
Abstract
The molecular mechanisms regulating the zonal distribution of metabolism in liver are incompletely understood. Here we use single nuclei genomics techniques to examine the spatial transcriptional function of transcription factor 7-like 2 (TCF7L2) in mouse liver, and determine the consequences of TCF7L2 transcriptional inactivation on the metabolic architecture of the liver and the function of zonated metabolic pathways. We report that while Tcf7l2 mRNA expression is ubiquitous across the liver lobule, accessibility of the consensus TCF/LEF DNA binding motif is restricted to pericentral (PC) hepatocytes in zone 3. In mice expressing functionally inactive TCF7L2 in liver, PC hepatocyte-specific gene expression is absent, which we demonstrate promotes hepatic cholesterol accumulation, impaired bile acid synthesis, disruption to glutamine/glutamate homeostasis and pronounced dietary-induced hepatic fibrosis. In summary, TCF7L2 is a key regulator of hepatic zonal gene expression and regulates several zonated metabolic pathways that may contribute to the development of fibrotic liver disease.
Similar content being viewed by others


Introduction
The liver is an essential metabolic organ that acts as a central hub for numerous physiological processes, including blood volume regulation, xenobiotic metabolism, detoxification, and lipid and cholesterol homeostasis. The liver is also vital for the maintenance of blood glucose levels through the release of stored glucose (glycogenolysis) and the de novo synthesis of glucose from non-hexose precursors (gluconeogenesis). Diseases of the liver account for approximately 2 million deaths per year worldwide, including from cirrhosis, hepatocellular carcinoma (HCC), acute hepatitis, and alcohol-use disorders (AUDs)1. The twin epidemics of obesity and type 2 diabetes (T2D) are intricately linked to the increased prevalence of metabolic dysfunction–associated fatty liver disease (MAFLD), which encompasses a range of pathologies from the relatively benign simple steatosis to metabolic dysfunction–associated steatohepatitis (MASH)2. The latter is characterized by hepatocellular injury and inflammation, which can lead to severe liver outcomes, including progressive hepatic fibrosis, cirrhosis, and end-stage liver complications. In patients with underlying T2D, the prevalence of MASH may be as high as 15%3, and MASH is now the leading etiology of cirrhosis in Western and industrialized countries4. Given the central role that the liver plays in the maintenance of whole-body homeostasis, and the high prevalence and morbidity of liver disease globally, it is important that we broaden our understanding of hepatic physiology with the goal of designing new treatment strategies that reduce the burden of liver disease.
The liver executes its diverse range of metabolic functions through the spatial division of labor. Liver lobules are divided into zones that regulate distinct, but often metabolically linked pathways. These zones are defined by the location of hepatocytes along the lobule relative to the portal triad (portal vein, bile duct, and hepatic artery) and the central vein (CV). This lobular structure exposes hepatocytes to various hormones, nutrients, morphogens, and other factors that facilitate the zonation of metabolic processes in the liver. It has been appreciated for decades that hepatocytes exhibit significant heterogeneity in their contribution to glucose metabolism and bile secretion5, and it is now recognized that most if not all, metabolic pathways in the liver display some degree of spatial heterogeneity in mammals. For example, de novo lipogenesis (DNL), β-oxidation, cholesterol biosynthesis, and amino acid metabolism all display zonation in rodent livers6,7. But despite considerable progress in our understanding of zonation, the molecular and physiological mechanisms regulating the spatial distribution of metabolic pathways remain incompletely understood. More importantly, the pathophysiological significance of metabolic zonation remains to be elucidated.
The Wnt/β-catenin signaling pathway is recognized as an important component of many pathological states8,9 and has emerged as a regulator of metabolic zonation10,11. Prior work has focused primarily on the role of β-catenin, a multifunctional protein that interacts with TCF/LEF transcription factors in the nucleus and functions as an intracellular transducer12,13. Liver-specific ablation of β-catenin eliminates the expression of pericentral (PC) hepatocyte genes around the CV and impairs liver regeneration14,15. Conversely, activation of Wnt signaling through the silencing of adenomatous polyposis coli (Apc) expands the PC hepatocyte zone16. Surprisingly, however, there are few published reports detailing the specific role of Transcription factor 7-like 2 (TCF7L2) in hepatic metabolic zonation in vivo. This is noteworthy because TCF7L2 is the primary transcriptional effector of the Wnt signaling pathway, and the TCF7L2 gene is the most significant type 2 diabetes (T2D) candidate gene to emerge from genome-wide association studies (GWAS) and whole-genome sequencing efforts17. While we and others have demonstrated in cellular and animal studies that TCF7L2 regulates pathways of hepatic glucose metabolism18,19,20,21,22,23, our experiments in cultured hepatocytes suggest that TCF7L2 may have a broad role in regulating multiple metabolic pathways that are typically zonated in vivo18.
In the current study, we evaluated the spatial transcriptional function of TCF7L2 in mouse liver and its overall role in metabolic zonation. Using a combination of single-nuclei RNA- and ATAC-Seq, we reveal that under normal chow-fed dietary conditions, Tcf7l2 gene expression is ubiquitous across the liver lobule in mice but that the transcriptional activity of TCF7L2 is restricted to PC hepatocytes located in zone 3 of the liver. Transcriptional inactivation of hepatic TCF7L2 in vivo disturbs key transcriptional regulators of hepatic zonation and eliminates the expression of most PC hepatocyte-specific genes. In a dietary model of MASH, liver-specific TCF7L2 mutant mice develop severe fibrosis that we mechanistically link to disruptions in bile acid synthesis and cholesterol homeostasis and disturbances in glutamine and glutamate balance across the lobule. Finally, we demonstrate that the expression of TCF7L2 is progressively reduced in human liver biopsies obtained from patients with increasing severity of fibrosis. The findings described here detail a hitherto unappreciated spatial transcriptional role for TCF7L2 in the liver that impacts multiple zonated metabolic pathways that may contribute to the development of fibrosis.
Results
Multimodal single nuclei genomics identifies zonated hepatocytes
We developed an experimental approach utilizing single-nuclei RNA-Seq and ATAC-Seq that facilitated the examination of the transcriptional regulation of spatially expressed genes by TCF7L2. Nuclei were isolated from samples of frozen liver harvested from chow-fed TCF7L2LoxP/LoxP mice that had been fasted overnight (14–16 h) and underwent multiomic sequencing library preparation using the 10X Genomics Chromium platform. The final control dataset for cluster analysis was derived from 5,652 nuclei that passed all QC filtering steps (Fig. 1A). The median unique molecular identifier (UMI) counts per nucleus was approximately 6383. All single nuclei data is available for download on the Gene Expression Omnibus database (accession number: GSE239480).

A Nuclei were extracted from samples of frozen mouse liver isolated from control mice (TCF7L2LoxP/LoxP, n = 3) and processed through the 10X Chromium Single Cell Multiome ATAC and Gene Expression pipeline. After QC and filtering, 5,652 nuclei were used for clustering analysis. B The RNA data of all samples was integrated using the reciprocal PCA approach via Seurat. C ATAC data were integrated using the reciprocal LSI approach via Signac, and (D) the Weighted Nearest Neighbor (WNN) approach was used for the integrative analysis of the two modalities. Each modality and the integrative analysis detected the major parenchymal and non-parenchymal cell populations in mouse liver. Hepatocyte nuclei were subdivided into periportal (PP) zone 1, mid-lobular zone 2, and pericentral (PC) zone 3 hepatocytes based on the expression of zonated marker genes, as detailed in the methodology. E Trajectory analysis was performed on hepatocyte nuclei only using Monocle3. The component of WNN hepatocyte-only clustering that was enriched for zone 1 PP marker gene expression was designated as the root node. We considered a gene zonally expressed if it was identified as a differentially expressed gene (DEGs) and was associated with the hepatocyte trajectory. F–H The expression (solid colored line) and accessibility (dashed line) of landmark zonated genes identified by Halpern et al.26. in C57BL/6 mice was examined in our control liver dataset. Shown is the average with the spread (shaded region) representing the SEM. I Pathway analysis revealed enrichment of classically zonated metabolic pathways in TCF7L2LoxP/LoxP livers, including bile secretion, cholesterol metabolism, and xenobiotic metabolism. Source data are provided as a Source Data file. Single nuclei data are available for download at GEO (accession: GSE239480).
To integrate the snRNA-Seq and snATAC-Seq datasets, we used weighted nearest neighbor (WNN) analysis, which calculates cell-specific modality weights and generates an integrated representation of multimodal data24. In both modalities and the integrated data, we identified the expected parenchymal and non-parenchymal cell populations in mouse liver (Fig. 1B–D). The non-parenchymal cells included several immune cell populations (B and T lymphocytes, Dendritic cells, Kupffer cells), hepatic stellate cells (HSCs), and endothelial cells (Fig. 1B– D). Hepatocyte nuclei formed a gradient that was governed by zone-specific regions of active chromatin and gene expression. We labeled hepatocytes using WNN clusters. The expression of carbamoyl phosphate synthetase 1 (Cps1) and arginase 1 (Arg1) distinguished hepatocytes in zone 1, glutamine synthetase (Glul) and axis inhibition protein 2 (Axin2) expression identified zone 3, and a group of clusters between the two was labeled as mid-lobular or zone 2 hepatocytes.
To quantitate zonally expressed genes, we performed pseudobulk differential gene expression analysis between the identified hepatocyte zones and completed multiomic pseudotime analysis on hepatocyte nuclei using Monocle325. For the latter, hepatocytes were clustered separately, the results of which are shown in Supplementary Fig 1. The component of WNN hepatocyte-only clustering that was enriched for zone 1 PP marker gene expression, was designated as the root node when calculating pseudotime. We considered a gene zonally expressed in the liver if it was identified as a differentially expressed gene (DEGs) and was associated with the hepatocyte trajectory (Fig. 1E). From this integrated approach, we identified a total of 526 zonally expressed genes in mouse liver. This analysis confirmed the zonal expression of genes known to exhibit spatial expression in the liver but also revealed the expression of genes that have not previously been described as zonated. For example, the transcriptional regulators PBX/Knotted 1 Homeobox 2 (Pknox2) and Thymocyte selection-associated HMG-box (Tox) were highly enriched in zone 3 and zone 1 hepatocytes, respectively (Fig. 1E).
To demonstrate that zonation in TCF7L2LoxP/LoxP mouse liver is comparable to zonation in wild-type C57BL/6 liver, we examined the expression and accessibility of several landmark PC (Fig. 1F), PP (Fig. 1G), and non-monotonic (Fig. 1H) genes identified by Halpern et al.26. All these landmark genes were similarly zonally enriched in TCF7L2LoxP/LoxP livers, except for the Major Urinary Protein 3 (Mup3) gene, which was expressed predominantly in zone 1. Pathway analysis of zonally expressed genes confirmed the enrichment of lipid, bile acid, cholesterol, and xenobiotic metabolism in zone 3, and pathways related to urea cycle and amino acid biosynthesis in zone 1 hepatocytes (Fig. 1I). Consistent with prior reports27,28, key genes involved in gluconeogenesis (i.e., Pck1), bile acid synthesis (Cyp7a1) and glycerolipid synthesis (i.e., Mogat1) also displayed the expected PP and PC distribution (Supplementary Fig 2). These data establish the utility of multimodal single nuclei approaches to quantify the expression and transcriptional regulation of zonated genes in mouse liver.
Tcf7l2 mRNA expression is ubiquitous across the liver lobule
Leveraging the approaches described above, we next examined the spatial expression of Tcf7l2 mRNA in our pseudobulked dataset. Tcf7l2 mRNA was found predominantly in the liver parenchyma, but variable Tcf7l2 expression was detected in some non-parenchymal cells, most notably Kupffer cells (KCs) (Fig. 2A and B). However, the expression of Tcf7l2 mRNA was ubiquitous across the liver lobule, and was not differentially expressed between the hepatocyte zones (Fig. 2B). To corroborate these snRNA-Seq data, we extracted RNA from zone 3 PC hepatocytes surrounding the CV and zones 1 and 2 hepatocytes in the mid-lobular and PP regions using LCM (Supplementary Fig. 3A and Fig. 3B). As expected, the expression of Glul was significantly enriched in PC hepatocytes, but consistent with our snRNA-Seq data, Tcf7l2 mRNA was not enriched in any hepatocyte population (Supplementary Fig. 3C). The expression of genes encoding related Wnt/β-catenin signaling proteins, including lymphoid enhancer-binding factor 1 (Lef1) and beta-catenin (Ctnnb1), also did not display zonal expression in our LCM experiments (Supplementary Fig. 3C).

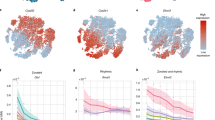
A, B The mRNA expression of Tcf7l2 is found in hepatocytes and several non-parenchymal cell populations, including Kupffer cells, hematopoietic stem and progenitor cells (HSPCs), and stellate cells. However, Tcf7l2 expression is not zonated and is found in nuclei isolated from hepatocytes across the liver lobule. C We detected 102 zonally enriched DNA binding motifs in our ATAC-Seq data, 50 of which are shown in this figure. These include highly similar motifs that are shared by transcription factors of the same family. Ordering hepatocyte nuclei by pseudotime highlights clear zonal preferences for the accessibility of several transcription factor motifs. D, E In our ATAC-Seq data, the accessibility of the conserved DNA binding motif for TCF7L2 (MA0253.1) was restricted to nuclei isolated from zone 3 hepatocytes and was largely inaccessible in non-parenchymal cell populations. F, G The presence of the TCF7L2 binding motif is enriched in linked ATAC-Seq peaks within 100 kb of the transcription start site (TSS) of zone 2 and 3 DEGs (FDR < 0.05), compared to non-zonated genes. In the plot, the lower and upper hinges correspond to the 25th and 75th percentiles, and the whiskers extend to ± 1.5 times the interquartile range (IQR) from each hinge. The notches extend 1.58*IQR / sqrt(n), which gives a roughly 95% confidence interval for comparing medians. Each small dot (gray) is an individual differentially expressed gene (DEG), and the large circles are those DEGs outside ± 1.5 times IQR. These data were derived from 3 independent TCF7L2LoxP/LoxP mouse livers. H In liver tissue isolated from TCF/LEF reporter mice, Wnt signaling pathway activity was restricted to zone 3 hepatocytes around the central vein (CV) and was not detected in zone 2 or periportal (PP) zone 3. The image shown is a representative image from five independent replicate experiments. Scale bar = 100 µm. Source data are provided as a Source Data file. Single nuclei data are available for download at GEO (accession: GSE239480).

A To excise the DNA binding domain (DBD) of TCF7L2, specifically in the liver, LoxP sites were placed either side of exon 11, and mice homozygous for this allele were bred with Alb-Cre mice. B, C In overnight fasted 32-week-old Hep-TCF7L2ΔDBD mice, a smaller TCF7L2 protein is expressed corresponding to the excision of the DBD. The Western blot image in (B) is representative of four independent experiments on at least three liver samples harvested from both control and Hep-TCF7L2ΔDBD mice. D Tcf7l2 transcripts containing exon 11 are significantly reduced in six-week-old (42-day) mice. E, F The expression of Axin2 and Lgr5, a direct TCF7L2 transcriptional target, are similarly reduced in 6-week-old Hep-TCF7L2ΔDBD mice. G–I The expression of Ccnd1, Lef1, and Tcf7 are not different in the livers of TCF7L2 mutant mice. J In control mice, glutamine synthetase (GS) protein was found exclusively around the central vein (CV) in the liver, but GS protein was not detectable in Hep-TCF7L2ΔDBD mice. K The mRNA expression of glutamine synthetase (Glul) also was undetectable in 6-week-old Hep-TCF7L2ΔDBD mice. Sample sizes for (D–I and K): Day 0 TCF7L2LoxP/LoxP n = 5, Hep-TCF7L2ΔDBD n = 4; Day 2 TCF7L2LoxP/LoxP n = 5, Hep-TCF7L2ΔDBD n = 8; Day 5 TCF7L2LoxP/LoxP n = 5, Hep-TCF7L2ΔDBD n = 5; Day 10 TCF7L2LoxP/LoxP n = 6, Hep-TCF7L2ΔDBD n = 5; Day 15 TCF7L2LoxP/LoxP n = 6, Hep-TCF7L2ΔDBD n = 5; Day 27 TCF7L2LoxP/LoxP n = 6, Hep-TCF7L2ΔDBD n = 5; Day 42 TCF7L2LoxP/LoxP n = 6, Hep-TCF7L2ΔDBD n = 5. Data were analyzed using a two-way ANOVA and Holm-Sidak multiple comparison tests, and are presented as mean values +/− SD. Source data are provided as a Source Data file.
The endogenous transcriptional activity of TCF7L2 is spatially regulated in liver
We hypothesized that the transcriptional activity of the TCF7L2 protein would be similarly ubiquitous in liver parenchyma. To test this, we first utilized our snATAC-Seq data to perform de novo transcription factor motif analysis in chromVAR, which was performed on hepatocytes in both their single-nuclei and pseudobulked states. To examine the pseudotime association with each motif, a new dataset was constructed via Monocle3 with chromVAR data for each nucleus. Motifs that were shared between the lists of trajectory-associated (q < 0.01) and differentially accessible motifs (q < 0.01 and a minimum absolute value of log2 fold change of 0.25 within any comparison) were considered zonally enriched.
We detected 102 zonally enriched DNA binding motifs, including highly similar motifs that are shared by transcription factors of the same family (Fig. 2C and Supplementary Fig. 4). Ordering hepatocyte nuclei by pseudotime demonstrated clear zonal preferences for several candidate transcription factors. For example, motifs for CCAAT-enhancer-binding proteins (C/EBP) and V-Maf Avian Musculoaponeurotic Fibrosarcoma Oncogene Homolog G (MAFG) were enriched in zone 3 PC hepatocytes (Fig. 2C), whereas binding motfis for the Aristaless-related homeobox gene (ARX) were enriched in zone 1 hepatocytes (Supplementary Fig 4). We also identified classes of transcription factors that have been recently described as regulators of zonal gene expression. Most notably, motifs for the T-box (TBX) family of transcription factors were enriched in zone 1 and zone 2 hepatocytes, but these motifs were inaccessible in zone 3 (Fig. 2C).
The TCF7L2 motif (MA0523.1; A-C/G-A/T-T-C-A-A-A-G) was amongst the most significantly enriched motifs in hepatocyte nuclei isolated from zone 3 but was predominantly inaccessible in zones 1 and 2 (Fig. 2C, E). Binding motifs of related TCF/LEF transcription factors were also highly enriched in zone 3 ATAC peaks, but it should be noted that these motifs are highly similar to the core TCF7L2 motif (Fig. 2C). Despite mRNA expression of Tcf7l2 in some non-parenchymal cells, we observed minimal enrichment of the TCF7L2 motif in these cells (Fig. 2E). To complement this analysis, we next quantitated the presence of TCF7L2 motifs in ATAC peaks significantly correlated to zonal DEGs within 100 kb of the transcription start site. The presence of a TCF7L2 motif was closely associated with the upregulation of genes expressed in Zone 3 and Zone 2 (Fig. 2F, G). Approximately 60% and 75% of the genes upregulated in Zone 3 and Zone 2 hepatocytes contained a TCF7L2 motif in a nearby linked peak, respectively, compared to 30% nearby genes that were not zonally expressed (Fig. 2G).
Finally, to directly visualize TCF7L2 activity in mouse liver in vivo, we examined GFP fluorescence in liver sections harvested from TCF/Wnt signaling reporter mice (TCF/Lef:H2B-GFP). These mice express an H2B-EGFP fusion protein under the control of six copies of the nuclear-localized TCF/Lef1 DNA binding site29 (Fig. 2H). A strong GFP signal was observed in just a few layers of zone 3 PC hepatocytes close to the central vein, but there was no detectable GFP signal in zones 1 or 2 (Fig. 2H).
Transcriptional inactivation of TCF7L2 in mouse liver
Given the zonal transcriptional activity of TCF7L2 in liver, we utilized our unique mouse model to specifically disrupt TCF7L2 DNA binding. TCF7L2LoxP/LoxP mice were bred with Alb-Cre mice (Hep-TCF7L2ΔDBD) to eliminate exon 11 (Fig. 3A), which leads to the expression of a TCF7L2 protein that lacks the DBD (Fig. 3B and C). To validate this strategy, we quantitated Tcf7l2 mRNA transcripts containing exon 11 in the liver during the postnatal period. In TCF7L2LoxP/LoxP mice, the expression of Tcf7l2 mRNA transcripts containing exon 11 significantly increased in liver during the postnatal period (Fig. 3D). However, in Hep-TCF7L2ΔDBD mice, the expression of these transcripts was significantly reduced from postnatal day 2 and were almost undetectable 6-weeks after birth (Fig. 3D). It is well established that DNA excision in liver by Alb-Cre occurs progressively with postnatal age30. The efficiency of recombination is typically 40% immediately after birth but increases to 60% by 1 week, 75% at weaning, and is complete at 6 weeks of age30. We confirmed transcriptional inactivation of TCF7L2 by examining the expression of several candidate target genes in the liver. In control livers, the expression of Axin2, which we and others have shown is a direct TCF7L2 target18,31, increased markedly in the postnatal period, but was undetectable in 6-week-old Hep-TCF7L2ΔDBD mice (Fig. 3E). Similar results were obtained for Leucine-rich repeat-containing G-protein coupled receptor 5 (Lgr5) expression (Fig. 3F), but we did not detect any changes in the expression of Cyclin D1 (Ccnd1) in Hep-TCF7L2ΔDBD mice (Fig. 3G), which was expressed at low levels in mature liver. In TCF7L2 mutant livers, no changes in the expression of related Wnt transcription factors Tcf7 (Fig. 3H) and Lef1 (Fig. 3I) were observed.
TCF7L2 inactivation eliminates zone 3-specific hepatocyte gene expression
We next examined whether the transcriptional inactivation of TCF7L2 disrupted PC hepatocyte marker gene expression. The expression of Glul, a classic zone 3 hepatocyte marker, was unaffected in the livers of newborn Hep-TCF7L2ΔDBD mice, but both Glul mRNA and glutamine synthetase (GS) protein levels were abolished in livers isolated from 6-week-old (42 days) Hep-TCF7L2ΔDBD mice (Fig. 3J, K).
To determine the impact of TCF7L2 inactivation on hepatic zonal gene expression, we employed our multiomic single nuclei approach in a new analysis that combined both the control and mutant datasets. Using snRNA- and snATAC-Seq and the integrated WNN data, we detected both parenchymal and non-parenchymal cell types in control and mutant livers (Fig. 4A–C). Consistent with the analysis detailed above in the control livers only, hepatocyte nuclei formed a gradient based on the zonal expression of genes and regions of active chromatin linked to these genes. Because the zone 3 subcluster was substantially diminished in TCF7L2 mutant livers (Fig. 4A–C), in hepatocyte-only clustering, we combined nuclei from zones 2 with any remaining nuclei from zone 3 in the TCF7L2 mutant mice to perform DEG and pseudotime analysis (Supplementary Fig 5). This analysis revealed that TCF7L2 inactivation eliminated the expression of the majority of zone 3-specific genes compared to control mice but had a minimal impact on zone 1 gene expression (Fig. 4D). Using an FDR of <0.01, we detected 75 and 189 DEGs in zone 1 and 2 hepatocytes, respectively, but 607 DEGs in zones 3 hepatocytes, compared to the corresponding zones in control mice (Fig. 4E). Most zone 2 (75%) and zone 3 (60%) DEGs were downregulated in mutant mice, consistent with positive regulation of these genes by native TCF7L2 (Fig. 4E).

Multiomics analysis was performed on nuclei isolated from the livers of TCF7L2LoxP/LoxP (n = 3) and Hep-TCF7L2ΔDBD (n = 3) mice. A Single nuclei RNA-Seq (snRNA-Seq), (B) single nuclei ATAC-Seq (snATAC-Seq), and (C) integrated weighted nearest neighbor (WNN) are shown. In Hep-TCF7L2ΔDBD mice, nuclei corresponding to zone 3 were largely absent and instead clustered with zone 2 nuclei. D Heatmap of gene expression following trajectory analysis reveals the clear absence of zone 3 gene expression in Hep-TCF7L2ΔDBD. The color gradient above the heatmap is for visual purposes only. E Differential expression analysis confirms that the loss of TCF7L2 transcription had minimal impact on zone 1 and 2 genes, but significantly impacted the expression of genes in zone 3 nuclei. For this DEG analysis, TCF7L2 mutant zone 3 nuclei were identified based on their clustering relative to zone 3 nuclei in the control mice. F In snATAC-Seq analysis, accessibility of the TCF7L2 binding motif was disrupted in zone 3 nuclei isolated from Hep-TCF7L2ΔDBD mice. Mutant zones 2 and 3 nuclei were combined for statistical comparison with respective control nuclei, as detailed in the text. G TCF7L2 loss of function alters the motif accessibility of several transcription factors that may contribute to the maintenance of metabolic zonation. Heatmap of chromVAR regression scores of motifs that were associated with the hepatocyte trajectory and displayed significant differences in accessibility between zones. H Analysis of snATAC-Seq data using Signac highlights the reduction of transcription factor (TF) motif accessibility in zones 2 and zones 3. A heatmap of fold enrichment for motif binding sites within sets of differentially less accessible chromatin regions between TCF7L2 and control nuclei in each hepatocyte zone. Source data are provided as a Source Data file. Single nuclei data are available for download at GEO (accession: GSE239480).
Transcriptional coregulators of zonation are altered in TCF7L2 mutant livers
Loss of TCF7L2 DNA binding in Hep-TCF7L2ΔDBD mice significantly restricted the accessibility of the TCF7L2 binding motif in zone 3 and zone 2 hepatocytes, but did not impact the accessibility of the motif in zone 1 (Fig. 4F–H). However, TCF7L2 loss of function also altered the motif accessibility of several transcription factors that may contribute to the maintenance of metabolic zonation (Fig. 4G, H). Consistent with our snRNA-Seq data, this was more pronounced in zone 3 compared to zone 1 nuclei, where we detected 77 and 11 differentially accessible motifs, respectively (Fig. 4G and H).
Recently published research using single nuclei multiomic techniques combined with deep learning computational methodology identified TBX3 and TCF7L1 as key transcriptional repressors of PC and PP hepatocytes, respectively32. Given that TCF7L1 is a member of the same TCF/LEF family of transcription factors as TCF7L2 and that TBX3 acts as a tissue-specific component of Wnt/β-catenin signaling33, we focused on the zonal expression and motif accessibility of these two factors in Hep-TCF7L2ΔDBD livers. Interestingly, we found that the accessibility and expression of Tbx3 and Tcf7l1 were inversely correlated. In control mice, Tcf7l1 expression was enriched in zones 1 and 2 (Fig. 5A, B), but the accessibility of the TCF7L1 motif was highest in zone 3 hepatocytes (Fig. 5C). Conversely, Tbx3 mRNA expression was restricted to zone 3 in control mice (Fig. 5D, E), but the accessibility of the TBX3 motif was highest in zone 1 (Fig. 5F). Negative associations between TBX3/TCF7L2 motif accessibility and Tbx3/Tcf7l1 expression may occur nearby genes that are negatively correlated with these transcriptional regulators32. Consistent with this, in Hep-TCF7L2ΔDBD mice we observed that the accessibility of the TBX3 motif was elevated more than 2-fold in PC hepatocytes but that its expression was significantly reduced in the same nuclei (Fig. 5D–F). In contrast, Tcf7l1 expression was upregulated in mutant zone 3 hepatocytes, where accessibility of the TCF7L1 motif was concomitantly diminished (Fig. 5A–C). These findings are consistent with the zonal repressor function of TBX3 and TCF7L1 and highlight that the coordinated action of these transcriptional regulators participates in the downregulation of PC gene expression in Hep-TCF7L2ΔDBD mice (Fig. 5G).

A, B snRNA-Seq demonstrates that the expression of Tcf7l1 is found primarily in zone 1 nuclei isolated from TCF7L2LoxP/LoxP mice, but is significantly enhanced in zone 2 and zone 3 nuclei in Hep-TCF7L2ΔDBD mice. C Conversely, the accessibility of the TCF7L1 motif is reduced in zone 3 nuclei isolated from Hep-TCF7L2ΔDBD mice. D and E The expression of Tbx3 is highest in zone 3 nuclei in TCF7L2LoxP/LoxP mice, which is ameliorated in Hep-TCF7L2ΔDBD mice. F The accessibility of the TBX3 binding motif is elevated in zone 3 nuclei isolated from TCF7L2 mutant livers. Default DESeq2 statistical testing methodology was used (Wald test) with Benjamin and Hochberg multiple comparison correction. Mutant zones 2 and 3 nuclei were combined for statistical comparison with respective control nuclei, as detailed in the text. G Summary of the proposed role of TBX3 and TCF7L2 in PC hepatocytes lacking TCF7L2. The release of TCF7L2 from the DNA enhances TCF7L1 expression and activity, which suppresses the expression of pericentral (PC) hepatocyte genes. Conversely, TBX3 expression and chromatin occupancy is reduced, allowing other transcription factors (TF) to access this site and activate the expression of periportal (PP) genes. Single nuclei data are available for download at GEO (accession: GSE239480).
Diet-induced hepatic fibrosis is exacerbated in Hep-TCF7L2ΔDBD mice
Important questions remain about the metabolic and pathophysiologic significance of disrupted zonation. Using our Hep-TCF7L2ΔDBD mice as a unique model of zone 3 hepatocyte disruption, we examined the phenotype of Hep-TCF7L2ΔDBD mice with a primary focus on liver health. Male and female Hep-TCF7L2ΔDBD mice that were weaned and monitored for 12 weeks developed normally on a chow diet and had similar body weights compared to controls (Supplementary Fig 6A). We placed a cohort of male 8-week-old mice on a regular chow diet for an additional 6 months and monitored their basic metabolic phenotype. There was no effect of hepatic TCF7L2 inactivation on bodyweight, food intake, body composition, glucose tolerance, or insulin sensitivity (Supplementary Fig 6B–G). However, there was a small but significant reduction in liver weight in Hep-TCF7L2ΔDBD mice (Supplementary Fig 6H). While chow-fed Hep-TCF7L2ΔDBD mice had normal levels of hepatic triglycerides, they exhibited significantly increased hepatic cholesterol (Supplementary Fig 6I, J).
To test the impact of TCF7L2 inactivation on the susceptibility to diet-induced liver pathology, we next fed mice either the GAN diet for 24 weeks or the CDAHFD diet for 8 weeks (Fig. 6A) and monitored lipid metabolism and the development of hepatic fibrosis. Body weight and food intake were not different between control and Hep-TCF7L2ΔDBD mice fed the CDAHFD, but both were lower in TCF7L2 mutant mice fed the GAN diet (Supplementary Fig 7). Consistent with chow-fed mice, liver weights were smaller in TCF7L2 mutant livers fed either diet (Fig. 6B). Total (Fig. 6C) and free (Fig. 6D) hepatic cholesterol levels were markedly increased in Hep-TCF7L2ΔDBD mice following both diets, but there were no differences in hepatic triglycerides (Fig. 6E). Circulating serum cholesterol concentrations were significantly lower in Hep-TCF7L2ΔDBD mice fed either diet (Fig. 6F), but serum triglycerides were elevated in Hep-TCF7L2ΔDBD mice after the CDAHFD only (Fig. 6G). No changes in circulating FFA levels were observed (Fig. 6H).

A Schematic outlining the two dietary models of MASLD and MASH. Mice were placed on the choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD) or the Gubra-Amylin NASH (GAN) diet for 8- and 24 weeks, respectively. B Liver weights were significantly lower in Hep-TCF7L2ΔDBD mice following both diets (CDAHFD diet: TCF7L2LoxP/LoxP n = 6, Hep-TCF7L2ΔDBD n = 11; GAN diet: TCF7L2LoxP/LoxP n = 12, Hep-TCF7L2ΔDBD n = 15). C Total and (D) free hepatic cholesterol was elevated in Hep-TCF7L2ΔDBD mice, but there were no differences in hepatic triglycerides (E). Amongst serum lipids, cholesterol (F) was consistently lower in Hep-TCF7L2ΔDBD mice fed either diet (sample sizes for C–F: CDAHFD: TCF7L2LoxP/LoxP n = 11, Hep-TCF7L2ΔDBD n = 10; GAN: TCF7L2LoxP/LoxP n = 12, Hep-TCF7L2ΔDBD n = 15). Triglycerides (G) were elevated in mutant mice fed the GAN diet (CDAHFD: TCF7L2LoxP/LoxP n = 13, Hep-TCF7L2ΔDBD n = 11; GAN: TCF7L2LoxP/LoxP n = 12, Hep-TCF7L2ΔDBD n = 15), but no changes in free fatty acids (FFA) (H) were detected (CDAHFD: TCF7L2LoxP/LoxP n = 11, Hep-TCF7L2ΔDBD n = 11; GAN: TCF7L2LoxP/LoxP n = 13, Hep-TCF7L2ΔDBD n = 15). I Hepatic fibrosis in mice fed the CDAHFD was examined using Sirius Red staining and was elevated in Hep-TCF7L2ΔDBD mice (quantitated in M). J The expression of genes involved in fibrogenesis and inflammation was elevated in Hep-TCF7L2ΔDBD mice fed the CDAHFD diet (TCF7L2LoxP/LoxP n = 12, Hep-TCF7L2ΔDBD n = 15). K Hepatic fibrosis in mice fed the GAN diet was examined using Sirius Red staining and was not different between control and Hep-TCF7L2ΔDBD mice (quantitated in M). L However, the expression of genes involved in fibrogenesis and inflammation was also elevated in Hep-TCF7L2ΔDBD mice fed the GAN diet (TCF7L2LoxP/LoxP n = 12, Hep-TCF7L2ΔDBD n = 15). N The expression of hepatic Tcf7l2 is reduced in mice fed the CDAHFD and GAN diet (CHOW n = 11, CDAHFD n = 11, GAN n = 12), and (O) in mice fed a fast-food diet (FFD) for 11 months CON n = 8, FFD n = 9). This diet consisted of a commercially available high-fat, high-cholesterol diet (Research Diets Inc, D12079B) and drinking water containing 23.1 g fructose and 17.2 g glucose per 1000 mL of water. P In deidentified and anonymized data obtained from the analysis of human liver biopsies, TCF7L2 expression is progressively reduced as fibrosis severity increases, an effect not modified by male (M) or female (F) sex (F0: males n = 5, females n = 5; F1: males n = 10, females n = 13; F2 males n = 11, females n = 10; F3: males n = 27; females n = 26; F4: males n = 21, females n = 19). In the plot, the box extends from the 25th to the 75th percentile, and the line across the box represents the median value. Whiskers extend to minimum and maximum values. Data are presented as mean values +/− SD and were analyzed using unpaired two-sided Welch t tests with (panels B–H, J, and M) or without (panel O) Holm-Sidak correction for multiple comparisons or one-way ANOVA with Holm-Sidak correction for multiple comparisons (panel N). Human biopsy data (panel P) were analyzed using a two-way ANOVA with Holm-Sidak multiple comparison correction. Source data are provided as a Source Data file.
Hepatic fibrosis was increased approximately 2-fold in Hep-TCF7L2ΔDBD mice fed the CDAHFD diet (Fig. 6I, M). This was confirmed by increased expression of markers of hepatic fibrosis, collagen synthesis, and inflammation in Hep-TCF7L2ΔDBD mice (Fig. 6J). In mice fed the GAN diet, we detected minimal fibrosis in control animals and no differences between Hep-TCF7L2ΔDBD and control mice (Fig. 6K and M). Interestingly, however, the expression of many pro-fibrogenic genes was also elevated in Hep-TCF7L2ΔDBD mice fed the GAN diet (Fig. 6L). We quantified the expression of Tcf7l2 in control mice fed either the CDAHFD or GAN diet and found that the expression of hepatic Tcf7l2 was significantly reduced (Fig. 6N). Confirming these findings, hepatic Tcf7l2 mRNA was also reduced in wild-type mice fed a pro-fibrogenic FFD for 11 months34. This diet led to marked increases in steatosis, fasting glucose and insulin, and hepatic fibrosis (Supplementary Fig 8). To determine which cell type might account for diet-induced reductions in Tcf7l2 expression, we re-analyzed a publicly available single-cell dataset from mice fed a high fructose high-fat diet (HFHFD) for 15 and 30 weeks35. This analysis demonstrate that Tcf7l2 expression was reduced in both hepatocytes (zone 1 and 2), and KCs following the HFHFD (Supplementary Fig 9).
To translate these findings to humans, we obtained data on the expression of TCF7L2 in baseline liver biopsies from patients with increasing severity of fibrosis (F0 – F4) that participated in the selonsertib and simtuzumab clinical trials36,37. Consistent with our pre-clinical data, the expression of TCF7L2 progressively decreased as fibrosis worsened, an effect that was not modified by sex (Fig. 6P).
Pericentral metabolic pathways are depleted in Hep-TCF7L2ΔDBD mice
To determine the mechanisms underlying the increased susceptibility to diet-induced fibrosis in Hep-TCF7L2ΔDBD mice, we performed gene set enrichment analysis using Enrichr38 to uncover the metabolic pathways disturbed in the livers of these mice. Consistent with a primary role for TCF7L2 in zone 3, multiple pathways typically active in this region were depleted in Hep-TCF7L2ΔDBD mice (Supplementary Fig 10A). For example, “Bile secretion” pathways, which includes key enzymes involved in the synthesis of bile acids from cholesterol (i.e., CYP7A1) as well as the transport of cholesterol from hepatocytes to into the bile canaliculus (i.e., ABCG5 and ABCG8), were significantly depleted in Hep-TCF7L2ΔDBD livers. Disturbances in glutamine and glutamate metabolism were also clearly represented in our pathway analysis. The “Glutamatergic pathway” is enriched in mutant zone 3 livers and includes the central enzyme glutaminase (GLS), which converts glutamine to glutamate and generates ammonium for urea synthesis. Several lipid metabolism pathways, including sphingolipid and glycerolipid metabolism, were similarly downregulated in mutant mice, as were Wnt/β-catenin and PPAR signaling pathways (Supplementary Fig 10B–E). As expected from our DEG analysis, there were minimal differences in the metabolism of zone 1 hepatocytes between Hep-TCF7L2ΔDBD and control mice (Supplementary Fig 10F, G).
Hepatic glutaminolysis is enhanced in Hep-TCF7L2ΔDBD mice
To explore these pathway results in more detail, the effect of hepatic TCF7L2 inactivation on the urea cycle and glutamine metabolism was quantitated. This pathway involves distinct enzymatic steps in zone 1 and zone 3 hepatocytes, and constitutes the two major systems for ammonia detoxification in mice39 (Fig. 7A). The significance of this pathway in liver fibrosis was recently demonstrated in studies where rate-limiting enzymes of glutamine catabolism (glutaminolysis) were upregulated40. Moreover, hepatic-specific ablation of GS in mice exacerbates HCC development driven by β-catenin and c-Met41. Immunohistological analysis of ASS1 revealed that expression of this enzyme was restricted to zones 1 and 2 in control mice fed any diet, as has been previously reported26,42 (Fig. 7B). However, in TCF7L2 mutant mice, ASS1 staining was also evident in zone 3 irrespective of the diet. In control mice fed the CDAHFD, we detected an expansion of GS staining in zone 3 hepatocytes, but GS expression remained undetectable in Hep-TCF7L2ΔDBD mice fed the CDAHFD or GAN diet (Fig. 7B).
The loss of GS in Hep-TCF7L2ΔDBD mice did not alter hepatic glutamine or glutamate in chow-fed mice (Fig. 7C, D). However, following both the CDAHFD and GAN diets, hepatic glutamine levels were significantly reduced in Hep-TCF7L2ΔDBD mice (Fig. 7C). Hepatic glutamate concentrations were reduced in control mice fed the CDAHFD and GAN diets, but in Hep-TCF7L2ΔDBD mice fed the CDAHFD glutamate levels remained elevated (Fig. 7D). These changes led to a marked increase in the hepatic glutamate/glutamine ratio in Hep-TCF7L2ΔDBD mice fed the CDAHFD (Fig. 7E). The circulating glutamate/glutamine ratio was elevated in chow- and GAN-fed Hep-TCF7L2ΔDBD mice compared to controls, but not in mice fed the CDAHFD (Fig. 7H). This was due to similar increases in serum glutamine and glutamate in control and TCF7L2 mutant mice fed the CDAHFD (Fig. 7F, G). The conversion of glutamine to glutamate in liver is catalyzed by two mitochondrial glutaminases (GLS and GLS2), which are found in periportal hepatocytes (Fig. 7A). The mRNA expression of both Gls and Gls2 was significantly elevated in Hep-TCF7L2ΔDBD mice fed either diet (Fig. 7I). Taken together, these data highlight a regulatory role for TCF7L2 in pathways of urea cycle, ammonia detoxification and glutamine/glutamate metabolism, and demonstrate the importance of maintaining zone 3 hepatocyte function in liver.

A The two primary pathways of ammonia [NH4]+ detoxification in the liver are urea and glutamine synthesis. Urea synthesis in zone 1 PP hepatocytes is a system of low affinity for ammonia detoxification. The glutaminase (GLS) enzyme is stimulated by [NH4]+ and regulates the flow of nitrogen derived from glutamine through the urea cycle. In zone 3 PC hepatocytes, glutamine synthetase (GS) uses [NH4]+ and glutamate to synthesize glutamine and, thus, acts as a high-affinity scavenger for [NH4]+ that escapes zone 1 detoxification. B In mouse liver, we used staining of argininosuccinate synthase 1 (ASS1) and GS to distinguish zones 1 and 2 from zone 3. In chow-fed TCF7L2LoxP/LoxP mice, ASS1 was not observed in zone 3 hepatocytes (top panel, red arrowheads), whereas GS staining was found in only a few layers of hepatocytes around the central vein (CV) (top panel, black arrowheads). Following the GAN diet, GS staining was more diffuse and expanded into mid-lobular zone 2 hepatocytes (middle panel, black arrowheads). In TCF7L2 mutant livers, GS staining is completely absent, and centrilobular ASS1 staining is observed. Scale bar = 100 µm. C Hepatic glutamine levels were unchanged in chow-fed Hep-TCF7L2ΔDBD mice but were significantly reduced following the CDAHFD (blue bars) and GAN (pink bars) diets. D Glutamate concentrations in the liver were reduced following both diets in control mice, but remained elevated in Hep-TCF7L2ΔDBD mice fed the CDAHFD. E An elevated hepatic glutamate:glutamine ratio was observed in TCF7L2 mutant mice fed the CDAHFD, indicative of glutaminolysis (sample sizes for panels C–E: CHOW TCF7L2LoxP/LoxP n = 11, CDAHFD TCF7L2LoxP/LoxP n = 11, GAN TCF7L2LoxP/LoxP n = 12; CHOW Hep-TCF7L2ΔDBD n = 15, CDAHFD Hep-TCF7L2ΔDBD n = 10, GAN Hep-TCF7L2ΔDBD n = 15). F, G Serum glutamine and glutamate concentrations were augmented following the CDAHFD, and a significant increase in the circulating glutamate:glutamine ratio (H) was observed in chow and GAN diet-fed mice, but not in CDAHFD fed mice (sample sizes for panel F–H: CHOW TCF7L2LoxP/LoxP n = 11, CDAHFD TCF7L2LoxP/LoxP n = 13, GAN TCF7L2LoxP/LoxP n = 10; CHOW Hep-TCF7L2ΔDBD n = 14, CDAHFD Hep-TCF7L2ΔDBD n = 11, GAN Hep-TCF7L2ΔDBD n = 15. I The mRNA expression of Gls and glutaminase 2 (Gls2) was increased in TCF7L2 mutant mice fed both diets (CDAHFD: TCF7L2LoxP/LoxP n = 11, Hep-TCF7L2ΔDBD n = 10; GAN: TCF7L2LoxP/LoxP n = 11, Hep-TCF7L2ΔDBD n = 16). Data are presented as mean values +/−SD and were analyzed using a two-way ANOVA (panels C–H) or Welch t tests (panel I), with Holm-Sidak correction for multiple comparisons. Source data are provided as a Source Data file.
Zonal TCF7L2 regulates hepatic cholesterol through bile acid synthesis
A consistent finding in Hep-TCF7L2ΔDBD mice is the accumulation of cholesterol in the liver, which may contribute to the development of fibrosis under certain conditions. Therefore, we explored the regulation of cholesterol homeostasis by TCF7L2, and focused on bile acid synthesis in mice, as this occurs primarily in zone 3 hepatocytes (Fig. 8A). Analysis of the total (whole-body) bile acid pool by mass spectrometry revealed a significant reduction in total bile acids in Hep-TCF7L2ΔDBD mice that was driven by reduced hepatic and intestinal bile acids, which is the largest contributor to the bile acid pool (Fig. 8B, C). Stool bile acids also were reduced (Fig. 8B). However, there were minimal to no changes to the relative composition of the bile acid pool (as a percentage of total) (Fig. 8D). The absolute reduction in hepatic and intestinal bile acids was characterized by reduced taurine-conjugated primary bile acids, although there were reductions in the murine-specific primary bile acid α-muricholic acid (α-MCA) in both tissues (Figs. 8E, F).

A Primary bile acids (i.e., cholic acid) are synthesized from cholesterol in zone 3 PC hepatocytes, following which they are secreted into the lumen of the intestine and modified by gut bacteria to produce secondary bile acids (i.e., deoxycholic acid). B, C In Hep-TCF7L2ΔDBD mice fed the CDAHFD, both hepatic and intestinal bile acids were significantly reduced. In a separate cohort of mice, stool bile acids were similarly lower in Hep-TCF7L2ΔDBD mice fed the CDAHFD. However, the composition of the bile acid pool (D) was not different between the two groups of mice. Example of hepatic (E) and intestinal (F) conjugated and unconjugated bile acid that were significantly reduced in Hep-TCF7L2ΔDBD mice. G The cholesterol 7 alpha-hydroxylase (CYP7A1) enzyme is the rate-controlling step in the synthesis of bile acids from cholesterol in the liver, and its activity can be monitored through the quantitation of the intermediate metabolite 7α-hydroxy-4- cholesten-3-one (C4). H Total hepatic C4 was reduced in TCF7L2 mutant mice (sample sizes for panels B–H: TCF7L2LoxP/LoxP n = 8, Hep-TCF7L2ΔDBD n = 8). I A TCF7L2 ChIP-Seq peak is observed in the proximal promoter of the mouse Cyp7a1 gene and overlaps a region of open chromatin. J The mRNA expression of hepatic Cyp7a1 was significantly reduced in Hep-TCF7L2ΔDBD mice (TCF7L2LoxP/LoxP n = 11, In Hep-TCF7L2ΔDBD n = 10). K The expression of ileal Fgf15 mRNA was reduced in Hep-TCF7L2ΔDBD mice, suggesting that feedback inhibition by elevated circulating FGF15 cannot explain the downregulation of Cyp7a1 in Hep-TCF7L2ΔDBD mice (TCF7L2LoxP/LoxP n = 7, In Hep-TCF7L2ΔDBD n = 8). Data are presented as mean values +/− SD and were analyzed using two-sided unpaired Welch t tests. To correct for multiple comparisons in bile acids analysis (panels E and F), a false discovery (FDR) approach was used with a q-value ≤ 0.05 considered significant. Source data are provided as a Source Data file. Mass spec data are available for download at Metabolomics Workbench (accession: ST003641).
To confirm a specific impairment in bile acid synthesis from cholesterol in Hep-TCF7L2ΔDBD mice, we quantitated hepatic levels of 7α-hydroxy-4-cholesten-3-one (C4), which is a stable intermediate that accurately reflects cholesterol 7α-hydroxylase (CYP7A1) activity and bile acid synthesis (Fig. 8G)43,44. While not statistically significant (P = 0.07), there was a clear reduction in hepatic C4 levels in Hep-TCF7L2ΔDBD mice (Fig. 8H). Consistent with these findings, Cyp7a1 mRNA was downregulated in TCF7L2 mutant livers following the CDAHFD (Fig. 8I). To understand the mechanisms regulating CYP7A1, we examined mouse liver ChIP-Seq data from our recent publication45, and noted a TCF7L2 binding peak in the Cyp7a1 proximal promoter that overlaps with a region of open chromatin (Fig. 8I). Interestingly, ileum Fgf15 mRNA was lower in TCF7L2 mutant mice, indicating that reduced Cyp7a1 in these mice is not the result of negative feedback regulation by intestinal-derived FGF15.
Discussion
Since the discovery that single nucleotide polymorphisms (SNPs) in the TCF7L2 gene are significantly associated with T2D, many research groups have attempted to unravel the metabolic function of TCF7L2 in various tissues46,47,48,49. While several mechanistic studies have examined the metabolic function of TCF7L2 in the liver in vitro and in vivo, the zonal function of hepatic TCF7L2 has not previously been described. In the current manuscript, we reveal that the transcriptional function of TCF7L2 in mouse liver is spatially restricted and that the metabolic consequences of TCF7L2 inactivation in vivo largely reflect its zonal activity. Hepatic-specific inactivation of TCF7L2 disrupted pathways linked to ammonia detoxification, including the urea cycle and glutamine/glutamate homeostasis. We also demonstrate that TCF7L2 plays a unique role in the regulation of cholesterol homeostasis through the control of zonated pathways of bile acid synthesis. Under conditions of metabolic stress, disruptions to these pathways accelerate the development of hepatic fibrosis in mice, a finding that is supported by data demonstrating that Tcf7l2/TCF7L2 expression is reduced in several preclinical models of MASH and in the livers of human patients with severe fibrosis. These findings further detail the transcriptional function of TCF7L2 in the liver, and underscore the importance of TCF7L2 in the regulation of multiple zonated metabolic pathways.
A significant finding in the current manuscript is that Tcf7l2 mRNA expression is found across the liver lobule, but the transcriptional activity of TCF7L2 is limited to a small population of zone 3 hepatocytes surrounding the CV. Removal of the DNA binding domain of TCF7L2 eliminates the expression of PC hepatocyte-specific genes in zone 3 but has only minimal effects on PP hepatocyte-specific gene expression in zones 1 and 2. This spatial function of TCF7L2 is consistent with the zonal activity of the Wnt/β-catenin signaling pathway in liver50 and suggests that under the conditions examined here (chow-fed and overnight fasted mice), PP and mid-lobular TCF7L2 is transcriptionally inactive. The zonal function of TCF7L2 in the liver also mirrors its function in the intestine, where Wnt signaling is sequestered to the base of colonic crypts despite the expression of Tcf7l2 throughout the intestine51. In the Hep-TCF7L2ΔDBD mice, reductions in PC gene expression were accompanied by increased expression of genes typically excluded from zone 3 (i.e., Ass1). We identify the transcription factors TBX3 and TCF7L1 as playing important roles in driving zonal gene expression changes in the absence of TCF7L2. These factors have recently been identified as repressors of PC and PP hepatocyte gene expression, respectfully32. While TCF7L1 is closely related to TCF7L2 and is a member of the TCF/LEF family of transcription factors, TBX3 also is a downstream target of Wnt signaling, and liver-specific deletion of Tbx3 promotes hepatic fibrosis52. We extend these findings and suggest a model where increased accessibility of binding sites previously occupied by TBX3 drives PP-specific gene expression in zone 3 of TCF7L2 mutant livers. The direct and indirect transcriptional mechanisms regulating zonal gene expression in the presence and absence of TCF7L2 will require further study, but our data indicate that a complex network of factors involving TCF7L2 cooperate to regulate zonal gene expression in the liver.
We demonstrate that Tcf7l2 expression is reduced in several preclinical models of hepatic fibrosis, and in humans with MASH, a finding that is consistent with recent reports53 and suggests an association between TCF7L2 and hepatic fibrosis. While we cannot establish a causal role for TCF7L2 in human liver fibrosis in the current study, Hep-TCF7L2ΔDBD mice were more susceptible to fibrosis when fed the CDAHFD diet. This may be mechanistically linked to our finding that TCF7L2 regulates several zonated pathways, including bile acid synthesis, cholesterol homeostasis and urea metabolism, ammonia detoxification, and glutamine/glutamate balance. Elevated hepatic cholesterol was observed in Hep-TCF7L2ΔDBD mice when fed a chow diet, but both total and free hepatic cholesterol were elevated in mice fed the CDAHFD and GAN diets. A primary route of cholesterol efflux in the liver is through bile acid synthesis, which occurs in zone 3 hepatocytes54. In Hep-TCF7L2ΔDBD mice fed the CDAHFD, there were significant reductions in hepatic and intestinal bile acids, which we link to reduced hepatic CYP7A1 activity. These results differ from those described in mice lacking β-catenin in the liver. In these mice, while elevated hepatic cholesterol was observed following the MCD diet, this was associated with increased hepatic bile acids in the context of reduced Cyp7a1 expression55. The reason for this discrepancy is not clear, but it is possible that the improved sensitivity of our mass spec method to quantify the bile acid pool was an important factor. In addition, hepatic β-catenin knockout may have additional metabolic effects in the liver that are not observed in livers with targeted inactivation of TCF7L2.
Accumulation of cholesterol may have directly contributed to the increased fibrosis observed in Hep-TCF7L2ΔDBD mice fed the CDAHFD. Free cholesterol accumulation and cholesterol-metabolite-mediated toxicity directly result in hepatocyte dysfunction, primarily through disruptions to the fluidity of cell and organelle membranes56,57, and is a well-known risk factor for the development of MASH57. However, increased hepatic cholesterol was also observed in mice fed the GAN diet, but these mice did not develop significant fibrosis. One explanation for this is that fibrosis progression required additional metabolic abnormalities that were present in mice fed the CDAHFD. For example, while steatosis was observed in both GAN and CDAHFD-fed mice, hepatic triglycerides were approximately 2-fold higher in livers of mice fed the CDAHFD. Compared to a standard HFD, the CDAHFD diet also has been shown to promote greater mitochondrial dysfunction and oxidative stress and a distinct inflammatory gene expression profile58. Several inflammatory and non-parenchymal cell types play defining roles in the development and progression of hepatic fibrosis59, and cholesterol accumulation within KCs may contribute to the inflammation characteristic of NASH60. Activation of HSCs occurs via the release of fibrogenic cytokines from KCs, but these cells also accumulate free cholesterol, which can trigger their activation through toll-like receptor 4 (TLR-4)61.
Disturbances in enterohepatic bile acids may also have been responsible for the development of fibrosis in Hep-TCF7L2ΔDBD mice. A number of rare inborn metabolic disorders are caused by impairments in the bile acid synthesis pathway and are characterized by hepatic accumulation of abnormal bile acids, intermediary metabolites, and cholesterol62. The literature examining the association between bile acids and MASLD in humans is discordant, likely due to difficulties quantitating enterohepatic bile acids in humans. However, in one study, patients with T2D had significantly lower total bile acid levels63, whereas unconjugated bile acids were decreased in liver biopsy samples obtained from patients with MASH64. In rodents, a recent study evaluated the composition, metabolism, and signaling of bile acids in the enterohepatic compartment of two validated mouse models of MASH65. Consistent with our findings, mice with MASH had lower concentrations of bile acids in their portal blood and bile, and signaling through FXR and TGR5 was significantly reduced.
We also demonstrate that TCF7L2 regulates zonal pathways linked to ammonia detoxification in mice. The hepatic glutamate/glutamine ratio was increased, and the expression of glutaminase enzymes was also elevated in Hep-TCF7L2ΔDBD mice fed the CDAHFD. These data suggest that glutamine catabolism is enhanced in Hep-TCF7L2ΔDBD mice under conditions of metabolic stress. This is consistent with data demonstrating increased glutaminolysis in preclinical models of MASH40, and with recent observations that GS ablation in mouse liver promotes HCC development41. Knockout of hepatic GS was associated with multiple metabolic defects, including decreased hepatic glutamine and elevated glutamate levels, consistent with the phenotype of Hep-TCF7L2ΔDBD mice fed the CDAHFD in our study. Interestingly, the authors also used 15N-labeled ammonium to examine the metabolic fates of ammonia following hepatic GS ablation, which confirmed that the pool size and enrichment of glutamine and glutamate were reduced and increased, respectively. While we did not quantitate ammonia due to its instability in stored samples, these published data support the conclusion that the lack of GS in Hep-TCF7L2ΔDBD mice contributed to the development of fibrosis.
There are several important caveats and limitations to our study that should be considered. The regulation of metabolic zonation by TCF7L2 was examined using single nuclei RNA- and ATAC-Seq approaches in mice that were fed a chow diet but fasted overnight. The acute and chronic plasticity of metabolic zonation remains uncertain, but several zonated pathways, such as bile acid synthesis and gluconeogenesis are modified by feeding/fasting66. Given that the expression of hepatic Tcf7l2 may similarly be altered by feeding status21, in future studies it will be interesting to examine the zonal transcriptional function of TCF7L2 under different metabolic conditions and in different disease states. In contrast to prior in vivo studies20,23,53,67, we did not observe significant differences in glucose tolerance in Hep-TCF7L2ΔDBD mice, but this was only examined in overnight fasted mice fed a chow diet. Moreover, we did not directly quantitate gluconeogenesis using insulin clamps in Hep-TCF7L2ΔDBD mice but acknowledge prior in vitro evidence19,20 that TCF7L2 regulates gluconeogenic gene expression and hepatic glucose output. In a recent study, hepatic triglycerides were elevated in liver-specific TCF7L2 knockout mice when they were fed a prolonged high carbohydrate diet (HCD), but not a HFD53. The latter is consistent with our data demonstrating that hepatic triglycerides were not different in Hep-TCF7L2ΔDBD mice, but we did not feed mice a long-term HCD, so a direct comparison cannot be made. There are no perfect preclinical models of human MASLD, but the use of the CDAHFD and GAN diets in the present study allowed us to dissect whether TCF7L2 plays a more prominent role under different metabolic and inflammatory milieus, as has been recommended68. In our mouse studies Tcf7l2 was expressed in Kupffer, HSPCs, and stellate cells, and was reduced in Kupffer cells following the HFHFD. Therefore, the reduction in Tcf7l2 expression observed in fibrotic mouse and human liver may reflect changes in both hepatocyte and non-parenchymal cells. In Hep-TCF7L2ΔDBD mice, while inactivation of TCF7L2 occurred only in parenchymal cells of the liver, the development of hepatic fibrosis involves the coordinated activation of multiple non-parenchymal and inflammatory cells, including HSCs and myofibroblasts. As discussed above, we hypothesize that hepatocyte cholesterol accumulation and glutaminolysis are possible mechanisms linking hepatocyte TCF7L2 function and fibrosis. However, in humans we cannot be certain that reduced TCF7L2 levels play a causal role in the development of fibrosis since the necessary interventional studies are not possible. Finally, compared to studies in which Tcf7l2 has been knocked out, eliminating the DNA binding domain of TCF7L2 may not have completely inactivated TCF7L2 activity. However, we believe this is unlikely given that expression of known TCF7L2 target genes (i.e., Axin2, Lgr5, and Glul) is abolished in Hep-TCF7L2ΔDBD livers, and whole-body ablation of the DNA binding domain of TCF7L2 using the same floxed model produces an identical phenotype to the traditional TCF7L2 knockout mouse69.
We demonstrate that in mature mouse liver, the transcriptional function of TCF7L2 is focused on a small population of PC hepatocytes surrounding the CV. Transcriptional inactivation of TCF7L2 eliminates the expression of most PC hepatocyte-specific genes and disturbs key zonated metabolic pathways, including cholesterol metabolism, bile acid synthesis, and glutamine/glutamate homeostasis. Under conditions of dietary stress, these metabolic disturbances contribute to the development of hepatic fibrosis in mice lacking functional TCF7L2. These findings reveal an important function of TCF7L2 in the maintenance of the metabolic architecture of the liver and reveal insights into the role of zonated metabolic pathways to the development of hepatic diseases.
Methods
Animal studies
All rodent studies described in this manuscript were approved by the UT Health San Antonio Institutional Animal Care and Use Committee (IACUC, protocol number: 20130067AR). Mice with LoxP sites flanking exon 11 of the Tcf7l2 gene (TCF7L2LoxP/LoxP), which encodes part of the DNA binding domain of the TCF7L2 protein, have been described previously69,70. The original mouse line was maintained on a mixed C57BL/6 and 129 strain69, but was rederived upon entering the USA by The Jackson Laboratory and has been maintained in our laboratory on a C57BL/6 background for over 10 years. Approximately every 10 – 15 generations, the TCF7L2LoxP/LoxP colony was refreshed by backcrossing to C57BL/6 (The Jackson Laboratory, ME, USA). Albumin-Cre (Alb-Cre) mice were obtained from The Jackson Laboratory (Stock#: 003574). Mice were fed either regular chow (17% fat, 58% carbohydrate, 25% protein; Envigo, Cat#: 7012), a Gubra Amylin NASH (GAN) diet (40% fat, 20% fructose, 20% protein, 2% w/w cholesterol; Research Diets Inc, Cat#: D09100310) for 24-weeks, or a methionine and choline-deficient, HFD diet (CDAHFD, 62% fat, 20% carbohydrate, 18% protein, 0.1% methionine, no added choline; Research Diets, Cat#: A06071302) for 8-weeks. Tcf7l2 expression was examined in a cohort of mice fed a fast-food diet (FFD) for 11 months34. This consisted of a commercially available high-fat, high-cholesterol diet (40% fat, 43% carbohydrate, 17% protein, 1.5% cholesterol; Research Diets Inc, D12079B) and drinking water containing 23.1 g fructose and 17.2 g glucose per 1000 mL of water. The TCF/Lef:H2B-GFP reporter mouse was obtained from The Jackson Laboratory (stock # 013752) and was used to examine TCF/LEF activity in liver sections.
Mice were fed ad-libitum except for when fasted for in vivo physiological assays and/or tissue harvesting, details of which are provided below. Mice were provided ad-libitum access to acidified reverse osmosis water (pH 2.5 – 3.0) and nestlets and were housed in ventilated cages (Allentown Caging Equipment Company, cat # 75JAG) with irradiated Sani-Chip bedding in a room maintained at 75 ± 3 °F and on a 14 h light/10 h dark cycle. Littermate TCF7L2LoxP/LoxP controls were used in all experiments. For glucose and pyruvate tolerance tests (IPGTT and PTT), mice were fasted overnight for 14–16 h and injected with a fixed dose of glucose or pyruvate (50 mg) intraperitoneally. Insulin tolerance tests (ITT) were performed on 5-hour fasted mice with a dose of 1U/kg, injected intraperitoneally.
Single-nuclei multiomics
Nuclei were isolated from approximately 20 mg of frozen mouse liver that was harvested from chow-fed control (TCF7L2LoxP/LoxP) and liver-specific TCF7L2 mutant mice (Hep-TCF7L2ΔDBD) that were fasted overnight (14– 16 h). Briefly, samples were crushed to powder under liquid nitrogen and homogenized in lysis buffer containing 0.1% Tween and NP40, 0.01% digitonin, 1% bovine serum albumin and RNase inhibitor. Samples were lysed first using a Dounce homogenizer with a clearance of 0.0030-0.0050 inches, then with a second Dounce homogenizer with a clearance of 0.0005-0.0025 inches (Cat#: 885300-0002; DWK Life Sciences, NJ, USA). Following centrifugation and washing, nuclei were passed through a series of filters (70, 30, and 20 µm), washed, and resuspended in nuclei buffer (10X Genomics, CA, USA). Nuclei were assessed for quality and quantitated using a hemocytometer before proceeding to library preparation.
Multiome gene expression and Assay for Transposase-Accessible Chromatin (ATAC) libraries (10X Genomics) were prepared from approximately 5000 nuclei/mice at the Genome Sequencing Facility at the University of Texas Health at San Antonio (UTHSA). Liver samples were processed in pairs (one control and one mutant) on three consecutive days to minimize batch effects. Library quality and quantity were assessed using the 2100 Bioanalyzer and were sequenced on a NovaSeq 6000 (Illumina, CA, USA).
We performed two distinct analyses that required separate clustering steps. First, we used the control samples to establish the methodology of quantifying zonal gene expression and transcription factor occupancy, including TCF7L2. Second, we examined the consequences of TCF7L2 transcriptional inactivation on zonation in control vs. mutant livers.
Preprocessing and filtering of dataset
FASTQ files were generated from the raw base call (BCL) outputs with the Cell Ranger (10X Genomics) mkfastq pipeline. From this, we obtained a joint matrix of gene and peak counts for each nuclear barcode per sample using the Cell Ranger ARC (10X Genomics) count pipeline v.2.0.1. In this pipeline, default parameters were used to align reads to the GRCm38 (mm10) Mus musculus genome, with intron-aligned reads being counted as well. Sample-wise count results were then aggregated using Cell Ranger ARC aggr. The pipeline predicted 21,089 barcodes across all samples to represent intact single nuclei, and SoupX was run on the aggregated RNA count matrix using gene expression clusters provided by Cell Ranger to decontaminate nuclei of ambient RNA71. After a series of filtering steps, we generated a final set of 12,738 nuclei, of which a total of 7953 originated from hepatocytes (TCF7L2LoxP/LoxP = 5652 nuclei and 3498 hepatocytes; Hep-TCF7L2ΔDBD = 7,086 nuclei and 4555 hepatocytes).
Within each sample, various quality metrics per cell were analyzed, including the total numbers of unique transcripts (UMIs), detected genes, unique chromatin fragments, detected peaks, transcription start site (TSS) enrichment score, nucleosome signal, mitochondrial DNA percentage, RNA library complexity, and ATAC library complexity using the Seurat and Signac software packages, versions 4.3.0 and 1.9.0, respectively24,72,73. Library complexity for each assay was defined as the log of detected features divided by the log of unique captured molecules. Interquartile ranges from the sample-wise distributions of each metric were found, and outlier values were calculated according to the 1.5 × IQR rule, with nuclei having a low-end outlier value for the number of detected genes or peaks, the number of unique transcripts or fragments, TSS enrichment score, or library complexity for either assay being discarded. In addition, nuclei with a high-end outlier value for the percentage of mitochondrial DNA comprising their RNA libraries or a high-end outlier value for nucleosome signal were discarded. Following this step, the Seurat workflow was applied up to the step of cluster annotation on the RNA assay, specifically using default parameters but making sure to include all genes with a nonzero variance during the scaling and principal component analysis (PCA) steps. Following this, scDblFinder was run on the RNA data of each cell using sample and cluster annotations to predict doublets, and scType was then used to assign cell types by cluster from the liver subset of the provided cell type marker list74.
To ensure correct annotation of peaks prior to ATAC analysis, the MACS3 peak caller was applied separately for each fragment file provided by CellRanger ARC and for each predicted cell type by using the “group.by” parameter in the CallPeaks function in the Seurat/Signac multiomic workflow. Blacklisted/nonstandard regions were then removed prior to reconstructing a new peak by cell matrix, generating over 204,000 unique peaks. Term frequency-inverse document frequency (TFIDF) normalization and singular value decomposition (SVD) were performed on the refined ATAC data, and components 2 to 20 of the resulting latent semantic indexing (LSI) reduction were used to find cellular neighbors. ATAC cluster annotation was then performed using the smart local moving (SLM) algorithm, as recommended by the Signac vignette. Lastly, scDblFinder was run on the chromatin data of each cell using sample and ATAC cluster annotations while setting the “aggregateFeatures” parameter to true and the “processing” parameter to normalized features, as recommended by the scDblFinder vignette. With doublet annotations provided for each cell using both RNA and ATAC assays, nuclei that were predicted to be doublets in either assay were discarded prior to the process of batch correction/sample-wise integration.
Integration of nuclei across samples
The RNA data of all samples was integrated using the reciprocal PCA approach via Seurat, with the top 25% of all genes in terms of variance being selected as integration features and the first 20 principal components being used to generate an RNA anchorset and integrate normalized count values. The ATAC data of all samples was integrated using the reciprocal LSI approach via Signac, with all peaks being selected as integration features and LSI components 2 to 20 being used to generate an ATAC anchorset and integrate embeddings. Following this, the integrated RNA matrix was scaled and centered using the ScaleData function before a new PCA reduction was computed, with the first 30 principal components being used to find cellular neighbors and run UMAP dimensional reduction. The ATAC data was also visualized after using components 2 to 20 of the integrated LSI reduction to find cellular neighbors and run UMAP dimensional reduction, followed by weighted nearest neighbor (WNN) analysis and combined UMAP visualization as instructed by the multiomic Signac/Seurat vignette. Upon visualization, a set of nuclei was consistently located outside the main hepatocyte cluster in the gene expression UMAP, ATAC UMAP, and combined UMAP, and these nuclei had high mitochondrial DNA levels but low levels of any zonated or non-zonated hepatocyte markers. As a result, these nuclei were discarded by first finding clusters from each assay as well as the smoothed WNN graph, resulting in three sets of cluster labels, and removing the gene expression, ATAC, or combined clusters that contained these cells. Default settings, including setting the resolution to 0.8, in the FindClusters function generated cluster sets that identified these low-quality nuclei consistently on all three UMAP projections. Once the nuclei were removed, the process of sample-wise integration was repeated on the filtered RNA data and LSI reduction from the filtered ATAC data in a similar manner to the above procedure, this time with the first 30 principal components and LSI components 2 to 22 being used to generate RNA and ATAC anchor sets, respectively.
After scaling and running PCA on the newly created integrated RNA matrix, these same component intervals were used to guide cellular neighbor determination and UMAP dimensional reduction for each assay separately as well as both assays combined, with clusters from each assay separately and both assays combined being recalculated as well. Furthermore, scType was used to label cells again by their WNN clusters after rescaling all genes in the original normalized RNA matrix to provide an optimal set of annotations based on input from both the ATAC and RNA assays that was also free of potential miscalls from poor quality nuclei. The final clustering shown for all nuclei is a result of these dimensional reductions and annotations.
Hepatocyte-specific filtering and integration
Hepatocytes were isolated from the dataset to undergo sample-wise integration separately, This is because the distributions of scaled gene expression values and normalized peak accessibility values with all cells may compress the range of these values within cells of the same broad type, minimizing differences between cellular subtypes like the separate zones of hepatocytes. Thus, in order to maximize the visibility of differences between hepatocyte subtypes, not only were hepatocytes isolated, integrated by sample separately from all nuclei, and reclustered, but their original RNA count data was also subject to imputation by SAVER in an effort to denoise reported gene expression levels in a biologically relevant manner for all downstream analyses75. Genes with a total of at least 10 counts across all hepatocytes were corrected by the SAVER algorithm. The peak by cell matrix for hepatocytes also was renormalized separately from all nuclei using the RunTFIDF function in Signac with the default method of log(TF x IDF) as done previously, followed by SVD to generate a hepatocyte-specific LSI dimensional reduction. Integration features included the top 25% of variable genes and all peaks for the RNA and ATAC assays, respectively, with the former being scaled and subjected to PCA within each sample. The first 40 principal components were used to generate an RNA anchorset, while LSI components 2 to 14 were used to generate an ATAC anchorset, and the “k.anchor” parameter was set to 20 when finding anchors for either assay in the FindIntegrationAnchors function in Seurat. To generate a hepatocyte-specific clustering, the integrated RNA matrix was scaled and subjected to PCA, after which the first 30 principal components were used to find nearest neighbors and run UMAP dimensional reduction. Integrated LSI components 2 to 14 were used for the same steps to visualize the ATAC assay, and the combination of these component intervals was used to guide the WNN visualization process for hepatocytes as well. Following this, additional filtering was performed to remove nuclei that did not cluster in close proximity to the main hepatocyte cluster and/or had relatively high expression of mitochondrial DNA, with these nuclei being almost entirely contained within one GEX cluster and an overlapping WNN cluster derived from the smoothed WNN graph. The batch correction process was redone with the same parameters as described above, except for modified intervals of the first 20 principal components to integrate the RNA data and LSI components 2 to 13 to integrate the ATAC data. The nuclei were then reclustered using the first 30 principal components from the scaled integrated RNA matrix and integrated LSI components 2 to 13, with these assay-specific hepatocyte clusterings and the combination thereof being final and the set of nuclei contained within representing the set of all hepatocytes to undergo further analysis in the study. Following this, hepatocytes were labeled by the zone that they phenotypically fit using WNN clusters, with expression of carbamoyl phosphate synthetase 1 (Cps1) and arginase 1 (Arg1) being used to distinguish the periportal/zone 1 region, expression of glutamine synthetase (Glul) and axis inhibition protein 2 (Axin2) being used to identify the pericentral/zone 3 region, and a region of nuclei between the two zones expressing Cps1 in addition to various cytochrome C oxidase enzymes being labeled as mid-lobular/zone 2 hepatocytes. Hepatocytes that were removed from the hepatocyte-only dataset were then removed from the set of all nuclei, and zone annotations were transferred over.
Differential expression/accessibility analysis
Differential expression analyses was performed on raw counts of both genes and peaks via DESeq276 after pseudobulking hepatocytes by zone and sample using the AggregateExpression function in Seurat, with fold changes being shrunk using the apelgm package.6a Multiple comparisons were made between hepatocyte zones in the control cells and between controls and mutants, as detailed in the relevant results section. Pathway enrichment was performed on differentially expressed genes (DEGs) with log2-fold changes above 0.5 or below − 0.5 and adjusted p-values below 0.01 from all differentially expressed gene (DEG) sets as shown in the results using the clusterProfiler R package77 using KEGG signaling and metabolism pathways78. The same method was adopted with modifications for differential motif accessibility analysis based on chromVAR scores. After multiplying each hepatocyte’s chromVAR scores by its number of chromatin peaks, these scores were aggregated by zone and sample and added to the absolute value of the lowest pseudobulked score before rounding all values to the nearest whole number. In doing so, the minimum value for this assay became zero, and counts were made integers, fulfilling both requirements for the DESeq2 pipeline. To verify the validity of this analysis, differentially accessible regions (DARs) were also examined for motif binding sites according to the Signac motif analysis vignette, with sets of 50,000 background peaks being constructed solely from the ATAC data of the two groups of hepatocytes that underwent comparison to construct each DAR list.
Pseudotime analysis
The hepatocyte dataset was transformed into the “cell_data_set” class (cds) via Monocle325 to perform multiomic pseudotime analysis, with WNN clusters and the corresponding dimensional reduction from the existing Seurat object being transferred over prior to running the learn_graph and order_cells functions. Due to multiple nodes being displayed at sites populated by periportal hepatocytes but only one node being displayed at the end of the pericentral region, the latter was initially selected as the root node. The directionality was then reversed by subtracting the pseudotime of each cell from the maximum pseudotime in the dataset, ranking each hepatocyte along the continuum from zone 1 to zone 3. Genes that varied in expression in correlation to pseudotime were found using the graph_test function in Monocle3, with the “expression family” parameter being set to negative binomial for the most accurate results. Heatmaps of trajectory-associated genes that intersected with any set of DEGs was created using the Scillus R package, which is a derivative of the ComplexHeatmap R package79 and a wrapper for Seurat and ggplot2 visualization80. All candidates for the intersections between trajectory-associated genes and DEGs had a Benjamini-Hochberg adjusted p-value of less than 0.01, and the absolute value of shrunk log2-fold changes provided by DESeq2 for DEGs was greater than or equal to 1. Similarly, another cell_data_set object was created using chromvar data and examined for correlations between motif accessibility and pseudotime, after which heatmaps were constructed from trajectory-associated motifs that intersected with any set of differentially accessible motifs.
Control hepatocyte-specific clustering and gene-to-peak linkage
All nuclei (n = 5652) from control livers were then isolated and integrated by sample separately to produce a model dataset to showcase characteristics of zonation in genetically unaltered cells. The Seurat reciprocal PCA and Signac reciprocal LSI workflows were performed on control nuclei in a similar fashion to the iterations previously described. The top 25% of variable genes were selected as anchor features when integrating each sample’s RNA assay, the first 30 principal components derived from sample-wise RNA dimensional reduction were used to find integration anchors and create an integrated normalized RNA matrix, and components 2–14 derived from the LSI dimensional reduction performed on all control nuclei were used to find ATAC integration anchors prior to integrating nuclei embeddings. Peaks were linked to genes using the “LinksPeaks” function with “method = “spearman”, distance = 1e5” parameters, and links with positive correlation retained for downstream analysis.
TCF7L2 Binding site accessibility analysis
Motif analysis was done in all hepatocytes in both their single-nuclei and pseudobulked states according to the Signac motif analysis vignette. A list of position frequency matrices was loaded from the core collection of the JASPAR 2022 database81 after restricting the taxonomy group to vertebrates only, after which peaks were analyzed for motif presence through the “AddMotifs” function. The “RunChromVAR” function was then applied to quantify the binding site accessibility of each transcription factor with an individual z-score in each cell82. Similar to differential expression/accessibility analysis, the pseudobulked hepatocytes were also subject to differential motif activity testing via DESeq2 after adding a constant value to each chromVAR assay value and scaling it by a factor of 10000 to ensure a minimum value of 0 and integerized values across the board. A new cell data set was also constructed via Monocle3 with chromVAR data for each cell in order to test pseudotime associations with each motif. Resulting motifs that were shared between the list of trajectory-associated motifs (q < 0.01) and any of the aforementioned lists of differentially accessible motifs (q < 0.01 and a minimum absolute value of log-2-fold change of 0.25 within any comparison) were used to construct the heatmap(s) with hepatocytes being ordered by pseudotime.
Tissue processing and histological analyses
Mice were fasted overnight for 14–16 h, following which the liver was excised and prepared for downstream histological and molecular analysis. The left lateral lobe was placed in zinc formalin at 4 °C overnight, transferred to a tissue cassette in 70% ethanol, and paraffin-embedded. The median lobe was placed in 30% sucrose at 4 °C overnight and was frozen in OCT compound. The remaining liver was snap-frozen in liquid nitrogen for molecular analysis (i.e., RT-qPCR, Western blot). Hematoxylin and eosin (H&E) and Sirius Red staining were conducted on paraffin-embedded tissues sliced on a microtome to 5 μm thickness. The quantification of Sirius Red staining was performed using a custom CellProfiler pipeline83. Immunostaining for GFP (Cat#: ab13970, Abcam), glutamine sythetase (GS) (Cat#: 610517, BD BioSciences) and argininosuccinate synthase 1 (ASS1) (Cat#: 70720, Cell Signaling Technology) were carried out and staining was visualized using 3,3′-Diaminobenzidine (DAB) or using the SignalStain IHC Dual staining kit (Cell Signaling Technology, Cat#: 63084S). Images were captured on a BZ-X800 digital microscope (Keyence, IL, USA).
Western blotting and RT-qPCR
Real-time quantitative PCR (RT-qPCR) was performed as previously described18 using pre-designed TaqMan probes (Life Technologies) or PrimeTime SYBR green assays (Integrated DNA Technologies, IA, USA), which are listed in Supplementary Table 1. For analysis of Tcf7l2 transcripts containing exon 11, a custom PrimeTime probe assay was designed using PrimerQuest Design Tool (Integrated DNA Technologies). Immunoblot for TCF7L2 was performed using a rabbit monoclonal antibody (Cat#: 2569, Cell Signaling Technology) and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cat#: G9545, Sigma).
Human liver biopsy TCF7L2 expression
In the present study, we obtained deidentified and anonymized data on the expression of TCF7L2 mRNA in human liver biopsy tissue. No human liver samples were handled by the research team at UTHSA for this study. The TCF7L2 data were part of an RNA sequencing experiment that was performed by Gilead Sciences, Inc on a subset of biopsy samples from clinical trials GS-US-321-105 (ClinicalTrials.gov ID: NCT01672866)36, GS-US-321-106 (ClinicalTrials.gov ID: NCT01672879)36 and GS-US-384-1497 (ClinicalTrials.gov ID: NCT02466516)37. These were global multi-center phase 2 clinical trials that evaluated the efficacy of simtuzumab in patients with MASH. The primary outcome of these trials has been published previously36,37, and full details of the number and location of participating sites can be found in the ClinicalTrials.gov study record. The study protocol was approved by each local institutional review board (IRB) prior to study initiation, and each patient provided written informed consent, including for the transcriptomics analysis of liver biopsy samples. The studies were conducted in accordance with the International Conference on Harmonization Good Clinical Practice Guidelines and the Declaration of Helsinki.
In one parent study (GS-US-321-105, GS-US-321-106)36, the median age was 55 years (interquartile range [IQR] 48 to 59) for patients with bridging fibrosis and 57 years for those with compensated cirrhosis. Overall, 63% of patients were women, and 68% had diabetes. Median hepatic collagen content was 5.8% in patients with bridging fibrosis and 12.5% in those with cirrhosis. Of patients with cirrhosis, 40% had steatosis lower than 5%. At baseline, the median hepatic venous pressure gradient (HVPG) in patients with cirrhosis was 12 mmHg; 68% had clinically significant portal hypertension (CSPH), 42% had esophageal varices, and 21% were prescribed nonselective b-blockers.
In the other clinical trial (GS-US-384-1497)37, 72 patients with NASH and stage 2 or 3 fibrosis on biopsy were enrolled. 65% of patients had stage 3 fibrosis and a non-alcoholic steatohepatitis (NASH) activity score (NAS) ≥ 6 (71%) at baseline. Overall, 86% had severe hepatocellular ballooning. Approximately two-thirds (69%) of patients were female, the median age was 56 years, and 71% of patients had diabetes mellitus.
In the subset of data used in the current study for the analysis of TCF7L2 mRNA, 50% were from females (Supplementary Table 2). A two-way Anova was used to assess the effects of sex and fibrosis grade on hepatic TCF7L2 mRNA. The disaggregated date is provided in the source data file, and patient demographics are provided in Supplementary Table 2. Total RNA was isolated from three 13 μm FFPE curls per sample. All samples had > 100 ng of input RNA. RNA expression profiling was performed using Illumina TruSeq RNA Access chemistry on the Illumina sequencing platform with a 50 bp paired end strategy and a target of 40 million average reads. R packages edgeR and limma were used to normalize sequence count data and conduct differential gene expression analysis. Sequencing was performed by Q2 Solutions (Morrisville, North Carolina). The false discovery rate (FDR) was calculated using the Benjamini-Hochberg method.
Biochemical assays
Liver triglycerides were quantitated using Infinity Triglycerides reagent (ThermoFisher, MA, USA). Serum FFA levels were determined using a standard NEFA assay kit (Wako Chemicals USA, Inc., Richmond, VA, USA). Liver lysate cholesterol levels were measure using Cholesterol Fluorometric Assay (Cayman) or Cholesterol Quantitation Kit (Sigma-Aldrich, MO, USA) kits following the manufacturer’s instructions. Glutamine and glutamate were determined using the Glutamine/Glutamate-Go assay (Promega, WI, USA).
Laser capture microdissection (LCM)
The LCM protocol was adapted from previously published methods84. Briefly, multiple frozen sections (10 µm) were cut from OCT-embedded livers harvested from 12-week old wild-type mice. Hepatocytes around the CV and PV were excised using the Arcturus XT microscope (Life Technologies, USA). Multiple LCM caps for each sample were pooled together for RNA extraction using the Qiagen RNeasy Plus Micro Kit (Qiagen, MD, USA), and RNA quantity and integrity was examined using the 2100 Bioanalyzer (Agilent, CA, USA). Synthesis of cDNA was performed using SuperScript Vilo (ThermoFisher, MA, USA), and RT-qPCR carried out as described above.
Bile acid analysis
Liver (100 – 150 mg) was homogenized in 4 mL Optima LC/MS Grade methanol (ThermoFisher) containing 1 µl each of 10 mM G-UDCA (10 nmol) and G-CDCA (10 nmol). The whole intestine was homogenized in 9 mL Optima LC/MS Grade Methanol containing 2.25 ul of 100 mM G-UDCA and G-CDCA (each 225 nmol). Tissues were mechanically homogenized using a handheld Omni tissue homogenizer with a stainless-steel probe. Homogenates were vortexed then incubated at room temperature overnight. The next day, samples were centrifuged at 4000 rpm for 10 min, and the supernatant transferred to borosilicate glass tubes (Fisher Cat#: 14-959-25 A, Cat#: 14-959-25B).
Prior to analysis, bile was diluted 1:1000 into Optima LC/MS Grade Methanol, then further diluted 1:2 directly into autosampler vials. Liver supernatant was vortexed then filtered through a 0.45 µm syringe-driven filter unit (Millipore SLLHR04NL) directly into autosampler vials. The Intestine supernatant was thoroughly vortexed, then an aliquot centrifuged at 15,000 × g for 5 min. Samples were diluted 1:20 directly into autosampler vials. Liver (5 µL), bile, or intestine (2 µL) samples were injected via an Ultimate3000 (Thermo Fisher) autosampler module equipped with an ACE Excel 150 mm × 2.1 mm C18-PFP 1.7 µm UHPLC column (MAC-MOD Analytical, Cat#: EXL10101502U), maintained at 50 °C. To detect bile acids, a gradient method was used. Mobile phase buffer A was 10 mM ammonium acetate (Fisher Scientific, Cat#: A11450) in HPLC-grade water with 0.1% formic Acid (Fisher Scientific, Cat#: HB523-4), buffer B was 75% acetonitrile with 0.1% formic acid (Fisher Scientific, Cat#: HB9823-4): 25% Optima LC/MS grade methanol with 10 mM ammonium acetate, and buffer C was 100% Optima LC/MS grade methanol. At a flow rate of 0.375 µL/min the gradient method started with 26% buffer B, increasing incrementally to 27.5% by 5 min, 40% by 14 min, 85% by 22 min, 98% by 22.1 min then held at 98% buffer B for 3 min. At 26 min the flow rate was reduced to 0.2 ml/min with 100% buffer C to wash the column for 3 min. At 29 min the flow rate was increased to 0.3 ml/min 28.5% buffer B:71% buffer A, then 0.325 ml/min at 30 min, which was maintained until the end of the run (33 min). Sample ionization was performed using the following parameters: Polarity = Negative; Spray voltage = 3500 V; Vaporizer temp = 350 °C; Sheath and auxiliary gas (nitrogen) = 20 and 2 Arb resp.; ion transfer capillary temp = 300 °C; collision gas (argon) pressure = 1.5 mTorr; collision energy = 10–55 V. Bile acid concentrations were quantified in Skyline-daily software (MacCross Lab Software) from standard curves of each individual bile acid generated from a mix of purified bile acid standards.
We ran one technical replicate per sample. To generate the bile acid pool, we extracted bile acids from the gallbladder, liver, and intestine from CON and MUT mice (N = 8/treatment). Intestine and liver bile acids were normalized to spiked G-CDCA. A “blank control” was included, which consisted of the extraction mix alone (Optima Methanol + G-CDCA and G-UDCA spikes) and ran alongside liver and intestine samples
Hepatic 7α-hydroxy-4-cholesten-3-one (C4) quantitation
For the detection of hepatic C4, the LC-MS system was equipped with an ACE Excel SuperC18 UHPLC Column (MAC-MOD Analytical, EXL10111502U). Mobile phase buffer A was 80% HPLC Water with 0.1% formic acid (Thermo fisher): 20% Optima methanol, and buffer C was 100% Optima methanol. Using a flow rate of 0.3 mL/min, a gradient method starting at 60% Buffer C increased to 90% by 2 min, then 100% by 5 min. Buffer C was held at 100% until 9 min, then reduced to 60% by 10 min, and maintained at 60% until the sample run was terminated at 15 min. Equilibration for 0.5 min was performed using 60% Buffer C at the start of each sample run. Sample ionization was performed using the following parameters: Polarity = Positive; Spray voltage = 3500 V; Vaporiser temp = 350 °C; Sheath and auxiliary gas (nitrogen) = 20 and 2 Arb resp.; ion transfer capillary temp = 300 °C; collision gas (argon) pressure = 1.5 mTorr; collision energy = 10–25 V. Selected Reaction Monitoring (SRM) was performed using the following: Precursor m/z = 401.38; Product 1 m/z = 383.33; Product 2 m/z = 177.19; Explicit Collision Energy (Precursor, Product) = 10, 25; Retention time = 7.5 min. Liver samples (10uL), prepared and filtered for bile acid analysis, and standards (5uL) were injected into the LC-MS system via an autoinjector. C4 levels were quantified in Skyline-daily software (MacCross Lab Software) from a standard curve generated using C4 purified standards (Sigma, 52853). Data was exported with Molecule Setting set to MS level = 2.
Statistics
All results are represented as mean ± SD. The Student’s T-test or Welch’s T-test were used to compare two groups with equal or unequal variances, respectively. Data containing three or more groups was analyzed using one-, two- or three-way ANOVA with Holm-Sidak post-hoc testing accounting for multiple comparisons, where appropriate. The statistical approaches used in the single-cell multiomics analyses are detailed in the Supplementary Materials.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All single nuclei data is available for download from the Gene Expression Omnibus (GEO) repository (accession number: GSE239480; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE239480). Mass spectrometry data is available for download at Metabolomics Workbench (accession number: ST003641; https://www.metabolomicsworkbench.org/data/DRCCMetadata.php?Mode=Study&StudyID=ST003641). Any additional requests for information and/or data, including human liver biopsy RNA-Seq data, can be directed to and will be fulfilled by, the corresponding author. Source data are provided in this paper.
References
Asrani, S. K. et al. Burden of liver diseases in the world. J. Hepatol. 70, 151–171 (2019).
Rinella, M. E. et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. J. Hepatol. 78, 1966–1986 2023.
Harrison, S. A. et al. Prospective evaluation of the prevalence of non-alcoholic fatty liver disease and steatohepatitis in a large middle-aged US cohort. J. Hepatol. 75, 284–291 2021.
Collaborators, G.B.D.C. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 5, 245–266 (2020).
Gebhardt, R. Metabolic zonation of the liver: regulation and implications for liver function. Pharm. Ther. 53, 275–354 (1992).
Ben-Moshe, S. & Itzkovitz, S. Spatial heterogeneity in the mammalian liver. Nat. Rev. Gastroenterol. Hepatol. 16, 395–410 (2019).
Jungermann, K. & Kietzmann, T. Zonation of parenchymal and nonparenchymal metabolism in liver. Annu. Rev. Nutr. 16, 179–203 (1996).
Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 127, 469–480 (2006).
Sethi, J. K. & Vidal-Puig, A. Wnt signalling and the control of cellular metabolism. Biochem. J. 427, 1–17 (2010).
Preziosi, M. et al. Endothelial Wnts regulate beta-catenin signaling in murine liver zonation and regeneration: A sequel to the Wnt-Wnt situation. Hepatol. Commun. 2, 845–860 (2018).
Goel, C., Monga, S. P. & Nejak-Bowen, K. Role and regulation of Wnt/beta-Catenin in hepatic perivenous zonation and physiological homeostasis. Am. J. Pathol. 192, 4–17 (2022).
Behari, J. et al. beta-catenin links hepatic metabolic zonation with lipid metabolism and diet-induced obesity in mice. Am. J. Pathol. 184, 3284–3298 (2014).
Yang, J. et al. beta-catenin signaling in murine liver zonation and regeneration: a Wnt-Wnt situation! Hepatology 60, 964–976 (2014).
Sekine, S. et al. Liver-specific loss of beta-catenin results in delayed hepatocyte proliferation after partial hepatectomy. Hepatology 45, 361–368 (2007).
Sekine, S. et al. Liver-specific loss of beta-catenin blocks glutamine synthesis pathway activity and cytochrome p450 expression in mice. Hepatology 43, 817–825 (2006).
Benhamouche, S. et al. Apc tumor suppressor gene is the “zonation-keeper” of mouse liver. Dev. Cell 10, 759–770 (2006).
Grant, S. F. et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet. 38, 320–323 (2006).
Norton, L. et al. The mechanisms of genome-wide target gene regulation by TCF7L2 in liver cells. Nucleic Acids Res. 42, 13646–13661 (2014).
Norton, L. et al. Chromatin occupancy of transcription factor 7-like 2 (TCF7L2) and its role in hepatic glucose metabolism. Diabetologia 54, 3132–3142 (2011).
Ip, W. et al. Liver-specific expression of dominant-negative transcription factor 7-like 2 causes progressive impairment in glucose homeostasis. Diabetes 64, 1923–1932 (2015).
Ip, W. et al. The Wnt signaling pathway effector TCF7L2 is upregulated by insulin and represses hepatic gluconeogenesis. Am. J. Physiol. Endocrinol. Metab. 303, E1166–E1176 (2012).
Neve, B. et al. Alternative human liver transcripts of TCF7L2 bind to the gluconeogenesis regulator HNF4alpha at the protein level. Diabetologia, 2014.
Oh, K. J. et al. TCF7L2 modulates glucose homeostasis by regulating CREB- and FoxO1-dependent transcriptional pathway in the liver. PLoS Genet. 8, e1002986 (2012).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Halpern, K. B. et al. Erratum: Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 543, 742 (2017).
Schleicher, J. et al. Zonation of hepatic fatty acid metabolism - The diversity of its regulation and the benefit of modeling. Biochim. Biophys. Acta 1851, 641–656 (2015).
Hijmans, B. S. et al. Zonation of glucose and fatty acid metabolism in the liver: mechanism and metabolic consequences. Biochimie 96, 121–129 (2014).
Ferrer-Vaquer, A. et al. A sensitive and bright single-cell resolution live imaging reporter of Wnt/ss-catenin signaling in the mouse. BMC Dev. Biol. 10, 121 (2010).
Postic, C. & Magnuson, M. A. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 26, 149–150 (2000).
Jho, E. H. et al. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell Biol. 22, 1172–1183 (2002).
Bravo Gonzalez-Blas, C. et al. Single-cell spatial multi-omics and deep learning dissect enhancer-driven gene regulatory networks in liver zonation. Nat. Cell Biol. 26, 153–167 (2024).
Zimmerli, D. et al. TBX3 acts as tissue-specific component of the Wnt/beta-catenin transcriptional complex. Elife, 9, https://doi.org/10.7554/elife.58123 2020.
Charlton, M. et al. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol. 301, G825–G834 (2011).
Su, Q. et al. Single-cell RNA transcriptome landscape of hepatocytes and non-parenchymal cells in healthy and NAFLD mouse liver. iScience 24, 103233 (2021).
Harrison, S. A. et al. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology 155, 1140–1153 (2018).
Loomba, R. et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 67, 549–559 (2018).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Haussinger, D., Lamers, W. H. & Moorman, A. F. Hepatocyte heterogeneity in the metabolism of amino acids and ammonia. Enzyme 46, 72–93 (1992).
Du, K. et al. Increased Glutaminolysis Marks Active Scarring in Nonalcoholic Steatohepatitis Progression. Cell Mol. Gastroenterol. Hepatol. 10, 1–21 (2020).
Dai, W. et al. Glutamine synthetase limits beta-catenin-mutated liver cancer growth by maintaining nitrogen homeostasis and suppressing mTORC1. J. Clin. Invest. 132, https://doi.org/10.1172/jci161408 (2022).
Cunningham, R. P. & Porat-Shliom, N. Liver zonation - revisiting old questions with new technologies. Front. Physiol. 12, 732929 (2021).
Sauter, G. et al. Serum concentrations of 7alpha-hydroxy-4-cholesten-3-one reflect bile acid synthesis in humans. Hepatology 24, 123–126 (1996).
Galman, C. et al. Monitoring hepatic cholesterol 7alpha-hydroxylase activity by assay of the stable bile acid intermediate 7alpha-hydroxy-4-cholesten-3-one in peripheral blood. J. Lipid Res. 44, 859–866 (2003).
Mononen, J. et al. Genetic variation is a key determinant of chromatin accessibility and drives differences in the regulatory landscape of C57BL/6J and 129S1/SvImJ mice. Nucleic Acids Res. 52, 2904–2923 2023.
Nobrega, M. A. TCF7L2 and Glucose metabolism: Time to look beyond the pancreas. Diabetes 62, 706–708 (2013).
Jin, T. Current understanding on role of the Wnt signaling pathway effector TCF7L2 in glucose homeostasis. Endocr. Rev. 37, 254–277 (2016).
Grant, S. F. A. The TCF7L2 Locus: A genetic window into the pathogenesis of type 1 and type 2 diabetes. Diabetes Care 42, 1624–1629 (2019).
Fuchsberger, C. et al. The genetic architecture of type 2 diabetes. Nature 536, 41–47 (2016).
Wild, S. L. et al. The canonical Wnt pathway as a key regulator in liver development, differentiation and homeostatic renewal. Genes 11, https://doi.org/10.3390/genes11101163 (2020).
van de Wetering, M. et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 111, 241–250 (2002).
Jin, Y. et al. Wnt signaling regulates hepatocyte cell division by a transcriptional repressor cascade. Proc. Natl. Acad. Sci. USA 119, e2203849119 (2022).
Lee, D. S. et al. Tcf7l2 in hepatocytes regulates de novo lipogenesis in diet-induced non-alcoholic fatty liver disease in mice. Diabetologia 66, 931–954 (2023).
Twisk, J. et al. Heterogeneous expression of cholesterol 7 alpha-hydroxylase and sterol 27-hydroxylase genes in the rat liver lobulus. J. Clin. Invest. 95, 1235–1243 (1995).
Behari, J. et al. Liver-specific beta-catenin knockout mice exhibit defective bile acid and cholesterol homeostasis and increased susceptibility to diet-induced steatohepatitis. Am. J. Pathol. 176, 744–753 (2010).
Yeagle, P. L. Modulation of membrane function by cholesterol. Biochimie 73, 1303–1310 (1991).
Horn, C. L. et al. Role of cholesterol-associated steatohepatitis in the development of NASH. Hepatol. Commun. 6, 12–35 (2022).
Sugasawa, T. et al. One week of CDAHFD induces steatohepatitis and mitochondrial dysfunction with oxidative stress in liver. Int. J. Mol. Sci. 22, https://doi.org/10.3390/ijms22115851 (2021).
Kisseleva, T. & Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 18, 151–166 (2021).
Shono, S. et al. The immunologic outcome of enhanced function of mouse liver lymphocytes and Kupffer cells by high-fat and high-cholesterol diet. Shock 36, 484–493 (2011).
Teratani, T. et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 142, 152–164.e10 (2012).
Sundaram, S. S. et al. Mechanisms of disease: Inborn errors of bile acid synthesis. Nat. Clin. Pr. Gastroenterol. Hepatol. 5, 456–468 (2008).
Valanejad, L. et al. Dysregulation of Delta(4)-3-oxosteroid 5beta-reductase in diabetic patients: Implications and mechanisms. Mol. Cell Endocrinol. 470, 127–141 (2018).
Lake, A. D. et al. Decreased hepatotoxic bile acid composition and altered synthesis in progressive human nonalcoholic fatty liver disease. Toxicol. Appl. Pharm. 268, 132–140 (2013).
Gillard, J. et al. Bile acids contribute to the development of non-alcoholic steatohepatitis in mice. JHEP Rep. 4, 100387 (2022).
Martini, T., Naef, F. & Tchorz, J. S. Spatiotemporal metabolic liver zonation and consequences on pathophysiology. Annu. Rev. Pathol. 18, 439–466 (2023).
Boj, S. F. et al. Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic response to perinatal and adult metabolic demand. Cell 151, 1595–1607 (2012).
Gallage, S. et al. A researcher’s guide to preclinical mouse NASH models. Nat. Metab. 4, 1632–1649 (2022).
van Es, J. H. et al. A critical role for the Wnt effector Tcf4 in adult intestinal homeostatic self-renewal. Mol. Cell Biol. 32, 1918–1927 (2012).
Chen, X. et al. The diabetes gene and Wnt pathway effector TCF7L2 regulates adipocyte development and function. Diabetes 67, 554–568 (2018).
Young, M. D. & Behjati, S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience, 9, https://doi.org/10.1093/gigascience/giaa151 (2020).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019).
Stuart, T. et al. Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341 (2021).
Ianevski, A., Giri, A. K. & Aittokallio, T. Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nat. Commun. 13, 1246 (2022).
Huang, M. et al. SAVER: gene expression recovery for single-cell RNA sequencing. Nat. Methods 15, 539–542 (2018).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Korotkevich, G. et al. Fast gene set enrichment analysis. Preprint at https://doi.org/10.1101/060012 (2021).
Kanehisa, M. et al. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. (2016).
Castro-Mondragon, J. A. et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 50, D165–D173 (2022).
Schep, A. N. et al. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017).
Stirling, D. R. et al. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinforma. 22, 433 (2021).
Butler, A. E. et al. Recovery of high-quality RNA from laser capture microdissected human and rodent pancreas. J. Histotechnol. 39, 59–65 (2016).
Acknowledgements
This work was supported by grants from the NIDDK (R01DK128247 to L.N. and R01DK138340 to TQAV). The LCM data was generated with the assistance of the Bioanalytics and Single-Cell Core (BASiC) at the University of Texas Health San Antonio, which is supported by the Cancer Prevention Research Institute of Texas (RP150600) and the Office of Vice President of Research at UTHSA. Single nuclei 10X multiomics libraries were prepared with the assistance of the Genome Sequencing Facility (GSF) at UTHSCSA. We thank Zhai Lao at the GSF for technical assistance in completing the multiomics library preparation and sequencing. We are grateful to Gilead Sciences Inc. for the analysis of their human liver biopsy samples. Iriscilla Ayala was part of a Translational Science training program that was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant TL1TR002647 and T32 TR004544. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Contributions
The study was designed by L.N., and the data analysis, conclusions, and manuscript drafting were carried out by L.N. S.K.H., J.M., and S.H. analyzed the single-nuclei multiomics data and contributed to manuscript writing. I.A., C.E.S., I.V., M.F., T.M.B., A.Z., G.R., M.A., F.A.M., J.T.S., A.A.S., E.A.H., and L.C.F.L. contributed to the data generation and analysis. M.C.S. and T.Q.A.V. generated and analyzed the bile acid data. All authors contributed to the discussion of the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ayala, I., Hebbale, S.K., Mononen, J. et al. The Spatial Transcriptional Activity of Hepatic TCF7L2 Regulates Zonated Metabolic Pathways that Contribute to Liver Fibrosis. Nat Commun 16, 3408 (2025). https://doi.org/10.1038/s41467-025-58714-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58714-5
