognition pattern of tRNA-TyrRS. However, BMV TLS introduces an extra binding interface with TyrRS through its wobble loop, a feature absent in the classical tRNA structure. Compared to tRNA, this similar yet distinct recognition pattern between BMV TLS and TyrRS suggests that the TLS of plant viruses may have originated from tRNA molecules but evolved additional structural features for more efficient host aaRS hijacking. This also implies that TyrRS may employ the same molecular mechanism to catalyze the aminoacylation of TLS, although further investigation is required to fully elucidate this process.
Comparative analysis with other viral systems reveals that while the fundamental mechanisms of tRNA mimicry and aminoacylation are conserved, the specific structural adaptations and interactions can vary significantly. For instance, the structural features of BMV TLS, such as the extended B3 domain and its interactions with TyrRS, differ from those observed in Turnip Yellow Mosaic Virus (TYMV) and other plant viruses21,44. These differences highlight the evolutionary diversity among viruses in their strategies to exploit host cellular machinery.
Our study not only enhances our understanding of the structural and functional dynamics of BMV TLS but also sets the stage for future research aimed at targeting its interactions with host machinery for antiviral therapy. By elucidating the detailed mechanisms of TLS conformational plasticity and their implications for viral replication, we provide potential avenues for developing inhibitors that can disrupt the critical interactions. Future studies should focus on exploring the structural dynamics of other viral TLSs and their interactions with host factors to uncover universal or distinct targets for broad-spectrum antiviral strategies.
Methods
In vitro transcription, purification, and folding of RNAs
To initiate the process, we added a T7 promoter and two “GG” nucleotides in front of the BMV TLS DNA sequence and cloned this into the pUC19 vector. We amplified the double-stranded DNA fragments via PCR to serve as templates for in vitro transcription. During transcription, T7 RNA polymerase can exhibit heterogeneity, producing N + 1 or longer transcripts. To mitigate this, the reverse primer was designed with the two penultimate nucleotides at the 5′ end modified with 2′-O-methyl groups. The transcription mixture included 1 × transcription buffer [40 mM Tris-HCl, 1 mM spermidine, 0.01% (v/v) Triton X-100, pH 8.1], 10 mM DTT, 4 mM NTP, 45 mM MgCl2, 0.3 mg/mL T7 RNA polymerase, and 10 ng/µL DNA template. The reaction was conducted at 37 °C for 3 h followed by a 20-minute heat inactivation at 72 °C. The reaction was terminated by adding EDTA to a final concentration of 50 mM to chelate magnesium ions, followed by precipitation with three volumes of ethanol and NaCl, and stored at −80 °C overnight to precipitate RNA. RNA was purified using 8% denaturing PAGE, and eluted into ultrafiltration centrifuge tubes (Millipore). Then, 2 M NaCl and DEPC-treated water were added separately to the ultrafiltration centrifuge tubes to replace the buffer.
The purified RNA was then subjected to in vitro folding as follows: The RNA was transferred to an RNase-free PCR tube, and 50 mM HEPES-Na (pH 8.0) was added. The RNA was heated in a PCR machine set to 90 °C for 3 min. Afterward, the RNA was allowed to sit at room temperature for 10 min before adding 5 mM MgCl2. The RNA was then returned to the PCR machine, set at 50 °C for 20 min. Following a 3-minute rest at room temperature, the RNA was stored on ice for further complex preparation.
BMV TLS (2′–3′ cP) was engineered by adding a 69-nt hepatitis delta virus (HDV) self-excising ribozyme to the DNA template of TLS to generate a 2′-3′ cyclic phosphate at the TLS 3′ end, preventing its aminoacylation by TyrRS. TLS mutants were generated from the wild-type TLS DNA template through site-directed mutagenesis on the primers, and linearized fragments with mutations were obtained via PCR (oligonucleotides are provided in Supplementary Data 1). Then phosphorylation and blunting of linearized ends were introduced using a mutagenesis kit (Takara BioInc, Kusatsu, Shiga, Japan), followed by circularization using Solution I. The purification and folding of BMV TLS (2′–3′ cP) and mutant TLS were carried out similarly to the wild-type.
Cloning, expression, and purification of TyrRS constructs
The sequence of tyrosyl-tRNA synthetase (TyrRS) from Phaseolus vulgaris (common bean) was obtained from the UniProt database (gene: PHAVU_002G027700g) and codon-optimized for expression in E. coli. The gene was cloned into the p28 vector, which adds a 6-His tag to the N-terminus of the protein. The TyrRS plasmid was transformed into E. coli BL21(DE3) cells and cultured overnight at 37 °C in LB medium containing kanamycin. The culture was scaled up until the optical density at 600 nm (OD600) reached 0.8. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was then added to a final concentration of 0.5 mM, and the cells were incubated on a shaker at 16 °C for 20-24 h. Cells were collected by centrifugation at 5000 × g for 10 min at 4 °C. The cell pellet was resuspended in Binding Buffer (20 mM Tris, 1 M NaCl, pH 7.5) and lysed using a high-pressure homogenizer. The lysate was centrifuged at 10,000 × g for 30 min at 4 °C to collect the supernatant, which was then subjected to initial purification using a nickel column (GE Healthcare). Further purification was performed using a HiLoad 16/60 Superdex 200 column (GE Healthcare). The final protein was stored in 20 mM HEPES, 100 mM NaCl, pH 7.5. The construction method for TyrRS mutant plasmids was similar to that used for the TLS mutant plasmids (oligonucleotides are provided in Supplementary Data 1), and the expression and purification methods of TyrRS mutant proteins were the same as those of the wild-type.
In vitro aminoacylation
The in vitro aminoacylation reaction volume was 20 µL, consisting of 13 µL of water, 2 µL of 10 × reaction buffer, 2 µL of 1.2 µM RNA, 2 µL of 1 µM TyrRS dimer solution, and 1 µL of 3H-labeled L-tyrosine (40 – 60 Ci/mmol). The 10 × reaction buffer included 20 mM ATP, 300 mM KCl, 50 mM MgCl2, 50 mM DTT, and 300 mM HEPES-KOH at pH 8.0. The reaction was carried out at 30 °C for 1 h.
After the reaction, 100 µL of water-saturated phenol (Sangon Biotech, Shanghai, China) was added, followed by vigorous vortexing. The mixture was then centrifuged at 16,000 × g at 4 °C for 10 min. From this, 40 µL of the aqueous phase containing RNA was transferred to a new Eppendorf tube and mixed with 40 µL of chloroform:isoamyl alcohol (24:1, Solabao Technology, Beijing, China). After thorough mixing, three volumes of pre-chilled anhydrous ethanol were added and the sample was left to stand at −40 °C for 1 h. It was then centrifuged at 16,000 × g at 4 °C for 30 min, the supernatant was discarded, and the pellet was washed once with 75% ethanol. The supernatant was discarded again, and the ethanol was allowed to evaporate. Then 10 µL of DEPC-treated water was added to dissolve the RNA pellet. This 10 µL RNA solution was transferred to a vial containing 5 mL of scintillation fluid and placed into a liquid scintillation counter to detect the radioactivity associated with the purified RNA.
Electrophoretic mobility shift assay (EMSA)
In the EMSA, five or ten samples were set up, each with a reaction volume of 10 µL. Initially, TLS was folded in vitro as described above. To each sample, TLS was added to a final concentration of 0.5 µM. The first sample contained no protein, while the remaining samples were supplemented with proteins at increasing concentrations. The samples were then brought to a total volume of 10 µL using a buffer composed of 20 mM HEPES-Na, pH 7.8, 150 mM NaCl, 5 mM MgCl2, and incubated at 4 °C for 30 min. Following incubation, 2.5 µL of non-denaturing loading buffer was added to each sample. The samples were then subjected to electrophoresis on a 6% non-denaturing PAGE gel at a constant voltage of 120 V for approximately 20 min. Post-electrophoresis, the gel was stained with GelRed for 1 min, rinsed three times with water, and subsequently visualized using a gel imaging system.
Complex reconstitution for cryo-EM analysis
To obtain a high-purity BMV TLS-TyrRS complex (pre-1a state), BMV TLS was first subjected to in vitro folding. TyrRS and BMV TLS were then incubated together at 4 °C for 2 h at a molar ratio of 1:1.5. The complex was purified using a HiLoad 10/300 Superdex 200 increase column (GE Healthcare) with a buffer consisting of 20 mM HEPES-Na, 150 mM NaCl, 5 mM MgCl2, pH 7.8. Fractions from the peak of the complex were collected and concentrated to 4 mg/mL for preparation of cryo-EM samples.
To study how the substrates ATP and Tyr participate in the catalytic process involving TyrRS and BMV TLS, we prepared a BMV TLS-TyrRS-ATP-Tyr complex (post-1a state). Following the purification steps described above, additional 10-fold molar excess of ATP and Tyr was added before the sample was frozen.
To further obtain the BMV TLS-TyrRS-ATP-Tyr complex in the catalysis state, the reaction time was shortened and 1 mM CaCl2 to slow down the reaction. TyrRS, BMV TLS, ATP, and Tyr were incubated at room temperature for 5 to 10 min, and the sample was immediately prepared for analysis.
Cryo-EM grid preparation and data collection
For cryo-EM sample preparation of pre-1a state, post-1a state, and catalysis state, the sample concentration was maintained at 4 mg/mL. Quantifoil R2/1 200-mesh grids were subjected to glow discharge using a plasma system with a cleaning time of 60 s and a hold time of 15 s. Using tweezers, the grids were placed into an FEI Vitrobot, maintained at 4 °C and 100% humidity. Then 3 μL of the sample was applied to the grids, with a blot time of 4 s and a wait time of 5 s before rapidly freezing them in liquid nitrogen. Notably, similar with the previous study23, the preferential orientation issue also occurred on our pre-1a state samples. To address this issue, two different methods were tried: first, BMV TLS-TyrRS was concentrated to 10 mg/mL and 0.05% octyl glucoside (OG) was added before freezing; second, 5 μL of 1% poly-L-lysine was adsorbed onto the grids for 90 s, followed by two rinses with 5 μL deionized water (ddH2O) each, allowing the grids to dry completely before freezing. The grids were screened using a Glacios cryo-electron microscope (Thermo Fisher Scientific) operated at 200 kV. Then they were imaged in a Titan Krios cryo-electron microscope (Thermo Fisher Scientific) operated at 300 kV at a magnification of 105,000 × (corresponding to a calibrated sampling of 0.82 Å per pixel). Micrographs were recorded by EPU software (Thermo Fisher Scientific, version 2.7) with a Gatan K3 Summit direct electron detector. Finally, a total of 7726 movie stacks for pre-1a state, 5418 movie stacks for post-1a state, and 4560 movie stacks for catalysis state were collected with a defocus range of −1.3 to −3.3 μm.
Cryo-EM data processing
Data processing was performed using the software package cryoSPARC. Micrographs were motion corrected with patch motion correction. The CTF parameters of micrographs were estimated using patch CTF estimation. All particles were autopicked using the template picker in cryoSPARC. After 2D classification, 100,000 random particles in good 2D classes were used for ab initio map generation followed by 3D heterogeneous refinement. The particles of the good 3D classes were then refined into higher resolution by non-uniform refinement. Resolution for the final map was estimated with the 0.143 criterion of the Fourier shell correlation curve. The resolution map was calculated in cryoSPARC using the “Local Resolution Estimation” option. The processing details are shown in Supplementary Figs. 2–4 and listed in Supplementary Tables 1–3.
Model building and refinement
For the model building of the BMV TLS, the atomic model 7SAM23 was rigidly fitted into the cryo-EM map and flexibly fitted using MDFF. The resultant atomic model was refined using phenix.real_space_refine.
Model building was then performed on the TLS-TyrRS complex in the pre-1a state. The atomic model 7SCQ23 was used as the starting model, whose TLS and TyrRS were rigidly fitted into the cryo-EM map, respectively. Several rounds of molecular dynamics flexible fitting (MDFF)45 were then applied to flexibly fit the atomic model into the map until no noticeable changes were observed. The resulting atomic model was optimized with Coot46 and phenix.real_space_refine47. The bound ATP was generated and manually adjusted using Coot, and refined by phenix.real_space_refine.
For the model building of the TLS-TyrRS complex in the post-1a state, the final model of the pre-1a state was used as the starting model. MDFF was then applied to flexibly fit the atomic model into the map, followed by optimization with Coot and phenix.real_space_refine. The bound AMP-Tyr was generated and manually adjusted using Coot, and refined by phenix.real_space_refine.
For the model building of the TLS-TyrRS complex in the catalysis state, the atomic model 7SCQ was rigidly fitted into the cryo-EM map and flexibly fitted using MDFF. The resultant atomic model was refined using phenix.real_space_refine. The final models were evaluated by MolProbity48. Statistics of the model building are summarized in Supplementary Tables 1–3. All figures were prepared using ChimeraX49.
Biotinylated RNA pull-down and mass spectrometry
The biotinylated RNA transcription system included 1 × transcription buffer [40 mM Tris-HCl, 1 mM spermidine, 0.01% (v/v) Triton X-100, pH 8.1], 10 mM DTT, NTP mix (10 mM ATP, 10 mM CTP, 10 mM GTP, 6.5 mM UTP, 3.5 mM biotin-16-UTP), 0.3 mg/mL T7 RNA polymerase, and 10 ng/μL DNA template. The reaction was carried out at 37 °C for 3 h. The transcribed RNA was purified using the GeneJET RNA Cleanup and Concentration Micro Kit (Thermo Fisher Scientific).
Leaf tissue from Nicotiana tabacum L. was ground thoroughly in liquid nitrogen and lysed with a lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 0.5% Triton X-100, pH 7.4) and a protease inhibitor cocktail. The cell lysate was sonicated, and the supernatant was incubated with biotinylated RNA. The RNA pulldown kit (BersinBioTM) was used to capture proteins interacting with the biotinylated RNA. The eluted proteins were digested with trypsin at 37°C overnight and then analyzed by mass spectrometry using the timsTOF Pro 2 (Bruker). The MS settings were 100 m/z to 1700 m/z, positive ion mode, and PASEF scan mode. The TIMS settings ranged from 0.75 V·s/cm² to 1.3 V·s/cm². The data acquired from the timsTOF Pro 2 was analyzed using DataAnalysis. All the raw files were searched against the UniProt database using Parallel Search Engine in Real-Time (PaSER). DDA search basic parameter were CID/HCD fragmentation method, mono precursor mass type and mono fragment mass type. Precursor and peptide mass tolerance was 20 ppm. Tryptic specificity was both end and two missed cleavages were allowed. Carbamidomethylation on cysteine was set as a fixed modification. Cysteine was set as a fixed modification, oxidation on methionine and acetylation on lysine were set as variable modification. GO enrichment analysis was carried out using The Database for Annotation, Visualization, and Integrated Discovery (DAVID).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Cryo-EM structures and atomic models generated in this study have been deposited in the wwPDB OneDep System under EMD accession codes EMD-60247, EMD-60248, EMD-60249, EMD-60250 and PDB ID codes under accession codes 8ZMH, 8ZMI, 8ZMJ, 8ZMK, respectively. The mass spectrometry proteomics data reported in this paper have been deposited in the iProX repository under accession codes IPX0010110000 and PXD057815. Source data are provided with this paper.
References
Kao, C. C. & Sivakumaran, K. Brome mosaic virus, good for an RNA virologist’s basic needs. Mol. Plant Pathol. 1, 91–97 (2000).
Rao, A. L. N. & Cheng Kao, C. The brome mosaic virus 3’ untranslated sequence regulates RNA replication, recombination, and virion assembly. Virus Res. 206, 46–52 (2015).
Rietveld, K., Pleij, C. W. & Bosch, L. Three-dimensional models of the tRNA-like 3’ termini of some plant viral RNAs. EMBO J. 2, 1079–1085 (1983).
Sherlock, M. E., Hartwick, E. W., MacFadden, A. & Kieft, J. S. Structural diversity and phylogenetic distribution of valyl tRNA-like structures in viruses. RNA 27, 27–39 (2021).
Matsuda, D. & Dreher, T. W. The tRNA-like structure of Turnip yellow mosaic virus RNA is a 3’-translational enhancer. Virology 321, 36–46 (2004).
Sun, J. H., Adkins, S., Faurote, G. & Kao, C. C. Initiation of (-)-strand RNA synthesis catalyzed by the BMV RNA-dependent RNA polymerase: synthesis of oligonucleotides. Virology 226, 1–12 (1996).
Ni, P., Vaughan, R. C., Tragesser, B., Hoover, H. & Kao, C. C. The plant host can affect the encapsidation of brome mosaic virus (BMV) RNA: BMV virions are surprisingly heterogeneous. J. Mol. Biol. 426, 1061–1076 (2014).
Kwon, S.-J. & Rao, A. L. N. Emergence of distinct brome mosaic virus recombinants is determined by the polarity of the inoculum RNA. J. Virol. 86, 5204–5220 (2012).
Barends, S. et al. tRNA-like structure regulates translation of Brome mosaic virus RNA. J. Virol. 78, 4003–4010 (2004).
Fechter, P., Rudinger-Thirion, J., Florentz, C. & Giegé, R. Novel features in the tRNA-like world of plant viral RNAs. Cell Mol. Life Sci. 58, 1547–1561 (2001).
Vieweger, M., Holmstrom, E. D. & Nesbitt, D. J. Single-molecule FRET reveals three conformations for the TLS domain of brome mosaic virus genome. Biophys. J. 109, 2625–2636 (2015).
Fechter, P., Giegé, R. & Rudinger-Thirion, J. Specific tyrosylation of the bulky tRNA-like structure of brome mosaic virus RNA relies solely on identity nucleotides present in its amino acid-accepting domain. J. Mol. Biol. 309, 387–399 (2001).
Jaafar, Z. A. & Kieft, J. S. Viral RNA structure-based strategies to manipulate translation. Nat. Rev. Microbiol. 17, 110–123 (2019).
Rubio Gomez, M. A. & Ibba, M. Aminoacyl-tRNA synthetases. RNA 26, 910–936 (2020).
Giegé, R. & Eriani, G. The tRNA identity landscape for aminoacylation and beyond. Nucleic Acids Res. 51, 1528–1570 (2023).
Mirande, M. The Aminoacyl-tRNA synthetase complex. Subcell. Biochem. 83, 505–522 (2017).
Arnez, J. G. & Moras, D. Structural and functional considerations of the aminoacylation reaction. Trends Biochem. Sci. 22, 211–216 (1997).
Ibba, M. & Söll, D. The renaissance of aminoacyl-tRNA synthesis. EMBO Rep. 2, 382–387 (2001).
Rybak, M. Y. & Gagnon, M. G. Structures of the ribosome bound to EF-Tu-isoleucine tRNA elucidate the mechanism of AUG avoidance. Nat. Struct. Mol. Biol. 31, 810–816 (2024).
Smirnova, J. et al. Structure of the actively translating plant 80S ribosome at 2.2 Å resolution. Nat. Plants 9, 987–1000 (2023).
Dreher, T. W. Role of tRNA-like structures in controlling plant virus replication. Virus Res. 139, 217–229 (2009).
Hammond, J. A., Rambo, R. P., Filbin, M. E. & Kieft, J. S. Comparison and functional implications of the 3D architectures of viral tRNA-like structures. RNA 15, 294–307 (2009).
Bonilla, S. L., Sherlock, M. E., MacFadden, A. & Kieft, J. S. A viral RNA hijacks host machinery using dynamic conformational changes of a tRNA-like structure. Science 374, 955–960 (2021).
Francklyn, C. S. & Mullen, P. Progress and challenges in aminoacyl-tRNA synthetase-based therapeutics. J. Biol. Chem. 294, 5365–5385 (2019).
Bonilla, S. L. & Kieft, J. S. The promise of cryo-EM to explore RNA structural dynamics. J. Mol. Biol. 434, 167802 (2022).
Tsunoda, M. et al. Structural basis for recognition of cognate tRNA by tyrosyl-tRNA synthetase from three kingdoms. Nucleic Acids Res. 35, 4289–4300 (2007).
Yaremchuk, A., Kriklivyi, I., Tukalo, M. & Cusack, S. Class I tyrosyl-tRNA synthetase has a class II mode of cognate tRNA recognition. EMBO J. 21, 3829–3840 (2002).
Sakamoto, K. & Hayashi, A. Synthetic tyrosine tRNA molecules with noncanonical secondary structures. Int. J. Mol. Sci. 20, 92 (2018).
Kobayashi, T. et al. Structural basis for orthogonal tRNA specificities of tyrosyl-tRNA synthetases for genetic code expansion. Nat. Struct. Biol. 10, 425–432 (2003).
Dreher, T. W. Viral tRNAs and tRNA-like structures. Wiley Interdiscip. Rev. RNA 1, 402–414 (2010).
Griffey, R. H. et al. 15N-labeled Escherichia coli tRNAfMet, tRNAGlu, tRNATyr, and tRNAPhe. Double resonance and two-dimensional NMR of N1-labeled pseudouridine. J. Biol. Chem. 260, 9734–9741 (1985).
Sherlock, M. E., Langeberg, C. J. & Kieft, J. S. Diversity and modularity of tyrosine-accepting tRNA-like structures. RNA 30, 213–222 (2024).
Ward, W. H. & Fersht, A. R. Asymmetry of tyrosyl-tRNA synthetase in solution. Biochemistry 27, 1041–1049 (1988).
Ward, W. H. & Fersht, A. R. Tyrosyl-tRNA synthetase acts as an asymmetric dimer in charging tRNA. A rationale for half-of-the-sites activity. Biochemistry 27, 5525–5530 (1988).
Bosshard, H. R., Koch, L. E. & Hartley, B. S. Aminoacyl-tRNA synthetases from Bacillus stearothermophilus. Asymmetry of substrate binding to tyrosyl-tRNA synthetase. Eur. J. Biochem. 53, 493–498 (1975).
Bastin, M. & Hall, T. C. Interaction of elongation factor 1 with aminoacylated brome mosaic virus and tRNA’s. J. Virol. 20, 117–122 (1976).
Dreher, T. W., Uhlenbeck, O. C. & Browning, K. S. Quantitative assessment of EF-1alpha.GTP binding to aminoacyl-tRNAs, aminoacyl-viral RNA, and tRNA shows close correspondence to the RNA binding properties of EF-Tu. J. Biol. Chem. 274, 666–672 (1999).
Chen, X. et al. Binding between elongation factor 1A and the 3’-UTR of Chinese wheat mosaic virus is crucial for virus infection. Mol. Plant Pathol. 22, 1383–1398 (2021).
Andersen, G. R., Nissen, P. & Nyborg, J. Elongation factors in protein biosynthesis. Trends Biochem. Sci. 28, 434–441 (2003).
Djumagulov, M. et al. Accuracy mechanism of eukaryotic ribosome translocation. Nature 600, 543–546 (2021).
Brick, P., Bhat, T. N. & Blow, D. M. Structure of tyrosyl-tRNA synthetase refined at 2.3 A resolution. Interaction of the enzyme with the tyrosyl adenylate intermediate. J. Mol. Biol. 208, 83–98 (1989).
Wells, T. N., Knill-Jones, J. W., Gray, T. E. & Fersht, A. R. Kinetic and thermodynamic properties of wild-type and engineered mutants of tyrosyl-tRNA synthetase analyzed by pyrophosphate-exchange kinetics. Biochemistry 30, 5151–5156 (1991).
Bonnefond, L. et al. Crystal structure of human mitochondrial tyrosyl-tRNA synthetase reveals common and idiosyncratic features. Structure 15, 1505–1516 (2007).
Colussi, T. M. et al. The structural basis of transfer RNA mimicry and conformational plasticity by a viral RNA. Nature 511, 366–369 (2014).
Trabuco, L. G., Villa, E., Mitra, K., Frank, J. & Schulten, K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure 16, 673–683 (2008).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 66, 213–221 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 66, 12–21 (2010).
Pettersen, E. F. et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Acknowledgements
We thank the Cryo-EM Center at the University of Science and Technology of China (USTC) for their assistance with the experiments. We also thank Dr. Jinzhong Lin lab for providing the EF1α and EF2 samples. This work was supported by the National Key R&D Program of China (2022YFC2303700 to K.Z. and S.L. and 2022YFA1302700 to K.Z.), the National Natural Science Foundation of China (32301044 to S.L., 32371345 to K.Z., and 32371310 and 32071221 to Q.G.), Anhui Provincial Natural Science Foundation (2308085QC80 to S.L.), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0490000 to K.Z.), the Center for Advanced Interdisciplinary Science and Biomedicine of IHM (QYPY20220019 to K.Z. and QYPY20220004 to Q.G.), the Fundamental Research Funds for the Central Universities (WK9100000032 and YD9100002044 to S.L. and YD9100002048 to K.Z.).
Author information
Authors and Affiliations
Contributions
S.L., Q.G., and K.Z. conceived the study and designed the experiments. W.Y. prepared the samples and performed cryo-EM sample preparation, screening, data collection, and image processing. R.Y. helped with sample preparation and cryo-EM screening. J.Y. helped with sample preparation. Y.G. helped with data collection. K.Z. conducted final structure determination. S.L. built and refined the models. W.Y., S.L., Q.G., and K.Z. analyzed the data. S.L. wrote the manuscript with input from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, W., Yi, R., Yao, J. et al. Structural insights into dynamics of the BMV TLS aminoacylation. Nat Commun 16, 1276 (2025). https://doi.org/10.1038/s41467-025-56612-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56612-4
