
Abstract
Ion suppression is a major problem in mass spectrometry (MS)-based metabolomics; it can dramatically decrease measurement accuracy, precision, and sensitivity. Here we report a method, the IROA TruQuant Workflow, that uses a stable isotope-labeled internal standard (IROA-IS) library plus companion algorithms to: 1) measure and correct for ion suppression, and 2) perform Dual MSTUS normalization of MS metabolomic data. We evaluate the method across ion chromatography (IC), hydrophilic interaction liquid chromatography (HILIC), and reversed-phase liquid chromatography (RPLC)-MS systems in both positive and negative ionization modes, with clean and unclean ion sources, and across different biological matrices. Across the broad range of conditions tested, all detected metabolites exhibit ion suppression ranging from 1% to >90% and coefficients of variation ranging from 1% to 20%, but the Workflow and companion algorithms are highly effective at nulling out that suppression and error. To demonstrate a routine application of the Workflow, we employ the Workflow to study ovarian cancer cell response to the enzyme-drug L-asparaginase (ASNase). The IROA-normalized data reveal significant alterations in peptide metabolism, which have not been reported previously. Overall, the Workflow corrects ion suppression across diverse analytical conditions and produces robust normalization of non-targeted metabolomic data.
Similar content being viewed by others


Introduction
Metabolite levels reflect metabolic function and the integrated output of genomics, epigenomics, transcriptomics, and proteomics, including inputs from lifestyle and environment1,2,3. Hence, metabolomics is an effective approach for elucidating candidate drug targets4, candidate biomarkers of disease progression5, candidate biomarkers of therapeutic response6,7, mechanism(s) of drug sensitivity8, mechanism(s) of drug resistance9, and mechanisms of drug toxicity10. Unfortunately, rigorous, reproducible quantitation of metabolites is difficult. Standardization across laboratories, biological matrices, and analytical conditions is a major challenge for both research and clinical implementation of metabolomics.
Ion suppression is a type of matrix effect in mass spectrometry (MS) and a major contributor to those challenges (Fig. 1). The authors of a recent perspective article on best practices in metabolomics noted: “While there is no universal solution to the ion suppression problem, assessing the effects of ion suppression affords greater confidence in the accuracy of the results.”11 Indeed, until now, no universal solution has existed to counteract the negative effects of ion suppression across all analytes in a non-targeted metabolite profiling study. We present such a solution.

The red line represents observed, suppressed signal, which can be caused by the listed factors. The green line represents suppression-corrected signal, which can be realized by the techniques described in this paper. Representative examples of factors that affect the quantitative accuracy of MS-based measurements are listed and have been reviewed elsewhere12,13.
The mechanisms of ion suppression (e.g., in plasma, urine, cell culture, or tumor) are reviewed in detail elsewhere11,12,13; the type of ionization source5,14,15,16,17, mobile phase composition18, gas temperature, and physicochemical properties (e.g., pKa, polarity/aromaticity, hydrophobicity/ lipophilicity) of analytes and matrix components are examples of factors that can contribute to ion suppression12,19,20.
Ion suppression for small numbers of analytes (e.g., in targeted MS) can be addressed to some degree by diluting samples, modifying chromatographic or MS conditions to eliminate interferences, conducting a sample cleanup procedure such as solid phase extraction, and/or adding a chemically matched stable isotope-labeled internal standard11,21. However, because the source and magnitude of ion suppression can vary extensively across metabolites and samples11,22, counteracting ion suppression across all analytes and all samples in a non-targeted profiling study remains an unsolved challenge11,23.
Stable isotope-labeled internal standards can correct for variability in ionization efficiency and ion suppression. However, isobaric isotopologs (e.g., the M + 0 isotopolog of lactate and the M + 1 isotopolog of alanine) are difficult to distinguish. That has been a barrier to the effective use of stable isotope mixtures. Isotopic Ratio Outlier Analysis (IROA) protocols24,25,26,27 solve that problem by generating clearly identifiable isotopolog patterns. IROA also facilitates removal of non-biological signals, which are common artifacts in MS data. Notwithstanding, IROA can also be adapted for studies in exposomics or pharmacometabolomics in which non-biological signals are of interest. By adjusting the IROA concentration and data analysis parameters, researchers can differentiate between endogenous metabolites and exogenous compounds, allowing for the simultaneous detection and quantification of both biological and non-biological molecules.
Here, we introduce here a IROA Workflow that effectively corrects ion suppression and uses a Dual-MSTUS (MS Total Useful Signal) normalization algorithm to improve the quantitative accuracy, precision, and sensitivity of metabolomic data across diverse origins and analytical conditions.
Results
Conceptual overview
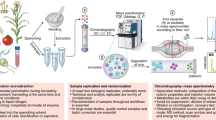
The Workflow presented here is based on the IROA Internal Standard (IROA-IS) and a chemically identical but isotopically different Long-Term Reference Standard (IROA-LTRS) (Fig. 2). The Workflow identifies each molecule in any type of sample based on a unique, formula-specific isotopolog ladder (Fig. 2c, d) created by (i) a low 13C (natural abundance or 5%) signal from the isotopologs of the molecule in question at the low mass end of the ladder and (ii) a 95% 13C signal for the isotopologs at the high mass end of the ladder. It is useful to call the collection of lower mass peaks in the isotopolog ladder the 12C channel and the collection of higher mass peaks in the ladder the 13C channel. IROA-LTRS (Fig. 2d, red/yellow samples) is a 1:1 mixture of chemically equivalent IROA-IS standards at 95% 13C and 5% 13C. The combination produces the IROA-LTRS isotopic pattern illustrated in Fig. 2d Metabolite 12C and 13C isotopologs co-elute, and the signature IROA peak pattern distinguishes real metabolites from artifacts, which lack the IROA pattern.

In this protocol, the experimental plasma samples (a) are prepared and dried and example spectra are shown (a′). They are then reconstituted with a solvent containing the IROA-IS (b) and its example spectral pattern (b′) to yield the analytical samples (c) and its spectral pattern (c′). The analytical samples are randomized and injected within a sequence that starts and ends with injections of the IROA-LTRS (d) & and its spectral pattern (d′), which is also injected approximately every 10 injections. In IROA MS/MS fragmentation, the IROA peaks retain their patterns (d2′) because wide windows are used. Based on the presence of the IROA-IS, each sample can be suppression-corrected and normalized despite significant differences in sample input (original sample aliquot volume prior to dry down) (e). Source data are provided as a Source Data file. Data are shown as mean ± s.d. (e).
Since metabolites in the Internal Standard are spiked into samples at constant concentrations, the loss of 13C signals due to ion suppression in each sample can be determined and used to correct for the loss of corresponding 12C signals. To model ion suppression, we created a single methanol extract of plasma, divided it into aliquots ranging from 50 to 1500 µL, dried the aliquots, and reconstituted them with a fixed volume and concentration of IROA-IS. We developed Eq. 1 to calculate and correct ion suppression as described in Methods.
Biologically relevant signals are considered to be those observed in both the IROA-LTRS and analytical samples as an IROA signature isotopolog ladder of signals with regular M + 1 spacing, decreasing amplitude signal in the 12C channel, and increasing amplitude signal in the 13C channel (from low to high mass) (Fig. 3a). An initial goal of the Workflow for quantitative metabolomic profiling was to calculate an AUC-12Csuppression-corrected value for each metabolite (Eq. 1). Without ion suppression, endogenous (12C) metabolite AUCs should increase linearly with aliquot volume, and internal standard (13C) levels should remain constant (Fig. 3b). However, the MSTUS values28 calculated for both [MSTUS-12C] (blue lines) and [MSTUS-13C] (green lines) deviated from proportionality because of increasing ion suppression with increasing sample concentration. Nevertheless, because ions in the 12C and 13C isotopolog channels experience equal degrees of suppression with increasing sample volume, IROA-IS enabled improved precision, relative quantitation, and correction of ion suppression for each metabolite.

a Detection and identification of metabolites by their isotopolog ladder patterns in IROA-LTRS. b Calculation of MSTUS (MS Total Useful Signal) values was performed as described in the “Methods” section using plasma sample. The illustrated data are from RPLC-MS with cleaned ionization source. 13C SC (Suppression Corrected) = 13C SC MSTUS; 13C raw = 13C raw MSTUS; 12C SC = 12C suppression corrected MSTUS; 12C raw = 12C raw MSTUS.
Ion suppression correction in non-targeted metabolomics
As expected, IROA-IS-based suppression-corrected values for MSTUS-13C were constant across sample input volumes, and 12C values increased proportionally to sample input (Fig. 3b). To explore the generalizability of the IROA Workflow, we performed a detailed characterization of ion suppression in both positive and negative ion modes for three highly used chromatographic systems: ICMS, RPLC-MS (C18-MS), and HILIC-MS with unclean and clean electrospray ionization (ESI) source conditions (Fig. 4a). Negative ionization mode detected fewer ions than did positive ionization mode (Fig. 4b), but extensive ion suppression was evident in both polarities and all chromatographic systems. Ion suppression calculation and corrections were performed automatically by ClusterFinder software (version 4.2.21, 64-bit, IROA Technologies) using Eq. 1. As expected, uncleaned ionization sources demonstrated significantly greater levels of ion suppression than cleaned ionization sources (Fig. 4c–k).

a The IROA ion suppression correction workflow. b Number of MSTUS peaks detected across analytical conditions. c–k Raw MSTUS-12C (blue lines) and suppression-corrected MSTUS-12C (red lines) values are shown for: c HILIC positive mode, uncleaned source; d HILIC positive mode, clean source; e HILIC negative mode, uncleaned source; f HILIC negative mode, clean source; g RPLC positive mode, uncleaned source; h RPLC positive mode, clean source; (i) RPLC negative mode, uncleaned source; j RPLC negative mode, clean source; and k IC negative mode, cleaned source. l Ratio of raw MSTUS-12C to suppression-corrected MSTUS-12C peak intensity across chromatographic methods and experimental conditions. m Raw and suppression-corrected phenylalanine values in RPLC positive ionization mode with cleaned source. n Raw and suppression-corrected pyroglutamylglycine values in IC negative ionization mode. o Identified chemical composition in entire RPLC clean dataset, as an example. p Kohonen Self Organizing Maps (SOM) show suppression patterns in the RPLC Clean raw dataset for all 539 compounds. q Density map shows compounds associated with each of the patterns discovered in (o). r Raw MSTUS-12C (blue lines) and suppression-corrected MSTUS-12C (red lines) values are shown for RPLC positive mode and RPLC negative mode (s) for urine. t Ratio of raw MSTUS-12C to suppression-corrected 12 C peak intensity for urine matrix in positive and negative ion modes. u Plasma and urine MSTUS-12C signals for 4 common metabolites before and after suppression correction. 12C SC = 12C suppression corrected MSTUS; 13C raw = 13C raw MSTUS. Colors represent percent peak intensity as indicated by the color bar. Source data are provided as a Source Data file. Data are shown as mean ± s.d. (c–n, r, s).
Strikingly, all tested chromatographic systems and conditions exhibited up to nearly 100% ion suppression, which was effectively corrected by the IROA Workflow, which yielded a linear increase in signal with increasing sample input (Fig. 4l, Supplementary Data 1, and Supplementary Figs. 1, 2). Of course, if a metabolite peak were 100% suppressed in the 12C or 13C channel, it would not yield the IROA ladder pattern and, therefore, would appear fully suppressed and absent from the output. But even if we attempted to include such a metabolite, the additional unknown in Eq. 1 would prevent its correction. Hence, the greatest weakness of the Workflow is that the output includes only metabolites that are detected in both the 12C and 13C channels (i.e., at most 99% suppressed). Absence from the 12C or 13C channel could be caused by 100% suppression, true absence in the IROA-IS, or true absence from the sample. Future versions of the ClusterFinder software will facilitate identification of fully suppressed analytes.
Historically there has been a tradeoff between injecting a large sample volume to increase assay sensitivity and injecting a smaller volume to decrease matrix effects. Using the IROA Workflow, analysts can err on the side of injecting larger sample volumes to ensure robust measurement of low-abundance analytes while simultaneously performing ion suppression correction to achieve more accurate results. The Workflow produces accurate concentration values for most analytes, even in highly concentrated samples. For example, phenylalanine (M + H) exhibited 8.3% ion suppression in RPLC positive mode with a cleaned ionization source, and suppression correction restored the expected linear increase in signal with increasing sample input (Fig. 4m). In a more extreme example, pyroglutamylglycine (M − H) exhibited up to 97% suppression in ICMS negative mode, and the IROA workflow corrected for that suppression as well (Fig. 4n).
The IROA TruQuant Workflow facilitated identification and measurement of 539 different metabolites across the entire sample set (Fig. 4o). An average of 422 metabolites were observed in each sample, with 216 being common to all samples. Unexpectedly, the total number of peaks observed decreased as a function of sample input, likely due to the suppression driving some compounds’ peaks to extinction. The workflow demonstrated broad metabolic coverage, successfully identifying and quantifying compounds across major biochemical categories including amino acid metabolism (143 compounds), nucleotide metabolism (71 compounds), peptide metabolism (89 compounds), carbohydrate metabolism (52 compounds), secondary metabolism (22 compounds), lipid metabolism (14 compounds), and various other pathways such as vitamins, cofactors, and redox metabolism (Supplementary Fig. 3 and Supplementary Data 2). This comprehensive coverage of metabolites that can be produced through yeast cell metabolism indicates the workflow’s robust applicability in metabolomics research, particularly for quantitative studies where labeled matches produced by yeast metabolism enable reliable suppression correction. Kohonen Self Organizing Maps (SOMs) revealed patterns of increasing concentration with sample input, decreasing concentration with sample input, and various intermediate patterns (Fig. 4p, q and Supplementary Fig. 4). For instance, Figs. 4p-15, 4p-13, and 4p-09 indicate metabolite peaks that first increase and then decrease in amplitude as the input volume is increased. Analysis of the full dataset shows: i) for suppression levels up to 60%, the observed [AUC-12Csuppressed] value was weakly correlated with the expected [AUC-12Csuppression-corrected] value; from 60% to 80% suppression, the correlation was lost; and above 80% suppression, [AUC-12Csuppressed] values were negatively correlated with the expected [AUC-12Csuppression-corrected] value due to decreasing peak size with increasing sample input.
We next evaluated the performance of IROA-based ion suppression correction with urine samples, which notoriously exhibit high ion suppression5,16. We were pleased to find that the IROA Workflow produced comparable suppression-corrected data in both positive and negative ionization modes (Fig. 4r, s). The correction was robust even at high urine sample inputs for which the ion suppression effects were most severe (Fig. 4t and Supplementary Fig. 5). Common metabolites identified in both plasma and urine revealed that ion suppression correction yielded comparable results in the two matrices (Fig. 4u). Overall, the ability of the IROA Workflow to produce a linear increase in suppression-corrected signals consistently with increasing sample input demonstrates the broad applicability and versatility of the Workflow for ion suppression correction across diverse biofluids. Moreover, our results demonstrate that the Workflow successfully corrects ubiquitous ion suppression across diverse metabolic pathways, analytical techniques, and experimental conditions (Fig. 5a, b, c and Supplementary Fig. 6).

a Raw and suppression-corrected (SC) peak intensities across chromatographic systems and uncleaned and cleaned ionization source conditions. b Metabolic pathways determined by ICMS before and after ion suppression correction. c Metabolic pathways determined by RPLC-MS before and after ion suppression correction. Data were drawn in SBGN (system biology graphical notation), Process Description (PD) and Activity Flow (AF) languages or Simple Interaction Format (SIF). Metabolites are color-coded based on the percent peak intensity. Raw = 12C raw; SC = 12C suppression corrected; Uncleaned+ uncleaned positive ion mode, clean+ clean positive ion mode, uncleaned- uncleaned negative ion mode, cleaned- cleaned negative ion mode. BCAA branched chain amino acid, PPP pentose phosphate pathway. Colors represent percent peak intensity as indicated by the color bar.
Development of dual MSTUS for robust normalization of metabolomic data
The workflow presented in the previous section promises to improve quantitative accuracy and correct for a subset of factors that introduce variation in metabolomic assay sensitivity, but ion suppression correction alone cannot enable accurate comparison of metabolite levels across variable sample input, analytical batches, biological matrices, instruments, and many other factors. Inconveniently, accurate cross-matrix comparison requires absolute quantitation using matrix-matched external calibration curves. Dual MSTUS normalization provides a solution to those challenges.
MSTUS normalization determines a total useful signal value for each sample by excluding all clearly non-relevant signals and then sums the remaining data22. That relatively simple strategy has been successful in metabolomics. Here we extend the MSTUS strategy into a Dual MSTUS algorithm. Since the IROA-IS 13C values for each metabolite should be equal across all samples, adjusting each MSTUS-12C value to the corresponding MSTUS-13C value normalizes all 12C signals to produce accurate comparisons across variable conditions (Fig. 6). An important distinction here is that, unlike suppression correction, wherein each compound is corrected individually, MSTUS normalization calculates a Normalization Factor, NF, that is applied to all peaks in each sample (see Section “Methods” for detail).

Raw MSTUS-12C values (blue lines), normalized MSTUS-13C values (green lines), and suppression-corrected MSTUS-12C values (red lines) for indicated chromatographic systems and conditions in plasma. a, b HILIC positive mode, uncleaned and cleaned source; c, d HILIC negative mode, uncleaned and cleaned source; e, f RPLC (C18) positive mode, uncleaned and cleaned source; g, h RPLC (C18) negative mode, uncleaned and cleaned source; i ICMS negative mode, clean source. Dual MSTUS normalization for urine by RPLC uncleaned positive mode (j) and negative mode (k). l Percent coefficient of variation (%CV) for raw, suppression-corrected, and normalized data from uncleaned and cleaned source conditions across different sample matrices and chromatographic systems including IC, HILIC, and RPLC. Colors represent percent peak intensity as indicated by the color bar. Source data are provided as a Source Data file. Data are shown as mean ± s.d. (a–k).
When applied to the data presented in the previous section, Dual MSTUS normalization produced significantly improved precision across different chromatographic methods and conditions (Fig. 6a–k). Notably, metabolites lacking a matched IROA-IS could not be suppression-corrected but still benefited from Dual MSTUS normalization because NF is a sample-based value and not a metabolite-specific value (Supplementary Fig. 7 and Supplementary Data 3). Since those measurements were not corrected for ion suppression, the normalized data still reflected suppressed signal, but Dual MSTUS normalization improved the biological and statistical significance of comparisons across samples, analytical conditions, and matrices with dramatic decrease in error from 16% to <1% in plasma (Fig. 6l and Supplementary Data 4) and from >50% to <1% in urine (Fig. 6l and Supplementary Data 5). Note that we designed the experiment to include extreme variance in sample input (50–1500 µL plasma extract), which is not likely to occur in ordinary experiments. Under typical experimental conditions with the same chromatographic system and equal input across samples, the Workflow can be expected to improve biological significance, statistical significance, and insight generation.
Optimization of the IROA Workflow for metabolomics
To enable the use of the IROA Workflow in experiments with cultured cancer cell lines, we next optimized the ratio of sample input: IROA-IS for an ovarian cancer cell line. We used methanol:water = 80:20 to extract metabolites from 0.5 to 6 million cells, vacuum concentrated the extracts to dryness, and reconstituted each extract in 40 µL IROA-IS (Fig. 7a). The resulting raw MSTUS signal increased non-linearly with increasing sample input, but suppression-corrected MSTUS signal increased sharply at 2 million cells with very little increase at larger cell numbers (Fig. 7b, c). Those observations indicate that an input of 2 million cells struck the optimal balance between maximizing the output and minimizing ion suppression.

a Optimization of cancer cell count for the IROA-IS ion suppression correction workflow. Raw MSTUS-12C (blue lines), suppression-corrected MSTUS-12C (red lines), and DUAL-MSTUS normalized (green lines) values are shown for: b RPLC positive mode, cleaned source; and c RPLC negative mode, clean source. d Optimization of IROA-IS during extraction and during reconstitution for ion suppression correction workflow. e Identified chemical composition in OVCAR-8 cell using IROA-IS in extraction solvent for entire RPLC clean dataset by both positive and negative mode. f Identified chemical composition in OVCAR-8 cell using IROA-IS as reconstitute solvent for entire RPLC clean dataset by both positive and negative mode. g Identified chemical composition in OVCAR-4 cell using IROA-IS in extraction solvent for entire RPLC clean dataset by both positive and negative mode. h Identified chemical composition in OVCAR-4 cell using IROA-IS as reconstitute solvent for entire RPLC clean dataset by both positive and negative mode. i Percent coefficient of variation (%CV) for raw, suppression-corrected, and normalized data from OVCAR-8 cell using IROA-IS in extraction solvent for entire RPLC clean dataset by both positive and negative mode. j Percent coefficient of variation (%CV) for raw, suppression-corrected, and normalized data from OVCAR-8 cell using IROA-IS as reconstitute solvent for entire RPLC clean dataset by both positive and negative mode. k Percent coefficient of variation (%CV) for raw, suppression-corrected, and normalized data from OVCAR-4 cell using IROA-IS in extraction solvent for entire RPLC clean dataset by both positive and negative mode. l Percent coefficient of variation (%CV) for raw, suppression-corrected, and normalized data from OVCAR-4 cell using IROA-IS as reconstitute solvent for entire RPLC clean dataset by both positive and negative mode. Percent coefficient of variation (%CV) for non-IROA-driven raw, and IROA-driven raw, suppression-corrected, and normalized data from OVCAR-4 (m) and OVCAR-8 (n) cell lines, respectively, for entire RPLC clean dataset by both positive and negative mode. Colors represent percent peak intensity as indicated by the color bar. Source data are provided as a Source Data file. Data are shown as mean ± s.d. (b, c).
Next, we compared the use of IROA-IS as an extraction standard (i.e., spiked into the extraction solvent) and as an injection standard (i.e., spiked into the reconstitution solvent) (Fig. 7d). Unexpectedly, IROA-IS produced comparable ion suppression correction and normalization results as an extraction standard and as an injection standard (Fig. 7e–l), with slightly better performance as an injection standard. The observed weak and variable signals in the absence of IROA-IS confirm the significantly negative impact of ion suppression on metabolomic data (Fig. 7m, n). Moreover, the observation that IROA-IS as an extraction standard did not mitigate a greater degree of error than IROA-IS as an injection standard indicates that the major source of variance in a metabolomics experiment arises within the MS itself.
The IROA workflow unveils biological insight into mechanisms of l-asparaginase resistance
l-Asparaginase (ASNase) is an important component of the standard-of-care treatment regimen for pediatric acute lymphoblastic leukemia (ALL)29. However, it exhibits limited anticancer activity against solid tumors, and the mechanisms of resistance remain poorly understood. To elucidate such mechanisms, we applied the IROA Workflow to analyze the response of ovarian cancer cell lines to ASNase. We treated the OVCAR-8 (sensitive to ASNase) and OVCAR-4 (resistant to ASNase) cell lines with PBS (vehicle control) or 0.5 IU/mL ASNase (Spectrila®) for 24 h (Fig. 8a). We included a parallel set of samples processed without IROA-IS. The data were processed using Compound DiscovererTM 3.3 by Thermo Fisher Scientific.

a IROA-IS Workflows for demonstration of ovarian cancer cell metabolism with or without ASNase treatment. Heatmap demonstrate most altered metabolite networks includes amino acid and peptide metabolism using IROA-IS and no IROA-IS in OVCAR-8 (b, c) and OVCAR-4 cell (d, e), respectively. f Prospective mechanism of peptide elevation in ASNase resistant ovarian cancer cell (OVCAR-4). Colors represent percent peak intensity as indicated by the color bar.
Consistent with results discussed in previous sections, raw and suppression-corrected data exhibited weaker biological and statistical significance than IROA-normalized 12C metabolomic data (Fig. 8b–e). OVCAR-8 cells exhibited pronounced modulation of amino acid and peptide metabolism following ASNase treatment, but most of the effects became clear only after Dual MSTUS normalization (“12C Norm”). Clear effects on amino acid metabolism were observed in OVCAR-4 cells without IROA, but IROA improved measurement precision. The effects of ASNase on peptide metabolism in OVCAR-4 cells, however, were in opposing directions with and without Dual MSTUS normalization; without IROA, ASNase appeared to cause down-regulation of peptides, whereas the IROA Workflow revealed up-regulation of peptides (Fig. 8f).
It’s noteworthy to mention that the discrepancy between non-IROA data and 12C raw data stems from methodological differences. The IROA-LTRS generated built-in reference library allows for accurate 12C raw data peak annotation as the 12C raw data is based on the same IROA internal standard pattern, providing a more reliable foundation for metabolite identification and quantification. Conversely, non-IROA data lacks this validated standard, potentially leading to misidentification or inconsistent quantification. The absence of isotope labeling in non-IROA methods makes it challenging to distinguish true metabolite signals from background noise or contaminants.
Biologically, the IROA-enabled observations are consistent with previous reports that ASNase induces autophagy, which produces peptides via lysosomal recycling of proteins30,31. Our observations specifically suggest that a cell’s capacity to up-regulate peptide generation is associated with ASNase resistance, prompting the hypotheses that ASNase resistance is mediated by: (i) autophagy-induced peptide generation; (ii) proteolytic production of amino acids from peptides; and/or (iii) de novo peptide synthesis (Fig. 8f). Future studies are warranted to test those hypotheses and whether such mechanisms could be targeted to improve anticancer activity of ASNase toward solid cancer types.
Discussion
We present an IROA-based Dual MSTUS Workflow that is applicable across chromatographic systems, ion sources, and biological matrices. The Workflow, including suppression correction and Dual MSTUS normalization algorithms, facilitates accurate comparison of metabolomic profiling data across: (i) unclean and clean ionization sources; (ii) IC-, HILIC-, and RPLC-MS systems; and (iii) plasma and urine matrices. We also optimized the IROA-IS workflow for cancer cell metabolomics and evaluated the impact of IROA-IS as an extraction standard and an injection standard, which revealed no significant difference in the variance that arises between sample preparation and sample analysis. That finding was surprising, because pre-analytical variance is a significant contributor to error in metabolomics32. Nevertheless, our results suggest that instrument-specific variance including ionization and ion suppression are the greatest contributors to error in metabolomics. Importantly, the Workflow provides an almost universal antidote to the negative effects of ion suppression across all analytes in a non-targeted metabolite profiling study. By doing so, it can increase the sensitivity, accuracy, precision, and biological and statistical significance of such measurements.
The IROA Workflow has significant implications for the metabolomics field. Matrices like fine-needle aspirates and stool, which produce high, variable suppression can benefit most profoundly. Fine-needle aspirates (FNAs) are central to the diagnosis and management of breast cancer, cerebrospinal fluid (CSF) profiling at multi-omics level is key for children brain tumor33, and stool profiling is fundamental to microbiome metabolite profiling. Notably, uncleaned MS system components variably affect intra- and inter-instrument performance within and across laboratories, but the IROA Workflow produces equivalent data for equivalent samples processed across variable conditions. Since the very high attrition rate for clinical biomarker development is thought to be associated with hidden artifacts in the source data, inappropriate statistical methods, and unmanageable variation34,35, the improved rigor and reproducibility provided by the IROA Workflow could decrease metabolomics-based biomarker failure. Indeed, we explored mechanisms of ASNase resistance in ovarian cancer cell lines and unveiled that the Workflow produced robust and meaningful metabolic insights that were not observed without Dual MSTUS normalization.
The Workflow is highly effective but does have limitations. Chief among them is that the output includes only metabolites that are detected in both the 12C and 13C channels. One factor that can limit such detection is instrument sensitivity. Another factor is ion suppression. A third factor is low abundance in the IROA-IS. Ongoing advances in MS sensitivity and in the IROA-IS itself will translate into increased coverage of the metabolome and increased utility of the IROA TruQuant workflow. Our experience suggests that extraction methods may need to be customized by users according to their specific experimental parameters, prominently including matrix properties.
By addressing multiple sources of error, the IROA Workflow effectively corrects for a majority of the intrinsic inaccuracies of MS analyses and could help to move the metabolomics field toward standardization of non-targeted analyses.
Methods
Ethics
The study was approved by the institutional review board (protocol #2024-0411) and conducted according to the Declaration of Helsinki.
Plasma sample preparation
For each chromatographic system tested, triplicate 225 µL aliquots of human plasma with K2-EDTA as the anticoagulant (BioIVT lot #BRH1225104) were added to separate 15 mL polypropylene centrifuge tubes and combined with 5.4 mL dry ice-cooled methanol, and centrifuged at 16,100 × g at 4 °C for 10 min. The supernatant was transferred to a new tube from which triplicate aliquots of 1500, 1250, 1000, 750, 500, 250, 100, and 50 µL were prepared. The methanol extracts were then dried using a centrifugal vacuum concentrator (Thermo Fisher Scientific Savant RVT5105 SpeedVac) and stored at −80 °C. Immediately before analysis, samples were reconstituted in 40 µL of IROA-IS solution, prepared by dissolving the contents of an IROA-IS vial in 1.2 mL H2O, vortexing, and briefly centrifuging. Following the IROA TruQuant protocol (Fig. 2), the entire contents of the tubes were then transferred to polypropylene autosampler vials. IROA-LTRS, an isotopically-labeled complex standard composed of extracts universally labeled at both 5% and 95% U-13C in a 1:1 ratio, was prepared by dissolving the contents of an IROA-LTRS vial in 40 µL H2O. In addition to blank and process blank samples (deionized H2O, before and after SOP), injections of the 350 µL aliquot plasma extract sample (without IROA-IS) and the IROA-IS solution alone served as controls. A 3 µL injection of IROA-LTRS and 5 µL injection of each sample were analyzed.
Urine sample preparation
Urine samples were obtained from healthy volunteers under The University of Texas MD Anderson Cancer Center IRB protocol #2024-0411 and analyzed using reverse-phase liquid chromatography (RPLC)-coupled with high-resolution mass spectrometry in both positive and negative ionization modes with an uncleaned ionization source. Triplicate 100 µL aliquots of human urine samples were used for metabolomics analysis following the IROA workflow using the same protocol as used for plasma sample, as described in the previous section.
IROA-IS and LTRS
The IROA labeling process starts by growing yeast cells (Saccharomyces cerevisiae) in media containing either 5% or 95% 13C-labeled glucose as the primary carbon source. The yeast cells efficiently incorporate the labeled glucose into their biomass during growth. After multiple passages, the yeast cells are harvested, filtered, and extracted to obtain a 5% 13C-labeled yeast extract and a 95% 13C-labeled yeast extract. An equal combination of these two extracts generates the IROA Long-Term Reference Standard (LTRS), also called the pooled IROA extract. Additionally, the 95% 13C-labeled yeast extract alone serves as the internal standard, referred to as IROA-IS. Yeast is ideal for this isotopic labeling process because it preferentially utilizes glucose, enables cost-effective growth at scale, and has been widely studied. A brief workflow of the IROA 13C labeling process is presented in Fig. 2.
The IROA Workflow can be employed across a wide range of isotopic balances, and the optimal range for metabolomics (molecules containing between 3–50 carbons) is 95%:5%. At any isotopic balance, using the monoisotopic mass and number of carbons in the molecule we can calculate a unique molecular formula. However, the isotope balance and number of carbons shift the patterns in opposing ways, and therefore the optimal balance depends on the properties of the molecules with which it will be used.
For instance, at 5% the base peak shifts up from the monoisotopic every 20 carbons (to the M + 1, M + 2, M + 3, etc.) and the monoisotopic diminishes rapidly (as the probability of a molecule being 100% 13C gets smaller). At higher isotope percentages the shift happens earlier (at 10% the base peak shift happens at 10 carbons, and the monoisotopic peak diminishes even more rapidly to the point of being hard to find and to verify the molecular formula. At lower percentages this is less of a problem, but the measure of the height of the M + 1 relative to the monoisotopic is a step function for the number of carbons in the molecule and the 5% step function is large enough to be unambiguous, even with modern instrumentation error. At lower levels, such as ~3% 13C there is increased error.
Multiple charging complicates the computation so we tend not to use it in proteomics where it would work but the calculation of the shape of the clusters becomes more complex with the larger collection of charges overlaying the isotopolog patterns. Fortunately, metabolites tend to be almost exclusively singly or doubly charged.
We have also conducted stability studies of IROA-IS and LTRS samples and found that these samples remain stable for at least 5 years when stored at −80 °C36. We also provided the list of compound present in the IROA TruQuant Workflow as Supplementary Table 5.
Optimization of IROA-IS for cellular metabolomics
The OVCAR-8 cells were cultured in RPMI-1640 supplemented with 5% fetal bovine serum and 2 mM L-glutamine under 5% CO2 and 90% relative humidity at 37 °C. For harvesting, cells were detached with 0.05% trypsin, counted, and washed with PBS. The resulting cell suspension was divided into 18 aliquots, with 0.5, 0.75, 1.0, 1.5, 2.0, 4.0, and 6.0 million cells in triplicate. Cells were extracted by adding 500 mL of 8:2 methanol:water as previously reported33. The samples were then vortex-mixed and centrifuged. The supernatant was dried using a Thermo Fisher Scientific Savant RVT5105 SpeedVac and reconstituted with the 40 µL IROA-IS mixture.
Assessment of IROA-IS as extraction vs injection standard
OVCAR-8 cells were cultured in RPMI-1640 supplemented with 5% fetal bovine serum and 2 mM l-glutamine under 5% CO2 and 90% relative humidity at 37 °C. For harvesting, cells were detached with 0.05% trypsin, counted, washed with PBS, and aliquoted in triplicate with 2.0 million cells per tube. Metabolites were extracted by adding 500 µL methanol:water = 8:2 with or without 40 µL IROA-IS, vortex-mixed for 2 min, and centrifuged. The supernatant was dried using Thermo Fisher Scientific Savant RVT5105 SpeedVac and reconstituted with or without the 40 µL IROA-IS mixture.
ASNase treatment
OVCAR-4 and OVCAR-8 cells were cultured in RPMI-1640 supplemented with 5% fetal bovine serum and 2 mM l-glutamine under 5% CO2 and 90% relative humidity at 37 °C. Cells received ASNase treatment with PBS (vehicle control) or 0.5 IU/mL ASNase (Spectrila®) for 24 h. Each cell line was seeded at two different densities to ensure ~80% confluence at the end of treatments. At 48 h, medium was discarded, and fresh medium was added. Meanwhile, PBS or 0.5 IU/mL ASNase was added for 24 h. At 24 h after seeding, cells in the vehicle and treatment groups were harvested. All treatments were performed in triplicate. For harvesting, cells were detached with 0.05% trypsin, counted, and washed with PBS. Aliquots of cell suspension with 2 million cells were centrifuged, and the pellets were snap-frozen at -80°C.
Reversed-phase liquid chromatography (RPLC)
RPLC mobile phase A (MPA; weak) was water containing 10 mM ammonium acetate and 0.1% formic acid, and mobile phase B (MPB; strong) was methanol containing 10 mM ammonium acetate and 0.1% formic acid. The stationary phase was a Phenomenex Kinetex C18 column (2.6 µm particle size, 100 Å, 150 × 2.1 mm) with column compartment kept at 40 °C. The autosampler tray was chilled to 4 °C. The mobile phase flow rate was 200 µL/min, and the gradient elution program was: 0–5 min, 5% MPB; 5–30 min, 5–95% MPB; 30–40 min, 95% MPB; 40–45 min, 5% MPB. The total run time was 60 min.
Hydrophilic interaction liquid chromatography (HILIC)
HILIC mobile phase A (MPA) was 95/5 (v/v) water/acetonitrile containing 20 mM ammonium acetate and 20 mM ammonium hydroxide (pH~9), and mobile phase B (MPB) was acetonitrile. Thermo Vanquish LC system included a Xbridge BEH Amide column (3.5 µm particle size, 100 × 4.6 mm) with column compartment kept at 30 °C. The autosampler tray was chilled to 4 °C. The mobile phase flow rate was 300 µL/min, and the gradient elution program was: 0–3 min, 85% MPB; 3–10 min, 85–30% MPB; 10–20 min, 30–2% MPB; 20–25 min, 2% MPB; 26–30 min, 2–85% MPB. The total run time was 30 min.
Ion chromatography (IC)
IC mobile phase A (MPA; weak) was water, and mobile phase B (MPB; strong) was water containing 100 mM KOH. A Thermo Scientific Dionex ICS-6000+ system included a Thermo IonPac AS11 column (4 µm particle size, 250 ×2 mm) with column compartment kept at 35 °C. The autosampler tray was chilled to 4 °C. The mobile phase flow rate was 360 µL/min, and the gradient elution program was: 0-5 min, 1% MPB; 5–25 min, 1–35% MPB; 25–39 min, 35–99% MPB; 39–49 min, 99% MPB; 49–50, 99–1% MPB. The total run time was 50 min. To assist the desolvation for better sensitivity, methanol was delivered by an external pump and combined with the eluent via a low dead volume mixing tee.
Ultra-high-resolution mass spectrometry
For HILIC and RPLC chromatographic systems, a Thermo Scientific Orbitrap Fusion Lumos Tribrid mass spectrometer was operated in full scan mode using a scan range of 70–1400 m/z and a resolution of 240,000. Heated electrospray positive ionization used a spray voltage of 3500 (V) and vaporizer and capillary temperatures of 250 and 375 °C, respectively. The sheath, auxiliary, and sweep gas pressures were 35, 10, and 0 (arbitrary units), respectively. Identical parameters were used for negative ionization with the exception of −2800 (V) spray voltage. For ICMS, data were acquired using a Thermo Orbitrap IQ-X Tribrid Mass Spectrometer under ESI negative ionization mode with settings otherwise as noted above.
Data processing and calculations
Raw data files acquired by XCalibur software (Thermo Fisher) were converted to mzXML format using ProteoWizard’s msConvert (version 3.0.193336, 64 bit) using only the “peak picking by vendor” mode (both MS1 and MS2 scans were converted). The mzXML files were analyzed using ClusterFinder (version 4.2.21, 64-bit, IROA Technologies). IROA-LTRS mzXML files were analyzed in non-targeted mode to determine the identity, RT, and MS1/MS2 characteristics of the IROA-LTRS peaks. Those data were then used to create runtime libraries (Dual MSTUS Metabolite Lists), which were used as the basis for annotation and quantitative analyses of experimental samples. The quantitative data included raw, suppression-corrected, and normalized data for each peak in the analytical samples. Annotation data included the name of the compound, the reporting standard achieved, identity algorithm used (all inferred by comparison to the IROA-LTRS), retention time (RT), and m/z for both 12C and 13C monoisotopic peaks.
Analytical batch, quality control, and quality assurance
Analytical batches included HILIC clean positive mode, HILIC clean negative mode, HILIC unclean positive mode, HILIC unclean negative mode, RPLC clean positive mode, RPLC clean negative mode, RPLC unclean positive mode, RPLC unclean negative mode, and ICMS clean negative mode. “Clean” refers to a batch analyzed immediately following cleaning of the ionization source. Each analytical batch contained: (a) a distilled water blank, (b) a sample with an aliquot level of 350 µL extract that did not contain the Internal Standard mixture, and (c) a sample that contained the Internal Standard mixture (IROA-IS) but none of the experimental extract. Data quality was assessed by monitoring IROA-IS metabolite levels; high quality was apparent based on the observation of IROA-IS metabolite levels averaging less than 20% CV across all experimental samples in the batch. Solvent blanks provided an additional index of quality for carry-over assessment. The TruQuant Protocol also used IROA-LTRS as a Quality Assurance injected every 20 samples to ensure that instrument performance was within acceptable levels and to aid in peak annotation under current runtime conditions. The use of the LTRS and these additional sample types are further described in the Results and Discussion section.
Suppression-correction calculation
As part of the experimental design ion suppression was intentionally induced by increasing sample concentration. The following algorithm was employed for the correction of ion suppression.
In Eq. 1, the raw MS data provide values for [AUC-12Csuppressed] and [AUC-13Csuppressed]. [AUC-13Csuppression-corrected] can be approximated from the least-suppressed value in the dataset—the Internal Standard Only (IS-ONLY), internal standard sample. For optimal accuracy, it is critical that the AUCs include the sum of all of the isotopologs derived from the sample (12C end of the isotopolog ladder) and the internal standard (13C end of the isotopolog ladder). Whereas the sample isotopologs are generally limited to the 12C monoisotopic peak (M) and its M + 1, the IS will include the 13C monoisotopic peak (U-13C) and all of its isotopologs (M-1, M-2, etc.). We can solve Eq. 1 for [AUC-12Csuppression-corrected].
It is important to note that other values can be used in place of the “least-suppressed value” to estimate [AUC-13Csuppression-corrected]. For instance, if the concentration of the internal standard is held constant across experiments performed over a period of time, then the least-suppressed value could be a historically and more accurately determined value. The least-suppressed value could be either a quantitative molar quantity or a relative intensity (e.g., an AUC). The values determined from Eq. 1 can be used to calculate % suppression as (1-[AUC-12Csuppressed]/[AUC-12Csuppression-corrected]).
Dual MSTUS algorithm
We developed a modified version of the mass spectrometry total useful signal (MSTUS) algorithm as follows28. Because it uses two MSTUS values, i.e. that of the IROA-IS sample ([MSTUS-13C]) and the original natural abundance sample ([MSTUS-12C]), we refer to this as a “Dual MSTUS algorithm” (see Section “Results and discussion”).
Step 1: Calculate [MSTUS-12C] and [MSTUS-13C].
The Dual MSTUS Metabolite List for each sample includes only those peaks that are found in both the sample (12C) and IROA-IS (13C).
[MSTUS-12C] = the sum of the [AUC-12C suppression-corrected] values for all 12C peaks that are paired with a 13 C peak.
[MSTUS-13C] = the sum of the [AUC-13C suppression-corrected] values for all IS peaks that are paired with a 12 C peak.
Step 2: Calculate the sample-specific Normalization Factor (NF).
NF considers the total peak intensity in the sample relative to the total peak intensity in the IROA-IS to adjust the total content of the sample to the total content of the internal standard. Leveraging the statistical principal that variance decreases with increasing sample size, greater accuracy is achieved when a larger number of metabolites is used to establish the total content of each sample.
Step 3: Calculate normalized values for each metabolite in each sample.
Once NF has been determined for each sample, each [AUC-12C suppression-corrected] value (Eq. 1) is normalized by dividing by NF as indicated in Eq. 3. The calculated NF values were close to the values expected from the known aliquot sizes (Fig. 6).
The process for Dual MSTUS normalization is straightforward. It involves two main steps. First, NF is computed for each sample (Eq. 2) based on the Dual MSTUS Metabolite List, which includes peaks found in both the 12C and 13C channels of each sample. Second, the suppression-corrected 12C values (Eq. 1) of each metabolite in the Dual MSTUS Metabolite List are adjusted so that their sum is equal to the total [MSTUS-13C] value. That adjustment aligns with the original MSTUS rationale that normalizing over many metabolites will average out experimental variance, improving our ability to analyze and interpret the underlying biology accurately. By first suppression-correcting then applying the dual MSTUS NF, multiple sources of variability can be mitigated to provide greater rigor and reproducibility in non-targeted metabolomics. The total number of metabolites detected in each experimental condition determine the normalized linear dynamic range.
Data analysis and software
Calculations of ion suppression and Dual MSTUS normalization were performed automatically within ClusterFinder using the algorithms described in previous sections. Detailed peak picking settings are shown in Supplementary Data 6. Compound DiscovererTM 3.3 by Thermo Fisher Scientific was used for non-IROA data processing. Microsoft Office, Windows 10, R, Newt37, and GraphPad Prism 10.0.3 were used for data handling, processing, and visualization.
Data availability
All processed data are available in the article, Supplementary Information, and source data file. The data generated in this study have been deposited in the Zenodo database under accession code https://zenodo.org/records/14285171. Source data are provided with this paper.
References
Mahmud, I., Kabir, M., Haque, R. & Garrett, T. J. Decoding the metabolome and lipidome of child malnutrition by mass spectrometric techniques: present status and future perspectives. Anal. Chem. 91, 14784–14791 (2019).
Chen, L. et al. Metabolite discovery through global annotation of untargeted metabolomics data. Nat. Methods 18, 1377–1385 (2021).
Patti, G. J., Yanes, O. & Siuzdak, G. Metabolomics: the apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 13, 263–269 (2012).
Qiu, S. et al. Small molecule metabolites: discovery of biomarkers and therapeutic targets. Signal Transduct. Target. Ther. 8, 1–37 (2023).
Mahmud, I. et al. Rapid diagnosis of prostate cancer disease progression using paper spray ionization mass spectrometry. Anal. Chem. 93, 7774–7780 (2021).
Buergel, T. et al. Metabolomic profiles predict individual multidisease outcomes. Nat. Med. 28, 2309–2320 (2022).
Lee, B. et al. Integrated RNA and metabolite profiling of urine liquid biopsies for prostate cancer biomarker discovery. Sci. Rep. 10, 3716 (2020).
Weinstein, J. N. & Lorenzi, P. L. Discrepancies in drug sensitivity. Nature 504, 381–383 (2013).
Wishart, D. S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 15, 473–484 (2016).
Yao, C.-H. et al. Dose-response metabolomics to understand biochemical mechanisms and off-target drug effects with the TOXcms software. Anal. Chem. 92, 1856–1864 (2020).
Alseekh, S. et al. Mass spectrometry-based metabolomics: a guide for annotation, quantification and best reporting practices. Nat. Methods 18, 747–756 (2021).
Annesley, T. M. Ion suppression in mass spectrometry. Clin. Chem. 49, 1041–1044 (2003).
Volmer, D. & Jessome, L. L. Ion suppression: a major concern in mass spectrometry. LCGC N. Am. 24, 498–510 (2006).
Knochenmuss, R., Dubois, F., Dale, M. J. & Zenobi, R. The Matrix suppression effect and ionization mechanisms in matrix-assisted laser desorption/ionization. Rapid Commun. Mass Spectrom. 10, 871–877 (1996).
Taylor, A. J., Dexter, A. & Bunch, J. Exploring ion suppression in mass spectrometry imaging of a heterogeneous tissue. Anal. Chem. 90, 5637–5645 (2018).
Pinto, F. G., Mahmud, I., Harmon, T. A., Rubio, V. Y. & Garrett, T. J. Rapid prostate cancer noninvasive biomarker screening using segmented flow mass spectrometry-based untargeted metabolomics. J. Proteome Res. 19, 2080–2091 (2020).
Ismaiel, O. A., Halquist, M. S., Elmamly, M. Y., Shalaby, A. & Thomas Karnes, H. Monitoring phospholipids for assessment of ion enhancement and ion suppression in ESI and APCI LC/MS/MS for chlorpheniramine in human plasma and the importance of multiple source matrix effect evaluations. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 875, 333–343 (2008).
Guimaraes, G. J. & Bartlett, M. G. The critical role of mobile phase pH in the performance of oligonucleotide ion-pair liquid chromatography–mass spectrometry methods. Future Sci. OA 7, FSO753 (2021).
Mahmud, I., Sternberg, S., Williams, M. & Garrett, T. J. Comparison of global metabolite extraction strategies for soybeans using UHPLC-HRMS. Anal. Bioanal. Chem. 409, 6173–6180 (2017).
Mobile Phase Composition - an overview | ScienceDirect Topics. https://www.sciencedirect.com/topics/medicine-and-dentistry/mobile-phase-composition.
Vats, P. & Verma, S. M. Mechanistic evaluation of matrix effect on three different model of mass spectrometer by using a model drug. J. Anal. Bioanal. Tech. 7, 314 (2016).
Antignac, J.-P. et al. The ion suppression phenomenon in liquid chromatography–mass spectrometry and its consequences in the field of residue analysis. Anal. Chim. Acta 529, 129–136 (2005).
Taylor, P. J. Matrix effects: the Achilles heel of quantitative high-performance liquid chromatography–electrospray–tandem mass spectrometry. Clin. Biochem. 38, 328–334 (2005).
de Jong, F. A. & Beecher, C. Addressing the current bottlenecks of metabolomics: Isotopic Ratio Outlier AnalysisTM, an isotopic-labeling technique for accurate biochemical profiling. Bioanalysis 4, 2303–2314 (2012).
Clendinen, C. S. et al. An overview of methods using (13)C for improved compound identification in metabolomics and natural products. Front. Plant Sci. 6, 611 (2015).
Stupp, G. S. et al. Isotopic ratio outlier analysis global metabolomics of Caenorhabditis elegans. Anal. Chem. 85, 11858–11865 (2013).
Qiu, Y. et al. Isotopic ratio outlier analysis of the S. cerevisiae metabolome using accurate mass gas chromatography/time-of-flight mass spectrometry: a new method for discovery. Anal. Chem. 88, 2747–2754 (2016).
Warrack, B. M. et al. Normalization strategies for metabonomic analysis of urine samples. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 877, 547–552 (2009).
Chiu, M., Taurino, G., Bianchi, M. G., Kilberg, M. S. & Bussolati, O. Asparagine synthetase in cancer: beyond acute lymphoblastic leukemia. Front. Oncol. 9, 1480 (2020).
Lorenzi, P. L., Claerhout, S., Mills, G. B. & Weinstein, J. N. A curated census of autophagy-modulating proteins and small molecules: candidate targets for cancer therapy. Autophagy 10, 1316–1326 (2014).
Yu, M. et al. L-asparaginase inhibits invasive and angiogenic activity and induces autophagy in ovarian cancer. J. Cell. Mol. Med. 16, 2369–2378 (2012).
Kirwan, J. A. et al. Preanalytical processing and biobanking procedures of biological samples for metabolomics research: a white paper, community perspective (for “Precision Medicine and Pharmacometabolomics Task Group”—The Metabolomics Society Initiative). Clin. Chem. 64, 1158–1182 (2018).
Lee, B. et al. Medulloblastoma cerebrospinal fluid reveals metabolites and lipids indicative of hypoxia and cancer-specific RNAs. Acta Neuropathol. Commun. 10, 25 (2022).
Kern, S. E. Why your new cancer biomarker may never work: recurrent patterns and remarkable diversity in biomarker failures. Cancer Res. 72, 6097–6101 (2012).
Ioannidis, J. P. Biomarker failures. Clin. Chem. 59, 202–204 (2013).
iroatech. Compound Identification and Mass Spectrometry. IROA TECHNOLOGIES https://www.iroatech.com/compound-identification-and-mass_spectrometry/.
Balci, H. et al. Newt: a comprehensive web-based tool for viewing, constructing and analyzing biological maps. Bioinformatics 37, 1475–1477 (2021).
Acknowledgements
The Metabolomics Core Facility is supported by NIH grants S10OD012304-01 and P30CA016672.
Author information
Authors and Affiliations
Contributions
Experimental design was performed by I.M., L.V., C.B., and P.L.L. Data acquisition was performed by L.T., B.W., S.M., B.T., Y.L., J.D., Y.X., W.C., and L.V. Data analysis and interpretation were performed by I.M., L.V., R.A., F.D.J., and C.B. Manuscript composition was done by I.M., L.V., C.B., and P.L.L. Manuscript review and editing were done by I.M., J.N.W., R.A., F.D.J., C.B., and P.L.L.
Corresponding author
Ethics declarations
Competing interests
The authors I.M., B.W., L.V., L.T., S.M., B.T,. Y.L., J.D., Y.X., W.C., R.A., J.N.W., and P.L.L. declare no competing interest. C.B., F.D.J., and A.R. are employed by IROA Technologies.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mahmud, I., Wei, B., Veillon, L. et al. Ion suppression correction and normalization for non-targeted metabolomics. Nat Commun 16, 1347 (2025). https://doi.org/10.1038/s41467-025-56646-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56646-8
