
Abstract
The ring opening of cyclopropenes provides a compelling platform for the rapid synthesis of various polysubstituted acyclic alkenes. However, radical-mediated reactions of this type remain underexplored, and none of the existing methods have successfully produced tetrasubstituted olefins with high stereoselectivity. We present here an aminative ring-opening of cyclopropenes with iron-aminyl radical to afford tetrasubstituted alkenyl nitriles in a highly stereoselective manner. Computational studies indicate that both the substrate-directed radical addition and the following stereospecific ring-opening of cyclopropyl radical contribute to the extraordinary stereocontrol observed in the reaction. In addition, trisubstituted alkenyl nitriles could also be obtained using this method or via a base-promoted isomerization of the tetrasubstituted alkenyl nitriles, both with consistently high stereoselectivity.
Similar content being viewed by others
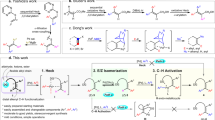
Introduction
Multisubstituted alkenyl nitriles are important scaffolds in bioactive molecules and organic materials. For example, entacapone is used together with levodopa and carbidopa to treat wearing-off symptoms of Parkinson’s disease; rilzabrutinib is currently under clinical investigation as advanced oral treatment for moderate-to-severe asthma (Fig. 1a)1,2,3. Thus, substantial efforts have been made to develop efficient methods for the synthesis of multisubstituted alkenyl nitriles4. However, highly stereoselective synthesis of these molecules, especially tetrasubstituted alkenyl nitriles, remains a formidable challenge5.

a Selected bioactive molecules containing polysubstituted alkenyl nitriles. b Radical-mediated ring opening of cyclopropenes. c This work: Stereoselective aminative ring opening of cyclopropene with iron-aminyl radical.
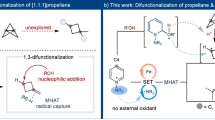
Cyclopropenes are versatile building blocks in organic chemistry, which could be potentially converted to various valuable structures, such as polysubstituted cyclopropanes, acyclic olefins, and other carbocycles6,7,8,9. Although transition metal (TM) catalysis is predominant in the chemistry of cyclopropenes, there is a growing interest in radical-mediated reactions due to their potential to broaden the range of incorporated functionalities and the unique selectivity that complements TM catalysis10. The existing radical-mediated transformations of cyclopropenes mainly lead to polysubstituted cyclopropanes via direct functionalization of cyclopropyl radical11,12,13,14,15,16,17,18,19,20,21, while selective ring-opening of the cyclopropyl radical to access valuable (multi)substituted acyclic olefins remains far less explored (Fig. 1b)22,23,24,25,26. These results might stem from the relatively higher kinetic barrier for the ring opening of cyclopropyl radicals compared to the direct radical functionalization under existing conditions21,27,28,29,30. More importantly, the isolated examples on radical-mediated ring-opening reactions provided the acyclic alkene products with low stereoselectivity23,24, possibly due to the insufficient stereo-chemical control during radical addition to the C = C double bond and/or ring-opening of cyclopropyl radicals (Fig. 1b).
Substrate-directed stereoselective metalation of cyclopropenes has been employed to generate numerous structurally diverse cyclopropanes with high diastereoseletivity6,7,8,9. These reactions typically proceed through coordination of R-M species with the basic functional groups in substrate, followed by selective alkene insertion to afford a cyclopropyl metal species. However, the leverage of such substrate-directed strategy for radical-mediated reactions remains elusive due to the lack of significant attractive interactions between the substrate and most radical species24. Recently, we disclosed an ortho-selective arene C–H amination with iron-aminyl radical, wherein iron is speculated to simultaneously bind with both the substrate and the aminyl radical, thereby imparting high regioselectivity for radical addition to arenes31,32,33,34. We proposed that the challenges associated with stereoselective radical addition to cyclopropenes could be addressed using iron-aminyl radical, as it possesses the potential to chelate with cyclopropene substrates and direct the radical addition. We report here an aminative ring-opening of cyclopropenes with iron-aminyl radical to afford tetra- and tri-substituted alkenyl nitriles with excellent stereoselectivity (Fig. 1c). Results from computational studies indicate that the facile ring-opening of cyclopropyl radical is attributed to an n-σ* interaction between the incorporated amino group and C–C bond, and both the substrate-directed stereoselective radical addition and the stereospecific outward disrotatory ring-opening of the cyclopropyl radical dictate the stereo-chemistry of alkene product.
Results
Reaction development
As noted above, the limited success in radical-mediated ring-opening functionalization of cyclopropenes might arise from the slow ring-opening of cyclopropyl radicals, which also prompted us to examine the kinetic barriers of different substituted cyclopropyl radicals using density functional theory (DFT) calculations. The preliminary results suggest that amino group may substantially accelerate the ring-opening of cyclopropyl radical in comparison to other functionalities, due to an n-σ* interaction between the lone electron pair on nitrogen and the anti-bonding orbital of the C2–C3 bond (Fig. 2). Notably, the kinetic barrier for ring-opening of azido-substituted cyclopropyl radical was also markedly reduced relative to the reaction where R = H or CF321, a finding that is also corroborated with the facile ring-opening reactivity observed by Waser and coworkers23,24.

The results clearly display that amino substituent could drastically reduce the kinetic barrier for ring-opening.
These results encouraged us to interrogate the substrate-directed stereoselective ring-opening reaction of cyclopropenes with iron-aminyl radical, aimed at synthesizing polysubstituted alkenyl nitriles1. The proposed reaction pathway is illustrated in Fig. 3a, which initiates with the selective formation of an iron-aminyl radical (int.-1) from Fe(II) and aminating reagent [N-O]31,32,35,36,37. Substrate chelation allows a stereoselective radical addition to the C = C double bond, generating cyclopropyl radical (int.-3)38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53. The subsequent ring-opening of int.-3, followed by single-electron oxidation, afforded the tetrasubstituted alkenyl iminium 2 and regenerated the iron catalyst. Finally, 2 was converted to the desired alkenyl nitrile via condensation with [N-O] and fragmentation.

a The proposed reaction pathway. b Initial trials for aminative ring opening of cyclopropenes. c Time courses. d Control experiments. e The overall reaction. Yields were determined by 1H-NMR analysis with CH2Br2 as internal standard.
Our studies commenced with the reaction of 3, using amide as chelating group, FeCl2 as catalyst and O-pivaloyl hydroxyl ammonium trifluoromethane sulfonate [N-O] as aminating reagent in fluorobenzene (Fig. 3b)54,55. Strikingly, tetrasubstituted alkenyl nitrile (4) was afforded in 47% yield with excellent Z selectivity. The proposed alkenyl iminum 2 and related aldehyde after hydrolysis was only detected with trace amount, indicating the facile condensation of 2 with [N-O]. The yield of 4 could be increased to 58% in the presence of 2.5 equiv. of [N-O]. Notably, reaction of 3 under conditions with azido radical could also furnish the same product, albeit with lower yield and a Z/E selectivity of 4:1 (see Fig. S3 of the Supplementary Information for details)23. Monitoring time course of the reaction indicated an induction period for product formation, as well as the formation of an oxime intermediate 5 (Fig. 3c). 5 was also isolated from the reaction and could be readily transformed to 4 in the presence of FeCl2 (73% yield) or under the standard conditions (96% yield, Fig. 3d). In addition, vinyl triflate 6 was produced in a yield of approximate 20%, also with an induction period (Fig. 3c). Of note, one equivalent of triflic acid (HOTf) will be released from the reaction by balancing the equation from 3 and [N-O] (2.0 equiv.) to 4 (Fig. 3e). We contributed the generation of 6 to the in-situ formation of HOTf. Indeed, 6 was generated in 41% yield by mixing HOTf with 3 in fluorobenzene (Fig. 3d)56.
These results prompted us to examine a series of additives to scavenge HOTf. However, the inclusion of inorganic bases into the reaction, such as Li2CO3, Na2CO3 and K2CO3, led to a decreased yield or even completely inhibited the reaction, in which cases the byproduct 6 was not observed either (Fig. 4, entry 1-4). These bases might rapidly react with the [N-O] reagent and release free O-pivaloyl hydroxylamine, which is proven inert for the present reaction. We speculated that a suitable base should meet the following criteria that include a) avoid the rapid neutralization reaction with [N-O] reagent, b) do not compete with substrate in coordinating with iron-aminyl radical, and c) consume HOTf effectively. We envisioned that N-Boc-O-pivaloylhydroxylamine 7 would be a suitable HOTf scavenger, producing the [N-O] reagent simultaneously57. Although the yield could be increased to 64%, 6 was still observed (18%, entry 5), which might be attributed to the possible slow reaction between 7 and HOTf. Inspired by the acid-mediated deprotection of tert-butyl carbamates, whose reaction with HOTf would ultimately lead to ammonium triflates, an array of carbamates was then examined (see Table S5 of the Supplementary Information for full optimization). Pleasingly, a yield of 67% yield was obtained, along with only 8% of 6, when N-ethyl carbamate 8 was used as additive (entry 6). Given that 6 appeared with an induction period under additive-free conditions (Fig. 3c), we tried to add the carbamate after the reaction was run for 0.5 h, which led to a slightly increased yield (71%, entry 7). In addition, a comparable yield was observed by replacing 8 with N-isopropyl carbamate 9 as additive (71%, entry 8). Finally, the best results were obtained when the reaction was run with 2.5 equiv. of [N-O] reagent and only 0.5 equiv. of 9 (77%, entry 9-10).

The additive effect on the reaction. aThe additive was added into the reaction mixture after 0.5 h. b2.5 equiv. of [N-O] was used. c0.5 equiv. of additive was used. All yields were determined by 1H-NMR analysis with CH2Br2 as internal standard.
Computational studies
DFT calculations were then performed to gain mechanistic insights into the reaction pathway and the origin of stereoselectivity (Fig. 5). Our previous investigations have demonstrated that the aminating reagent could coordinate with the Fe(II) catalyst, leading to metal-to-ligand electron transfer and the subsequent intramolecular proton transfer, and eventually resulting in an Fe(III)-aminyl radical31. We found that the present reaction proceeded via a similar pathway to afford a tetrahedral Fe(III)-aminyl radical (IM3), and the electron transfer and proton transfer occurred with very low kinetic barriers (ΔG≠ < 10 kcal/mol).). The involvement of nitrogen centered radical is also evidenced by electron paramagnetic resonance (EPR) analysis (see Fig. S4 of the Supplementary Information for details). IM3 was set as the reference point for constructing the reaction energy profile (Fig. 5a). Then, substrate chelation with IM3 generates IM4, and the subsequent intramolecular radical addition leads to the cyclopropyl radical IM5, where the amino and amide groups are positioned on the same side of the cyclopropane. The outward disrotatory ring-opening of IM5 took place via TS4 to afford IM6. Notably, our computational studies suggest that the [N-O] coordination with IM6 facilitates the following ligand-to-metal electron transfer, ultimately furnishing the iminium triflate salt and regenerating IM1. This electron transfer appears to occur throughout the course from IM5 to IM1, as evidenced by the spin density analysis of the intermediates (IM6 and IM7) and localized orbital bonding analysis (LOBA) of the iron center. In addition, reaction pathways at other spin states (triplet and septet) were also evaluated (Fig. 5a). The results indicate that all intermediates and transition states in the triplet state are significantly higher in energy compared to those in the quintet state. Furthermore, the intermediates and transition states in the septet state are generally less energetically favorable than those in the quintet state, with the exceptions of IM4 and TS2. The small energy differences observed between quintet and septet also suggest the possibility of spin-crossover events during the reaction58,59,60.

a The possible reaction pathways and associated energy profiles. b The origin of stereoselectivity for ring-opening of amino cyclopropyl radical.
The ring-opening of the cyclopropyl radical is expected to proceed in a disrotatory manner28. Our computational studies indicated that the outward disrotatory ring-opening of IM5 led to IM6 with a kinetic barrier of only 7.8 kcal/mol (Fig. 5a), while the transition state for the inward disrotatory ring-opening was not located. Given there is no significant interaction between the amino group and the iron center in TS4 (interatomic distance is 3.0 Å and Mayer bond order <0.1), ring-opening of the iron-free amino-substituted cyclopropyl radical IM8 was then evaluated to provide insights into stereoselective control (Fig. 5b). The optimized geometries of the cyclopropyl radicals (IM8 and IM9) reveal that the butyl substituent is positioned outside the plane of the cyclopropyl ring, and IM8 exhibits greater stability than the IM9 (ΔG = 1.4 kcal/mol). IM8 is prone to undergoing an outward disrotatory ring-opening, while the transition state of an inward disrotatory ring-opening is not located, potentially due to the poor overlap of the breaking σ-bond orbital with the radical orbital. In contrast, IM9 is likely to undergo an inward disrotatory ring-opening instead. However, the inward disrotatory ring-opening of IM9 is considerably less favorable than the outward disrotatory ring-opening of IM8 due to steric repulsion between the amino and amide groups in TS5 (Fig. 5b). These results are consistent with the excellent stereoselectivity observed in the present reaction.
Substrate scope
Efforts were then made to assess the reaction scope (Fig. 6). While the reaction with primary amide only generated the alkenyl nitrile product in 15% yield (10), moderate yield was observed for secondary amide (11, 51%). Replacing the methyl group in 3 with a longer carbon chain did not alter the reactivity and selectivity (4, 12-13). Unsymmetric amides are also suitable substrates for the aminative ring-opening reaction of cyclopropenes (14-15). The reaction is applicable to cyclic amides derived from pyrrolidine, piperidine, morpholine, 7-azabicyclo[2.2.1]heptane, azepane and piperazine, affording the desired product with moderate yields and excellent stereoselectivity (16-28). Functional groups including cyano, ketone, sulfonamide, pyridine and aryl chloride are compatible with the reaction. Configuration of the product was confirmed by X-ray crystallographic analysis of 23 and 27. However, amide is, by now, the only suitable directing group for the reaction, while other carbonyl groups, such as carboxylic acid, ester and ketone failed to give appreciable yield (see Fig. S5 of the Supplementary Information for details).

a The scope of amides. b The scope of alkyl group at C−1 position. c. Other suitable substrates. Isolated yields.
The existing methods for radical-mediated ring-opening of cyclopropenes not only suffer from low stereoselectivity, but also are limited to substrates bearing an aryl group at C-1 position22,23. We attributed the high reactivity of unactivated alkenes in the present reaction to the substrate chelation with iron-aminyl radical. Replacing the butyl group in 3 with other carbon chains did not exhibit detrimental effects on the reactivity, affording the tetrasubstituted alkenyl nitriles in moderate-to-good yields (29-37, Fig. 6b). Various functional groups, including alkyl chloride, thiophene, phosphate, furan, pyrrole, pyridine and alkyne, were well tolerated. Zhu, Wu and coworkers demonstrated that the cyclopropyl radical could undergo intramolecular 1,5-hydrogen atom transfer (HAT), whereby achieving a remote sp3 C–H bond (hetero)arylation reaction21. In contrast, such 1,5-HAT reactivity was not observed in the present reaction, underlying the facile ring-opening of cyclopropyl radical, as shown in Fig. 2. While the yields were only moderate, aryl substituted cyclopropenes are also suitable substrates for the present reaction (38 and 40). In addition, this method was applicable to the synthesis of trisubstituted alkenyl nitrile, in which cases the high stereoselectivity was consistently observed (41-42). Configuration of 42 was confirmed by X-ray crystallographic analysis.
Base-promoted isomerization
The tetrasubstituted alkenyl nitrile products could be utilized to synthesize trisubstituted alkenyl nitriles via base-promoted isomerization, and good stereoselectivity was also obtained (Fig. 7). For example, 43 was directly obtained from 3 in a yield of 60% following a two-step process without purification of 4. Other substrates, containing thiophene, phosphate, pyridine, ester, piperidine, ketone and electron-rich arene, were then examined using the two-step protocol to afford the desired trisubstituted alkenyl nitriles with moderate yield and excellent stereoselectivity (44-48).

Isolated yields. The Z/E ratio is determined by 1H-NMR analysis of the crude reaction mixture.
To conclude, we demonstrate here an aminative ring-opening of cyclopropenes with iron-aminyl radical that furnishes tetrasubstituted alkenyl nitriles in a highly stereoselective manner. The reaction is proposed to proceed through a substrate-directed stereoselective radical addition, followed by a stereospecific ring opening of the cyclopropyl radical. Computational studies indicate that the ring-opening of the cyclopropyl radical occurs via an outward disrotatory pathway. In addition, the resulting tetrasubstituted alkenyl nitriles can be readily converted into trisubstituted alkenyl nitriles through base-promoted proton migration. Future research will focus on the development of new catalytic method to accommodate broader scope of directing groups and the application of directed radical addition for other selective transformations.
Methods
General procedure
In a glovebox, a 4 mL glass vial was charged with FeCl2 (0.02 mmol, 10 mol%), aminating reagent [N-O] (0.50 mmol, 133 mg, 2.5 equiv.), followed by adding the solution of cyclopropylene (0.1 M, 2 mL, 1.0 equiv.) in PhF. The vial was sealed with a rubber septum or screwed access cap, and the reaction mixture was vigorously stirred at 30 °C for 0.5 h. Then, the carbamate iPrNHBoc (16 mg, 0.5 equiv.) was added into the reaction mixture and stirred at 30 °C for additional ca. 24 h. The vial was then cooled to room temperature (25 °C). The reaction mixture was added with saturated EDTA solution (disodium edetate dihydrate, 1 mL) and then solid NaHCO3 (slow addition) until it became basic. After stirring for an additional hour, the reaction mixture was extracted with EtOAc (20 mL) for three times. The organic phases were combined and the solvent was evaporated on a rotary evaporator. The residue was then separated through flash column chromatography to give the desired aniline products.
Data availability
Experimental procedures and characterization data are included in Supplementary Information. Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 2392830 (23), 2392699 (27), 2393053 (42). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. Source data are provided with this paper. All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Fleming, F. F., Yao, L., Ravikumar, P. C., Funk, L. & Shook, B. C. Nitrile-Containing Pharmaceuticals: Efficacious Roles of the Nitrile Pharmacophore. J. Med. Chem. 53, 7902–7917 (2010).
Janssen, G. V. et al. Development of Tyrphostin Analogues to Study Inhibition of the Mycobacterium tuberculosis Pup Proteasome System. Chembiochem 22, 3082–3089 (2021).
Zhao, Z. & Bourne, P. E. Med. Res. Rev https://doi.org/10.1002/med.22084 (2024).
Rappoport, Z. The Chemistry of the Cyano Group, (Wiley, London, 1970).
Flynn, A. B. & Ogilvie, W. W. Stereocontrolled Synthesis of Tetrasubstituted Olefins. Chem. Rev. 107, 4698–4745 (2007).
Rubin, M., Rubina, M. & Gevorgyan, V. Transition Metal Chemistry of Cyclopropenes and Cyclopropanes. Chem. Rev. 107, 3117–3179 (2007).
Dian, L. & Marek, I. Asymmetric Preparation of Polysubstituted Cyclopropanes Based on Direct Functionalization of Achiral Three-Membered Carbocycles. Chem. Rev. 118, 8415–9434 (2018).
Vicente, R. C–C Bond Cleavages of Cyclopropenes: Operating for Selective Ring-Opening Reactions. Chem. Rev. 121, 162–226 (2020).
Cohen, Y. & Marek, I. Regio- and Diastereoselective Carbometalation Reaction of Cyclopropenes. Acc. Chem. Res. 55, 2848–2868 (2022).
Royer, J., Lefebvre, G., Charron, O. & Meyer, C. Radical Addition Reactions to Cyclopropenes. Arkivoc 2, 202312099 (2024).
Yamago, S., Ejiri, S. & Nakamura, E. Hydrostannation of Cyclopropene. Strain-Driven Radical Addition Reaction. Chem. Lett 23, 1889–1892 (1994).
Nihei, T., Hoshino, T. & Konno, T. An Efficient Approach to gem-Difluorocyclopropylstannanes via Highly Regio- and Stereoselective hydrostannylation of gem-Difluorocyclopropenes and Their Unique Ring-Opening Reaction to Afford β-Fluoroallylic Alcohols. Org. Biomol. Chem 13, 3721–3731 (2015).
Ferjančić, Z., Čeković, Ž. & Saičić, R. N. Intermolecular Free Radical Additions to Strained Cycloalkenes. Cyclopropene and Cyclobutene as Radical Acceptors. Tetrahedron Lett. 41, 2979–2982 (2000).
Legrand, N., Quiclet-Sire, B. & Zard, S. Z. Radical Addition to Strained Olefins: a Flexible Access to Small Ring Derivatives. Tetrahedron Lett 41, 9815–9818 (2000).
Ueda, M. et al. Reaction of Cyclopropenes with a Trichloromethyl Radical: Unprecedented Ring-Opening Reaction of Cyclopropanes with Migration. Chem. Commun 51, 4204–4207 (2015).
Dange, N. S., Robert, F. & Landais, Y. Free-Radical Carbocyanation of Cyclopropenes: Stereocontrolled Access to All-Carbon Quaternary Stereocenters in Acyclic Systems. Org. Lett. 18, 6156–6159 (2016).
Dange, N. S., Jatoi, A. H., Robert, F. & Landais, Y. Visible-Light-Mediated Addition of Phenacyl Bromides onto Cyclopropenes. Org. Lett 19, 3652–3655 (2017).
Muriel, B., Gagnebin, A. & Waser, J. Synthesis of Bicyclo[3.1.0]hexanes by (3 + 2) Annulation of Cyclopropenes with Aminocyclopropanes. Chem. Sci. 10, 10716–10722 (2019).
Lefebvre, G., Charron, O., Cossy, J. & Meyer, C. Radical Addition of SF5Cl to Cyclopropenes: Synthesis of (Pentafluorosulfanyl)cyclopropanes. Org. Lett 23, 5491–5495 (2021).
Michalland, J., Casaretto, N. & Zard, S. Z. A Modular Access to 1,2- and 1,3-Disubstituted Cyclobutylboronic Esters by Consecutive Radical Additions. Angew. Chem. Int. Ed 61, e202113333 (2022).
Liu, C., Feng, T., Wu, X. & Zhu, C. Fe-Catalyzed Regioselective C(sp3)−H-Abstraction by Tertiary Cyclopropyl Radicals. ACS Catal 13, 8394–8401 (2023).
Liu, G. X. et al. Carbon-Centered Radical with Leaving Group-Mediated Ring Opening of Cyclopropenes via the Rearrangement of Cyclopropyl to the Allyl Radical: A General Access to Multisubstituted 1,3-Dienes. ACS Catalysis 13, 5307–5314 (2023).
Muriel, B. & Waser, J. Azide Radical Initiated Ring Opening of Cyclopropenes Leading to Alkenyl Nitriles and Polycyclic Aromatic Compounds. Angew. Chem. Int. Ed 60, 4075–4079 (2021).
Smyrnov, V., Muriel, B. & Waser, J. Synthesis of Quinolines via the Metal-free Visible-Light-Mediated Radical Azidation of Cyclopropenes. Org. Lett 23, 5435–5439 (2021).
Chandu, P., Biswas, S., Pal, K. & Sureshkumar, D. Organophotoredox Catalysis: Switchable Radical Generation from Alkyl Sodium Sulfinates for Sulfonylation and Alkylative Activation of C–C Bonds of Cyclopropenes. J. Org. Chem 89, 3912–3925 (2024).
Biswas, S., Das, D., Pal, K., Chandu, P. & Sureshkumar, D. Photocatalyzed Direct C(sp3)–H Alkenylation of Unactivated Alkanes via Tandem C–C Activation of Cyclopropenes. J. Org. Chem 89, 12421–12431 (2024).
Quapp, W., Bofill, J. M. & Aguilar-Mogas, A. Exploration of Cyclopropyl Radical Ring Opening to Allyl Radical by Newton Trajectories: Importance of Valley-Ridge Inflection Points to Understand the Topography. Theor. Chem. Acc. 129, 803–821 (2011).
Kramer, Z. C., Carpenter, B. K., Ezra, G. S. & Wiggins, S. Reaction Path Bifurcation in an Electrocyclic Reaction: Ring-Opening of the Cyclopropyl Radical. J. Phys. Chem. A. 119, 6611–6630 (2015).
Maley, S. M. et al. Machine Learning Classification of Disrotatory IRC and Conrotatory non-IRC trajectory Motion for Cyclopropyl Radical Ring Opening. Physical Chem. Phys. 23, 12309–12320 (2021).
Genossar, N. et al. Ring-Opening Dynamics of the Cyclopropyl Radical and Cation: The Transition State Nature of the Cyclopropyl Cation. J. Am. Chem. Soc 144, 18518–18525 (2022).
Ma, C. R. et al. Site-Selective Arene C–H Amination with Iron-Aminyl Radical. Nat. Catal 7, 636–645 (2024).
Wang, Z. L., Cheng, J. K. & Wang, F. Iron-Catalyzed C-7 Selective NH2 Amination of Indoles. Angew. Chem. Int. Ed 63, e202412103 (2024).
Minisci, F., Galli, R. & Cecere, M. Homolytic Amination of Aromatic Compounds by Redox Systems. Reactivity and Orientation. Tetrahedron Lett 6, 4663–4667 (1965).
Pratley, C., Fenner, S. & Murphy, J. A. Nitrogen-Centered Radicals in Functionalization of sp2 Systems: Generation, Reactivity, and Applications in Synthesis. Chem. Rev. 122, 8181–8260 (2022).
Chatterjee, S. et al. A Combined Spectroscopic and Computational Study on the Mechanism of Iron-Catalyzed Aminofunctionalization of Olefins Using Hydroxylamine Derived N−O Reagent as the “Amino” Source and “Oxidant”. J. Am. Chem. Soc. 144, 2637–2656 (2022).
Zhou, Y. et al. Mechanism and Reaction Channels of Iron-Catalyzed Primary Amination of Alkenes by Hydroxylamine Reagents. ACS Catal 13, 1863–1874 (2023).
Liu, Y. et al. Iron-Catalyzed Primary Amination of C(sp3)–H Bonds. J. Am. Chem. Soc. 146, 24863–24870 (2024).
Zard, S. Z. Recent Progress in the Generation and Use of Nitrogen-Centred Radicals. Chem. Soc. Rev. 37, 1603–1618 (2008).
Chen, J.-R., Hu, X.-Q., Lu, L.-Q. & Xiao, W.-J. Visible Light Photoredox-Controlled Reactions of N-Radicals and Radical Ions. Chem. Soc. Rev. 45, 2044–2056 (2016).
Xiong, T. & Zhang, Q. New Amination Strategies Based on Nitrogen-Centered Radical Chemistry. Chem. Soc. Rev. 45, 3069–3087 (2016).
Jiang, H. & Studer, A. Chemistry With N-Centered Radicals Generated by Single-Electron Transfer-Oxidation Using Photoredox Catalysis. CCS Chem 1, 38–49 (2019).
Xiong, P. & Xu, H.-C. Chemistry with Electrochemically Generated N-Centered Radicals. Acc. Chem. Res. 52, 3339–3350 (2019).
Kwon, K., Simons, R. T., Nandakumar, M. & Roizen, J. L. Strategies to Generate Nitrogen-centered Radicals That May Rely on Photoredox Catalysis: Development in Reaction Methodology and Applications in Organic Synthesis. Chem. Rev. 122, 2353–2428 (2022).
Jat, J. L. et al. Direct Stereospecific Synthesis of Unprotected N-H and N-Me Aziridines from Olefins. Science 343, 61–65 (2014).
Legnani, L. & Morandi, B. Direct Catalytic Synthesis of Unprotected 2-Amino-1-Phenylethanols from Alkenes by Using Iron(II) Phthalocyanine. Angew. Chem. Int. Ed. 55, 2248–2251 (2016).
Legnani, L., Prina-Cerai, G., Delcaillau, T., Willems, S. & Morandi, B. Efficient Access to Unprotected Primary Amines by Iron-Catalyzed Aminochlorination of Alkenes. Science 362, 434–439 (2018).
Makai, S., Falk, E. & Morandi, B. Direct Synthesis of Unprotected 2-Azidoamines from Alkenes via an Iron-Catalyzed Difunctionalization Reaction. J. Am. Chem. Soc. 142, 21548–21555 (2020).
Yu, D., Shing, K.-P., Liu, Y., Liu, H. & Che, C.-M. Ruthenium porphyrin catalysed intermolecular amino-oxyarylation of alkenes to give primary amines via a ruthenium nitrido intermediate. Chem. Commun. 56, 137–140 (2020).
Pozhydaiev, V., Vayer, M., Fave, C., Moran, J. & Lebœuf, D. Synthesis of Unprotected β-Arylethylamines by Iron(II)-Catalyzed 1,2-Aminoarylation of Alkenes in Hexafluoroisopropanol. Angew. Chem. Int. Ed. 62, e202215257 (2023).
Boullé, A., Doumbia, A., Mahy, J.-P. & Avenier, F. Unprotected Amine Transfer Performed by Non-Heme Iron(II) Complexes. Chem. Commun. 59, 79–81 (2023).
Pozhydaiev, V., Paparesta, A., Moran, J. & Lebœuf, D. Iron(II)-Catalyzed 1,2-Diamination of Styrenes Installing a Terminal NH2 Group Alongside Unprotected Amines. Angew. Chem. Int. Ed. 63, e202411992 (2024).
Chu, D. & Ellman, J. A. Stereospecific Synthesis of Unprotected, α,β-Disubstituted Tryptamines and Phenethylamines from 1,2-Disubstituted Alkenes via a One-Pot Reaction Sequence. Org. Lett. 25, 3654–3658 (2023).
Tu, W., Farndon, J. J., Robertson, C. M. & Bower, J. F. An Aza-Prilezhaev-Based Method for Inversion of Regioselectivity in Stereospecific Alkene 1,2-Aminohydroxylations. Angew. Chem. Int. Ed. 63, e202409836 (2024).
Jinan, D., Mondal, P. P., Nair, A. V. & Sahoo, B. O-Protected NH-Free Hydroxylamines: Emerging Electrophilic Aminating Reagents for Organic Synthesis. Chem. Commun 57, 13495–13505 (2021).
Gasser, V. C. M., Makai, S. & Morandi, B. The advent of electrophilic hydroxylAmine-Derived Reagents for the Direct Preparation of Unprotected Amines. Chem. Commun 58, 9991–10003 (2022).
France, S., Phun, L. & Aponte-Guzman, J. Acid-Catalyzed Ring-Opening Isomerizations of Cyclopropenes. Synlett 23, 2723–2728 (2012).
Makai, S. Preparation of O-Pivaloyl Hydroxylamine Triflic Acid. Org. Synth 97, 207–216 (2020).
Dzik, W. I., Böhmer, W. & de Bruin, B. Multiple Spin-State Scenarios in Organometallic Reactivity. In Spin States in Biochemistry and Inorganic Chemistry 2015, 103–129.
Shaik, S. Two-State Reactivity: Personal Recounting of its Conception and Future Prospects. Isr. J. Chem. 60, 938–956 (2020).
He, P. et al. Spin Effect on Redox Acceleration and Regioselectivity in Fe-Catalyzed Alkyne Hydrosilylation. Natl. Sci. Rev. 11, nwad324 (2024).
Acknowledgements
We are grateful to the financial support from the National Natural Science Foundation of China (Nos. 22101140, 22371143 and 22188101 for F.W.), the Fundamental Research Funds for the Central Universities (No. 63223009 for F.W.), Haihe Laboratory of Sustainable Chemical Transformations, Frontiers Science Center for New Organic Matter at Nankai University (No. 63181206 for F.W.). K.N.H. and J.-K. C. acknowledge the computational resources at UCLA (Hoffman2 Shared Cluster provided by UCLA Institute for Digital Research and Education’s Research Technology Group). We than Prof. Yin Yang (Nankai University) for her help on electron paramagnetic resonance (EPR) experiments and analysis.
Author information
Authors and Affiliations
Contributions
Experimental work was carried out by Q.W. with support from S.-X.T. DFT calculations were performed by J.K.C. and K.N.H. Project administration was done by F.W. Supervision was the responsibility of K.N.H. and F.W.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Buddhadeb Chattopadhyay, Zhijun Jia and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Q., Cheng, JK., Tang, SX. et al. Stereoselective synthesis of tetra- and tri-substituted alkenyl nitriles via aminative ring-opening of cyclopropenes with iron-aminyl radical. Nat Commun 16, 3168 (2025). https://doi.org/10.1038/s41467-025-58555-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58555-2
