
Abstract
Adverse drug-drug interaction events (DDIEs) pose serious risks to patient safety, yet rare but severe interactions remain challenging to identify due to limited clinical data. Existing computational methods rely heavily on abundant samples, failing to identify rare DDIEs. Here we introduce RareDDIE, a metric-based meta-learning model that employs a dual-granular structure-driven pair variational representation to enhance rare DDIE prediction. To further address the challenge of zero-shot DDIE identification, we develop the Biological Semantic Transferring (BST) module, integrating large-scale sentence embeddings to form the ZetaDDIE variant. Our model outperforms existing methods in few-sample and zero-sample settings. Furthermore, we verify that knowledge transfer from DDIE can improve drug synergy predictions, surpassing existing models. Case studies on antiplatelet activity reduction and non-small cell lung cancer drug synergy further illustrate the practical value of RareDDIE. By analyzing the meta-knowledge construction process, we provide interpretability into the model’s decision-making. This work establishes an effective computational framework for rare DDIE prediction, leveraging meta-learning and knowledge transfer to overcome key challenges in data-limited scenarios.
Similar content being viewed by others
Introduction
Drug safety remains a paramount concern in the medical field1. With the increasing complexity of clinical diseases and continuous scientific advancements, combination drug therapies offer significant promise for treating challenging conditions2. However, the potential for unseen drug-drug interaction events (DDIEs) among drug combination introduces additional risks to patient treatments3. Of particular concern are rare but severe adverse reactions, which can have serious consequences4. In the United States, adverse DDI events account for approximately 74,000 emergency room visits and 195,000 hospitalizations annually5. Despite rigorous risk assessments required before drug approval6, identifying rare adverse drug reactions (ADRs) caused by DDIs in clinical practice remains highly challenging due to the limited scale of experimental studies7. Therefore, early and effective detection of DDIEs, especially those that are rare and difficult to observe, is essential for protecting patient health and evaluating drug risks8,9.
However, traditional experimental methods are time-consuming and labor-intensive, and ADR data are often incomplet10, complicating the identification of uncommon DDIEs. Additionally, as clinical observations and drug development progress, novel DDIEs continue to emerge11. To expedite the discovery of potential DDIEs, numerous machine learning techniques have been developed in recent years9,12,13,14,15. For instance, DeepDDI uses drug chemical substructures to build a deep learning-based model for predicting DDIEs16. SSI-DDI introduces a co-attention module to capture intra- and inter-molecular substructure interactions for DDIE prediction17. PEB-DDI proposes a dual-view substructure learning framework, utilizing molecular graph substructures and molecular fingerprint information to predict DDIEs18. BioDKG-DDI employs a knowledge graph embedding method and integrates multimodal information for prediction19. Despite these advances, existing computational methods predominantly focus on predicting DDIEs with sufficient training samples, always disregarding rare DDIEs by excluding long-tail data to prevent overfitting13,20,21. This approach preserves predictive performance but limits the models’ ability to predict rare events and address newly emerging events. We visualized the long-tail distribution of DDIEs for two commonly used datasets in the Supplementary Fig. 1-2, highlighting the current models’ significant deficiency in predicting rare events.
Predicting rare but severe DDIEs has long been a formidable challenge in drug development22. Fortunately, the advent of meta-learning offers a promising approach by framing this issue as a few-shot prediction problem23. Traditional supervised learning models require substantial labeled data, significantly limiting their scalability to new tasks and few-shot scenarios. In contrast, meta-learning leverages support and query sets to train models on optimization and matching results with minimal samples, corresponding to the optimization-based24 and metric-based25 approaches, respectively. The metric-based approach, which allows for direct inference based on a few provided samples without parameter updates, forms the foundation of our proposed model in this work. However, applying this framework to rare DDIE prediction involves three major challenges. The first challenge is constructing generalizable drug representations that comprehensively capture various functional information intrinsic to the drugs. The second challenge is creating a relational metric space for drug pairs to form specific event descriptions and adapt to new events. The third challenge is ensuring interpretability in the construction process of meta-knowledge to aid subsequent applications and analyses.
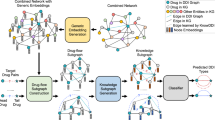
To address these challenges, we propose a metric-based model for rare DDIE prediction using dual-granular structure-driven drug-pair variational representation (RareDDIE). For the first challenge, we derive generalizable drug representations from dual-granular structures, as illustrated by the CSE and NAI components in Fig. 1a. The Chemical Substructure Information Extraction (CSE) module is designed to capture crucial chemical structure information of drugs based on graph neural network. Additionally, we introduce the Neighborhood Adaptive Integration with Task Guidance (NAI) module, which uses chemical structure information to build weak relations for task guidance and adaptively capture biological neighborhood structure information. Intuitively, this module autonomously aggregates features from neighboring nodes in the biological graph, constructing drug representations from a functional perspective to provide mechanistic insights into interaction types. Additionally, it enhances the model’s generalization ability, making it applicable to zero-shot scenarios such as novel drug interactions. The detailed NAI architecture is shown in Fig. 1b. For the second challenge, although individual drug features can be captured from dual-granular structures, representing drug pairs through simple concatenation can hinder transforming features into specific event descriptions. To overcome this, we propose the Pair Variational Representation (PVR) module based on an autoencoder, which maps pairwise data into a general relation metric space for predicting and autonomously forms medical semantic latent descriptions in an end-to-end manner. The details are represented in Fig. 1c. For the third challenge, our model ensures ample interpretability by focusing on critical molecular structures, biological neighborhood structures, and the mapping to the relation metric space. RareDDIE also includes a Feature Initialization (FIN) module and a Comparator Module, with detailed descriptions provided in the Methods section and Fig. 1a.

a The calculation process of RareDDIE based on our five constructed modules. Feature Initialization (FIN) utilizes the knowledge graph DRKG56 and the knowledge graph embedding method TransE59 to initialize features; Chemical Substructure Information Extraction (CSE) employs message passing mechanism and Self-Attention Graph Pooling (SAGPooling) to learn drug chemical granular structure information; Neighborhood Adaptive Integration with Task Guidance (NAI); Pair Variational Representation (PVR) and the Comparator Module. b The details of the NAI module, showing how weak event representations, built from chemical structure information, guide the aggregation of neighborhood information. This results in the creation of dual-granular structure features by integrating drug chemical structure information with biological neighborhood structure information. c The illustration of the PVR module, which uses a Variational Autoencoder (VAE)-based approach to construct an effective general relation metric space and automatically form latent event semantic information. Std represents the standard deviation, and N(0,1) denotes the standard normal distribution, which is used for noise sampling.
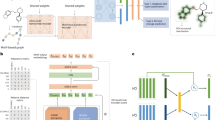
In our experiments, we compared the performance of our model with seven baseline methods and demonstrated its superior performance in a few-shot setting. Predicting DDI of new events is inherently a few-shot problem, as discovering a DDI of a new event in clinical practice is often accompanied by known drug usage. However, considering many undiscovered events may still exist, we frame the predicting DDI of new events as a zero-shot task. To deal with the zero-shot task, we introduce the Biological Semantic Transferring (BST) module and the LSEM (Large-scale Sentence Embedding Model) BioSentVec26,27, which occupies 21GB of memory, to extend RareDDIE into ZetaDDIE and validate its performance. The BST module aligns clinical semantic distributions, extracted from BioSentVec, with the general relation metric space to construct semantic information metrics for any event. The details are shown in Fig. 2b. Additionally, as shown in Fig. 2c, d, our model captures meta-knowledge of drug biological mechanisms, and previous studies have shown that different disease modules can simultaneously affect drug synergy and drug interactions28. Therefore, we find and validate the enhancement of drug synergy prediction performance by transferring DDIE meta-knowledge. Interestingly, this approach maintains competitive performance and even surpasses some leading drug synergy prediction models. Case studies on the DDIE of decreasing antiplatelet activities and drug synergy in non-small cell lung cancer further demonstrate the significant advantages of RareDDIE and its practical value. Analyzing the process of constructing meta-knowledge in the model also provides interpretability for the prediction results. RareDDIE is publicly available at https://github.com/MrPhil/RareDDIE.

a the training and testing processes in few-shot settings: the dataset is first partitioned by interaction type, ensuring that events appearing in the training set do not reoccur in the test set. Within the training set, samples for each event i are further divided into a support set and a query set. RareDDIE utilizes the sparse support set samples to construct a proxy feature zpairs, approximating the true sample distribution of the event. Meanwhile, query samples are mapped into the same pair variational representation space, producing corresponding zpairq. In the comparator, predictions for the query set are generated by a Multilayer Perceptron (MLP) that encodes the discrepancy between zpairs and zpairq, followed by loss computation. The overall loss Lfew includes the task-specific label loss Ls and the self-supervised loss Lv from the PVR module, weighted by a coefficient µ. Similarly, during testing, a limited number of known examples are assembled as a support set to guide inference on previously unseen query samples. FIN, CSE, NAI and PVR represent the four primary modules of RareDDIE, namely Feature Initialization, Chemical Substructure Information Extraction, Neighborhood Adaptive Integration with Task Guidance and Pair Variational Representation, respectively. b training and testing processes in zero-shot settings: the dataset is again partitioned by interaction type. However, in the absence of support set-based proxy features, we introduce LSEM (Large-Scale Sentence Embedding Model) BioSentVec26,27 and an adversarial learning-based module, BST (Biological Semantic Transferring), to align natural language descriptions of interaction events with the pair variational representation space. This alignment enables query set prediction using only textual descriptions. Specifically, LSEM maps event-related language descriptions into a biological semantic space Xbs, which is then perturbed with random noise Xr, weighted by a coefficient k, and fed into the Mapper to produce an aligned representation M(Xbs). A Discriminator is employed to assess the quality of the alignment and guide the optimization of the Mapper accordingly. The detailed training procedure is described in the Methods section. Notably, during training, a support set is constructed to assist in training BST’s alignment module (Mapper), whereas during testing, predictions are made without any support set guidance. c transfer learning with cross-domain testing for drug synergy: RareDDIE pre-trained on drug interaction data can be adapted and fine-tuned for drug synergy prediction. d interpretability, and case study examples: a series of visualization analyses and case studies explore drug interactions and drug synergy mechanisms. For a more detailed description of these experimental workflows, refer to the Methods section.
Results
Description of RareDDIE
In this study, we developed a deep learning framework called RareDDIE, formulating DDIE prediction as a meta-learning task to address the generalization problem of novel events in DDIE with or without a few known samples. Specifically, the meta-learning task is defined as predicting drug-drug interaction for specific events, where the mechanism meta-knowledge from common events can be transferred to other fewer and rare events by RareDDIE for enabling novel DDIE prediction. The flowchart of RareDDIE is illustrated in Fig. 1.
In detail, RareDDIE employs knowledge graph-based negative sampling29 as a data augmentation technique to address label imbalance. For each event, RareDDIE trains a DDIE prediction task. Within the few-shot learning framework, as shown in Fig. 2a, we randomly sample the query and support set for each event task to establish and train a metric space (details provided in the Methods section). The support set simulates a few known samples, while the query set is used to train the event-specific models. After multiple iterations of the event task and support set sampling training, we sum the loss of all tasks and optimize the model parameters with gradient descent. Additionally, the CSE and NAI modules perform dual-granular structure information fusion, enhancing the representation of each drug. By incorporating task-guided weak relationships, RareDDIE adaptively captures critical molecular substructures and biological neighborhood local structures, achieving interpretable results from a micro to macro perspective. Since the model analyzes drug pairs rather than individual drugs, we propose the PVR module, which leverages an encoding-decoding framework to project paired data into a generalized relational metric space. This design not only integrates dual-granular information but also encodes features into latent semantic representations. Furthermore, to address the potential for undiscovered events without related drug records, we extended RareDDIE with a Biological Semantic Transferring (BST) module to create ZetaDDIE, enabling the model to handle this zero-shot problem. Specifically, the BST aligns the clinical semantic distribution with the general relation metric space, thereby constructing a semantic information metric for aleatoric events without any known examples. ZetaDDIE synthesizes dual-granular structure properties and clinical semantic information. Notably, the zero-shot setting differs slightly from the few-shot setting, with the details provided in the Methods section and Fig. 2a, b.
RareDDIE outperforms existing methods in few-shot and zero-shot settings for DDIE prediction
To demonstrate the advantages of RareDDIE, we compared it with seven baseline methods using the evaluation criteria of AUC, ACC, and F1 on common, fewer, and rare event test sets, as well as an independent rare event test set. The construction process for these test sets is detailed in the Methods section. To ensure a fair comparison, we reproduced all the baseline methods and conducted training and testing based on the same data splits.
First, we investigated the predictive performance in few-shot scenarios. Our method was compared with four meta-learning baselines, containing META-DDIE30, GMatching31, MetaR-In32 and MetaR-Pre32, and three leading models of DDI prediction, including DSN-DDI33, MRCGNN34, and KnowDDI35. Detailed descriptions of these baselines are provided in the Supplementary Note 1. Since most DDI predicting methods focus on the events with a high number of interactions21, we evaluated performance not only on fewer and rare events but also on common events. Figure 3a illustrates the performance of all models across the three test sets when provided with one and five DDI samples, respectively. In the one-shot setting, RareDDIE achieves average AUC of 0.8492, 0.8655, and 0.9392 for the common, fewer, and rare event test sets, respectively, demonstrating a statistically significant advantage over the best-performing baseline method. In the five-shot setting, RareDDIE achieves average AUC of 0.9105, 0.9351, and 0.9878 for the common, fewer, and rare event test sets, respectively, demonstrating a statistically significant advantage over the best-performing baseline method. The specific p-values for all comparative experiments are reported in the Supplementary Table 1. Notably, to ensure a fair comparison, we optimized crucial hyperparameter for all baseline methods. The search results and detailed procedures can be found in Supplementary Figs. 3–16.

a The AUC (Area Under the Curve), ACC (Accuracy) and F1 scores of DDIE (Drug-Drug Interaction Event) prediction on the common, fewer and rare event test set using seven comparison methods under the few-shot setting. In the experiments, our proposed RareDDIE extracts mechanism meta-knowledge from known events DDIs and subsequently transfers this knowledge to new events. Consequently, the model can generalize to the DDIs of novel events with only a few support samples. However, since existing DDI prediction methods are not designed for predicting interactions in unknown events, we directly provided these models with a few DDI samples from new events for training. These samples were used as support samples of test set in our framework. We compared our method against META-DDIE30, GMatching31, MetaR-In32, MetaR-Pre32, DSN-DDI33, MRCGNN34, and KnowDDI35. Each experiment is conducted five times, with a distinct set of randomly selected support samples used for training and prediction in each iteration. b The analysis of the prediction capability on an independent rare event test set. The model was first trained on the common event samples from collected Dataset1 and Dataset2, and then predicted directly on the independent rare event test set without any fine-tuning. Due to the limitation of the independent dataset, only three meta-learning models are utilized for comparison. Five independent results are obtained from the models of five independent training. c The AUC, ACC and F1 scores of DDIE prediction on the common, fewer and rare event test set with three variants under the zero-shot setting. To extend the meta-knowledge of RareDDIE to zero-shot tasks, we introduced a BST (Biological Semantic Transferring) module to create ZetaDDIE. The BST aligns dual-granular structure information with biological semantic information, leveraging a large-scale sentence embedding model for semantic information acquisition. Three variants are constructed: (1) ZetaDDIE without BST, which removes the Biological Semantic Transferring module; (2) ZetaDDIE with BioBERT, which extracts semantic information through BioBERT language model; (3) ZetaDDIE with Premodel, which uses the trained parameters from the RareDDIE model to initialize the ZetaDDIE model before training; (4) ZetaDDIE with BioSentVec, which extracts semantic information through BioSentVec language model. Each experiment is conducted five times, with a distinct set of randomly selected support samples used for training and prediction in each iteration. The significance test results of all experiments based on the two-tailed t-test without adjustment are reported in Supplementary Tables 1–3. Error bars represent the mean standard deviation across the 5 independent experiments and apply to all the panels. Source data are provided as a Source Data File.
Our method consistently outperforms all comparison methods, particularly in the one-shot setting, demonstrating significant advantages in predicting DDIs for rare and common events. This demonstrates that our approach effectively learns mechanism meta-knowledge for predicting DDIs in rare events and holds substantial benefits for common events as well. We also observed better performance in the five-shot setting compared to the one-shot setting, which is expected as more support samples allow the model to infer a more accurate metric with the meta-knowledge. Moreover, our model consistently outperforms all meta-learning baselines across different numbers of support samples, highlighting the benefits of adaptive neighborhood selection based on dual-granular structure information to construct pair variational representation. When compared to the leading DDI prediction models, our method shows a more significant advantage in the one-shot setting. These comparison models use DDI samples from new events during training, which remain invisible to our model, resulting in their performance improving more rapidly as more such samples are provided.
Moreover, we observed that common events tend to perform worse than fewer events with same hyperparameter. However, directly comparing the results of common event with fewer event is inherently unfair, as a single sample accounts for approximately 2% of each common event but around 5% of each fewer event dataset. The primary reason for the poorer performance of common event under the same setting is the smaller proportion of support set samples, which provides insufficient reference information. To further investigate this, we conducted a performance comparison under the same support set sample ratio, with results reported in Supplementary Table 4. These findings suggest that in real-world applications, if sufficient data is available, traditional supervised learning methods can be directly applied, or the number of support set samples should be increased to mitigate performance degradation.
Next, considering the importance of model generalization across different data distributions, we further analyzed the prediction capability on an independent rare event test set. Specifically, as an out-of-distribution (OOD) problem, this test set consists of event types entirely independent of those in the training set. Therefore, we compared our model only with meta-learning baselines. Due to its event-specific training process and limited generalization ability, META-DDIE was excluded from this comparison. As shown in Fig. 3b, RareDDIE demonstrated AUC improvements ranging from 0.0296 to 0.0429 on Dataset136 (p-value = 0.0226, two-tailed t-test) and from 0.1006 to 0.1877 on Dataset220 (p-value < 0.0001, two-tailed t-test), both with statistical significance, compared to the baselines. The statistical significance of other evaluation metrics is provided in the Supplementary Table 2. This indicates the consistent generalization capabilities of our model and suggests the learned mechanism meta-knowledge possesses a degree of universality. Although GMatching, another meta-learning method based on local neighborhood information, was expected to perform well, it failed on both datasets. This failure is attributed to Gmatching’s focus on encoding individual drug entities, relying solely on single-granularity molecular structure information. Additionally, we observed that the model trained on Dataset2 outperformed the model trained on Dataset1. This improved performance is likely due to Dataset2 containing a more diverse range of events and a larger number of corresponding DDI samples.
Lastly, while discovering DDI of novel events in clinical treatment can be viewed as a few-shot prediction problem for rare events, there may still be other undiscovered events in clinical practice. Therefore, we investigated predictive performance in zero-shot scenarios. Given that our model is designed primarily for few-shot scenarios, we introduced a Biological Semantic Transferring (BST) module to leverage semantic information and align it with mechanism meta-knowledge, thereby enabling effective zero-shot problem solving. The underlying rationale is that in the absence of predefined known samples to construct a support set for obtaining interaction event representations, we aim to generate the corresponding vectors directly from natural language descriptions. These semantic vectors must then be mapped into the constructed pair representation space. Specifically, we leverage a large-scale sentence embedding model to learn semantic features describing interaction events. Subsequently, an adversarial training approach37 is employed to train a mapping module that aligns these semantic features with our representation space. Finally, all query set samples, along with the mapped semantic vectors, are fed into the Comparator module to complete the final prediction, as illustrated in Fig. 2b. To further clarify this process, a more detailed toy example is provided in Supplementary Fig. 17. Since no existing methods effectively predict DDIs in zero-shot scenarios, we evaluated the predictive performance of ZetaDDIE and its variants, containing ZetaDDIE without BST, ZetaDDIE with BioBERT38, ZetaDDIE with Premodel, and ZetaDDIE with BioSentVec26,27.
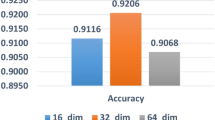
In the zero-shot setting, ZetaDDIE achieves AUCs of 0.6704, 0.6095, and 0.7915 in the common, fewer, and rare event test sets, respectively, demonstrating a statistically significant advantage over the best-performing baseline method. The specific p-values for all comparative experiments are reported in Supplementary Table 3. As shown in Fig. 3c, while the performance of ZetaDDIE is inferior to that in few-shot setting, it is generally superior to the three variant models. The variant without the BST module exhibits the worst performance, underscoring the importance of the BST module in aligning semantic and structural information. Additionally, in the common event test set, the variant initialized with RareDDIE parameters demonstrated better performance, suggesting that incorporating prior knowledge before alignment might be more effective for events with more samples. However, in the rare event test set, the choice of language model becomes more critical, as it directly determines the representation of classification labels. To further evaluate the embeddings from the two language models, we visualized and analyzed the embeddings of all events, reported in Supplementary Fig. 18.
Meta-knowledge of transfer-promoting drug synergy prediction
Through previous experiments, we demonstrated that our model can learn potential meta-knowledge of different drug functional mechanisms and universally generalize to new event types. Given the mechanistic similarities between drug interaction and drug synergy, both involving the regulation of protein networks by two drugs within a biological system, we hypothesize that the mechanism meta-knowledge can also be effectively transferred to drug synergy prediction tasks, enhancing prediction accuracy. To verify this hypothesis, we applied RareDDIE to cross-domain drug synergy predictions on the general dataset Dataset339. Specifically, drawing an analogy to DDIE prediction—where the goal is to determine whether a given drug pair will exhibit a specific interaction—we define drug synergy prediction as the task of predicting whether a given drug pair will exhibit synergistic effects in a specific cell line. A detailed definition can be found in the Methods section. We further compared RareDDIE with leading models specifically designed for drug synergy prediction and analyzed the critical role played by the transferred meta-knowledge.
We explored three different settings for RareDDIE in predicting drug synergy: de novo training RareDDIE with 1-shot setting (1-shot w/o transfer), fine-tuning RareDDIE with 1-shot setting (1-shot w/ transfer), and fine-tuning RareDDIE with 10-shot setting (10-shot w/ transfer). For fairness, we used the same data splits for comparison. Each experiment is conducted three times, with a distinct set of randomly selected support samples used for training and prediction in each iteration. As expected, the 10-shot w/transfer setting achieved the best performance, with an average AUC of 0.9047 and an average AUPR of 0.5887. The second-best was the 1-shot w/ transfer setting, with an average AUC of 0.7894 and an average AUPR of 0.2775. The unsatisfactory performance was observed in the 1-shot w/o transfer setting, with an average AUC of 0.5715 and an average AUPR of 0.1543. Figure 4a, b display the ROC and PR, respectively. These comparison results indicate that fine-tuning the model with transferred meta-knowledge outperforms de novo training the model. This suggests that the rich mechanism meta-knowledge derived from DDIE positively guides drug synergy prediction, thereby improving performance. Additionally, we observed that the performance improves with more support samples, similar to the DDIE prediction task, suggesting that the mentioned conclusions also apply to cross-domain drug synergy predictions.

a The ROC (Receiver Operating Characteristic) curves in drug synergy prediction based on three different settings: 1-shot w/o transfer, 1-shot w/ transfer, and 10-shot w/ transfer. 1-shot w/o transfer: directly training RareDDIE using the training set of the drug synergy dataset. 1-shot w/ transfer: using the pre-trained RareDDIE on the DDIE (Drug-Drug Interaction Event) dataset, followed by fine-tuning on the drug synergy test set in a 1-shot setting. 10-shot w/ transfer: using the pre-trained RareDDIE on the DDIE dataset, followed by fine-tuning on the drug synergy test set in a 10-shot setting. Each experiment is conducted three times, with a distinct set of randomly selected support samples used for training and prediction in each iteration. The shading represents the standard deviation. b The PR (Precision-Recall) curves for drug synergy prediction based on three different settings show the performance of 1-shot w/o transfer, 1-shot w/ transfer, and 10-shot w/ transfer. Each experiment is conducted three times, with a distinct set of randomly selected support samples used for training and prediction in each iteration. The shading represents the standard deviation. c The visualization of the embedding of the drug pair Topotecan with BEZ-235 alongside the embedding of its corresponding 8 cell lines. The model predicts synergy by assessing the distance between these embeddings. d The AUC (Area Under the Curve) on the training set during the training process for both transfer learning and de novo learning models. We recorded the average AUC values every 50 iterations over 12,000 iterations. This approach helps smooth out fluctuations caused by variations in data from different tasks in each iteration. e The AUC on the validation set during the training process for both transfer learning and de novo learning models. f The loss on the training set during the training process for both transfer learning and de novo learning models. Source data are provided as a Source Data File.
Next, we compared our method with leading models designed for drug synergy prediction, including PRODeepSyn40, DeepSynergy41, AudnnSynergy42, SVR43, and XGBoost44. Considering the limited availability of labeled samples in practice and the performance of the model, we used the 10-shot w/ transfer setting for RareDDIE. For fairness, we followed the guidelines from previous work40 to frame the drug synergy prediction as a classification task. Through benchmarking, RareDDIE demonstrates competitive predictive performance, with detailed comparative analyses provided in Supplementary Table 5. Notably, RareDDIE does not incorporate explicit cell line features during training. Instead, it relies entirely on meta-knowledge derived from the biological neighborhood structure and critical molecular substructures of support set samples, using these as reference information to distinguish cell lines. Comparison with the state-of-the-art method highlights RareDDIE’s ability to leverage rich drug interaction knowledge to enhance drug synergy prediction, despite the differing purposes and mechanisms between drug interactions and drug synergy. Moreover, fine-tuning the model with cross-domain knowledge within the RareDDIE framework markedly improves predictive accuracy compared to a model trained without such information. This is understandable, as the lack of explicit cell line features and the limited scale of available drug synergy datasets would otherwise constrain predictive performance without substantial prior mechanism meta-knowledge45.
To illustrate the model’s ability to distinguish the same drug pair under different cellular contexts, we visualized the embeddings of drug pairs known to exhibit distinct effects across cell lines, alongside reference embeddings extracted for corresponding cell lines. As a representative case, Fig. 4c shows the embedding relationship between the drug pair Topotecan–BEZ-235 and reference information from each cell line. The results indicate that when the Euclidean distance between their embeddings exceeds 1.2, the model predicts no synergy, whereas smaller distances correlate with synergy predictions. Crucially, RareDDIE does not rely on a fixed distance threshold but instead adapts its decision-making process dynamically. A more comprehensive explanation and full visualization results are provided in Supplementary Table 6. To further investigate whether the reference embeddings extracted from the support set capture aspects of the cellular environment, we analyzed the clustering patterns of drug pairs across different cell lines. Specifically, we computed cluster scores and compared performance before and after PVR-based feature extraction, as well as against randomly generated features. Results detailed in Supplementary Figs. 19–21 highlight that the model effectively integrates information from the support set as a proxy for cellular context, thereby competitive performance. Nevertheless, the absence of explicit cell line representations suggests potential areas for further optimization. Future work incorporating engineered cell representations could enhance model performance and improve generalization across diverse cellular contexts.
Our findings indicate that incorporating meta-knowledge of drug interactions into drug synergy prediction enhances model accuracy. To elucidate the performance differences between de novo learning and transfer learning, we analyzed the AUC values for the training and validation datasets as well as the training loss throughout the training process. The results are depicted in Fig. 4d–f. As shown in Fig. 4d, during the training process, AUC values for both models gradually increase and stabilize over iterations. However, the transfer learning model consistently exhibits significantly higher AUC values from the outset and maintains superiority throughout the entire training process. This suggests that the mechanism meta-knowledge inferred from DDIE provides a shortcut for drug synergy prediction, enabling the model to learn faster and more effectively, with enhanced fitting ability in cross-domain tasks. Figure 4e illustrates the performance of the validation set during training. Overall, AUC values for both models rise rapidly in the initial thousands of iterations and then stabilize with minor fluctuations. Similar to the training set results, the transfer learning model consistently outperforms de novo learning from the start, reaching higher AUC values more quickly. The main reason is the model has already acquired meta-knowledge containing mechanistic information from drug interactions, demonstrating strong generalization capability across different tasks. In Fig. 4f, we observe that the training loss values for both models decrease with more iterations, and the transfer learning model shows lower loss values overall and faster convergence. This aligns with the training and validation AUC performance, indicating that the transfer learning model, enriched with additional knowledge, is more stable and robust during training. Notably, there are some fluctuations during the training process. Around 6000 iterations, the loss value of the transfer learning model shows a significant spike, which coincides with fluctuations in the training and validation AUC values. This spike likely reflects the varying difficulty of tasks encountered during training, leading to temporary increases in loss on more challenging tasks. Subsequently, the loss quickly returns to a relatively low level, possibly due to our proposed NAI module, which uses task-guided weak relationships to adaptively capture the contributions of neighbors, thereby accelerating model convergence.
In summary, transferring drug interaction information to drug synergy prediction results in advantages in training speed, generalization ability, and training stability compared to de novo learning. This implies potential superiority and offers valuable insights for further research and practical applications.
Inferring mechanism meta-knowledge from dual-granular structure
Our findings indicate that our model effectively extracts mechanistic meta-knowledge of drug actions. This capability enables RareDDIE to generalize predictions to novel events and transfer this knowledge to cross-domain drug synergy prediction tasks, thereby enhancing prediction accuracy. To further investigate how the model constructs meta-knowledge, we conducted interpretability analysis from three perspectives: biological neighborhood, molecular structures, and relation metric space—corresponding to the three modules of our model. Additionally, we performed a contribution analysis to evaluate the role of each module in generating meta-knowledge. Furthermore, we evaluated the generalizability of the molecular representations by directly transferring the molecular features from the trained molecular representation module CSE of RareDDIE to molecular property prediction tasks.
Specifically, we analyzed the prediction process for the relatively important rare drug interaction event46, “The protein binding of #Drug2 can be decreased when combined with #Drug1”. Following the 1-shot setting, the model utilized the drug pair of DB01067-DB01032 (Glipizide-Probenecid) as the support set. Taking DB00252-DB01327 (Phenytoin-Cefazolin) as an example, we visualized the biological neighborhood background graph of these four drugs with software Gephi-0.9.2 in Fig. 5a. As shown in the figure, the network delineates the specific and correlated neighbors of four distinct drugs. Certain genes are uniquely associated with a single drug, endowing the respective drug with unique characteristics. These genes receive higher attention scores in the model, represented by thicker edges—for instance, Phenytoin-GABRA1 and Glipizide-PSMB8. Meanwhile, a shared set of genes is associated with all four drugs, establishing potential interconnections among them. This suggests functional overlap or common mechanisms of action, offering insights into potential synergy or competition between these drugs. For example, ALB is linked to all four drugs. Furthermore, different drugs and their combinations may establish connections with additional drug pairs that share analogous DDIEs through common neighbors. Leveraging a limited number of samples as reference, the NAI module in our model utilizes this shared neighbor information to construct meta-knowledge, enabling the inference of previously model-invisible event types.

a Visualization of biological neighborhood background graph for the support set drug pairs and query set drug pairs, where green nodes represent drugs and red nodes represent genes. Different edge colors denote various types of relationships, while edge thickness reflects the importance of the connections. Genes in the center are the common neighbors shared between the drug pairs. b Visualizations of the key molecular substructures captured by the chemical substructure information extraction (CSE) module for Phenytoin. Red regions indicate structures deemed important by the model, while green regions represent less significant structures. The intensity of the color reflects the degree of importance. c 2D interaction map illustrating the molecular interactions between Phenytoin and the residues of Albumin (P02768, PDB ID: 6YG9). The map highlights specific bonding interactions. The circle represents the names of residues, and different colors represent different bond types. d Docking conformation of Albumin with Phenytoin. Yellow lines indicate the three hydrogen bonds formed at the corresponding binding sites. The atomic coordinate data of the docking conformations is provided in Supplementary Data 1. e Detailed docking conformation of Albumin with Phenytoin, showing key interactions between critical residues and molecular substructures. The hydrogen bond distances are annotated to illustrate the binding tightness. The interaction energy calculated under CHARMm is −47.45 kcal/mol. f Feature distribution visualizations based on t-SNE50, which display all sample distributions at three stages: directly concatenated molecular structure features of drug pairs, concatenated dual-granular structure features of drug pairs, and variational drug pair features transformed into the relation metric space. Numeric labels indicate cluster centers for samples corresponding to specific events. Specific events mentioned in experiments: (1) Event 11: “#Drug1 may increase the central nervous system depressant (CNS depressant) and hypertensive activities of #Drug2”; (2) Event 12: “The risk of a hypersensitivity reaction to #Drug2 is increased when it is combined with #Drug1”; (3) Event 13: “#Drug1 may increase the photosensitizing activities of #Drug2”; (4) Event 17: “#Drug1 may increase the hypotensive and central nervous system depressant (CNS depressant) activities of #Drug2”.
Building on our analysis from the biological neighborhood perspective, we further investigated the relationships between the substructures of the four drugs and the key gene. Given ALB’s central role and its associations with all four drugs, we focused on its translated protein, Albumin (P02768, PDB ID: 6YG9). This protein is a crucial plasma protein in the human body, playing a pivotal role in drug transport, metabolism, and interactions. This protein is also implicated in the rare drug interaction events examined in this study. We first visualized the key molecular substructures identified by the CSE module using the similarity map47 implemented in RDKit. This module utilizes atom-level attention weights to highlight important local structures within the molecule. As shown in Fig. 5b, oxygen and nitrogen atoms within the cyclic structure of Phenytoin are prominently highlighted, suggesting that these substructures receive significant attention from the model. To further validate these findings, we conducted molecular docking48 experiments using Dock6.1249 to assess ligand-receptor docking conformations and grid scores. We also used Discovery Studio 2019 to perform docking simulations and calculated interaction energy (a measure of binding affinity, where lower values indicate stronger binding) using the CHARMm force field. Figure 5c–e illustrate the interaction between Phenytoin and key residues, alongside the docking conformation of the Albumin-Phenytoin complex. Additional docking results for the other three molecules with Albumin are provided in Supplementary Figs. 22–24. A detailed analysis revealed that the atom groups identified as important by our model correspond to protein binding sites, forming hydrogen bonds with key residues such as ARG-114, LEU-115, and ARG-117. These findings support the model’s ability to pinpoint molecular substructures relevant to protein interactions. This indicates that RareDDIE can adaptively identify critical structures for different molecules in an unsupervised manner, without relying on protein structure information. This capability allows the model to build molecular structure-level meta-knowledge that generalizes well across various molecules, thereby overcoming the limitations of similarity-based approaches.
Although our dual-granular structure can effectively represent chemical properties and potential biological functional relationship information of individual drugs, representing drug pairs by simple concatenation makes it difficult to transform features into specific event descriptions. Our designed PVR module learns meaningful representations of paired drugs, and we have observed that the constructed relation metric space can effectively describe latent medical semantic information. To demonstrate this, we employed t-SNE50 to reduce the dimensionality and visualize the feature distributions of all samples at three stages: directly concatenated molecular structure features of drug pairs, concatenated dual-granular structure features of drug pairs, and variational drug pair features transformed into the relation metric space. As shown in Fig. 5f, directly concatenated molecular structure features fail to distinguish between drug pair interaction events. When we incorporated background knowledge to construct dual-granular structure features, the identification of different event categories significantly improved. However, these features still lack a medical semantic representation. For instance, events 11 and 17, the most similar interaction events, are not the closest in the dual-granular structure feature distribution. In contrast, they are very close in the variational pairwise drug feature distribution, indicating that variational drug pair features could represent latent medical semantic information and automatically identify similar events. Similarly, events 12 and 13 are medically similar as they both involve similar immune response mechanisms, such as sulfonamides causing both systemic allergic reactions and photosensitivity. However, only variational pairwise drug features exhibited this proximity in embedded space, demonstrating that our model can infer high-level representations with hidden medical semantic information without similarity data supervision.
To further assess the generalization ability of RareDDIE in molecular property and function representation, we evaluated its performance on MoleculeNet51 benchmarks for molecular property prediction. Specifically, we directly applied the CSE module from RareDDIE, trained on common events with a 10-shot setting, to process all molecular SMILES and generate feature representation vectors. For a fair comparison, we employed standard machine learning algorithms such as Logistic Regression (LR) and Random Forest (RF) for classification. Seven datasets encompassing molecular property prediction and bioactivity tasks were selected to evaluate the model’s performance. The results demonstrate that our method consistently achieves either the best or second-best performance across most datasets, further indicating that the model effectively captures meaningful molecular features beyond explicitly trained drug interaction pairs. Detailed results and analyses are provided in Supplementary Table 7.
Case studies of severe rare DDI events and drug synergy prediction
When approving new drugs, regulatory agencies rely on randomized controlled trials (RCTs) submitted by applicants. However, patients are often too few to effectively detect rare adverse drug reactions7. Furthermore, predicting rare DDIEs is challenging due to the vast interaction space and limited data. To further validate the practical efficacy of our model, we conducted a case study using the RareDDIE model to predict a severe rare DDI event46,52: the decrease in antiplatelet activities. Based on the DrugBank database, we predicted whether this interaction event would occur when any two of 1706 drugs were used simultaneously. Specifically, we trained the model on the common event dataset and constructed the support set for the studied rare event using all seven available samples recorded in DrugBank: Rifampicin-Prasugrel, Omeprazole-Clopidogrel, Cangrelor-Clopidogrel, Esomeprazole-Clopidogrel, Erythromycin-Clopidogrel, Morphine-Clopidogrel, and Cangrelor-Prasugrel. The remaining drug pairs were used to evaluate their association with this rare event, and prediction scores were computed accordingly. Notably, model training was performed using interaction samples from all 63 common events in DrugBank. The case study focused on the event “the decrease in antiplatelet activities”, which does not belong to the common event category. Moreover, the seven known samples were provided as prior knowledge and were not subject to prediction. The complete prediction results, with scores greater than 0.1, are reported in Supplementary Data 2. Table 1 shows the top nine affected drugs, with ranks indicating the relative positions of the first occurrences of these drugs among the top 100 predicted drug interaction pairs.
As shown in the table, nearly all identified drugs are related to antiplatelet functions. Although Idarubicin is not directly related to antiplatelet activity, it is used to treat leukemia, which impairs normal hematopoietic function and reduces platelet production. Consequently, if Idarubicin’s efficacy of Idarubicin is compromised, it could indirectly affect platelet. This demonstrates that our model can identify potential indirect drug interaction events. Notably, among the seven available samples of this rare event used as prior knowledge, only two antiplatelet-related drugs—Clopidogrel and Prasugrel—were present. What is intriguing is that, as shown in Table 1, despite this limited information, our model successfully identifies additional antiplatelet agents, underscoring its robust generalization capacity in molecular function recognition. This success is primarily due to the effective capture of dual-granular structure information. Furthermore, to validate the predictive capability of the proposed model, we examined the top 10 prediction results.
As shown in Table 2, Most of the predicted results correspond to known DDI. For the decreasing in antiplatelet activities event, we observed that Manidipine and Lercanidipine can reduce the efficacy of Clopidogrel. Chlorpheniramine can reduce the metabolism of Vinorelbine, whose side effects are related to antiplatelet function. While some predictions remain unconfirmed, we cannot exclude the possibility of undiscovered mechanisms at present. Therefore, the predicted results from the model warrant further wet-lab experimental analysis.
Additionally, combination therapies for complex diseases like cancer are receiving significant attention. However, the combinatorial explosion presents a substantial challenge, necessitating more effective prediction methods. As previously demonstrated, transferring DDIE meta-knowledge can enhance drug synergy prediction performance. Therefore, we further investigated drug synergy prediction for non-small cell lung cancer cell lines. Specifically, based on the 5-shot setting with RareDDIE, we employed all samples from Dataset453 as the lung cancer test set, DDIE common events as the training set, and the drug combination dataset Dataset3 as the validation set. Table 3 shows the top 20 prediction results, with all results available in Supplementary Data 3.
As shown in Table 3, all predictions align with known facts, demonstrating the robust generalization capability of RareDDIE in predicting drug synergy. Notably, despite the absence of cell line features, the model leveraged reference information constructed from the support set as a proxy for the cellular environment, enabling it to successfully generate high-confidence predictions across 13 different cell lines. This underscores the model’s architectural strengths. While RareDDIE proves effective in drug synergy prediction, there remains room for improvement. The current model architecture, designed for DDIE prediction, does not incorporate cell-line features, and the constructed relation metric space may not fully capture the rich information inherent in cell lines. Future work should focus on integrating cell line information into RareDDIE to address these limitations and further enhance the model’s predictive performance.
Ablation experiments
To evaluate the contribution of individual modules in RareDDIE, we performed an ablation study to assess their effectiveness. The results indicate that the PVR module plays a particularly crucial role in our model. The details of RareDDIE with different variants and the corresponding comparative results are provided in Supplementary Note 2 and Supplementary Tables 8,9.
Discussion
This work chiefly focuses on predicting rare drug-drug interaction events and introduces a model of RareDDIE, based on the dual-granular structure-driven pair variational representation. RareDDIE addresses the challenges of constructing generalizable drug representations, creating a relation metric space for forming event descriptions, and ensuring interpretability, making it particularly suited for few-shot scenarios. Additionally, we introduce the Biological Semantic Transferring (BST) module and large-scale sentence embedding model to align clinical semantic distributions with the general relation metric space, allowing RareDDIE to extend to zero-shot prediction as its variant ZetaDDIE.
Our extensive experiments demonstrate that RareDDIE achieves superior performance in few-shot settings, while ZetaDDIE excels in zero-shot prediction scenarios. This success is attributed to the model’s ability to effectively capture crucial chemical substructures and adaptively aggregate neighborhood information. Furthermore, we verified that transferring DDIE knowledge significantly enhances drug synergy predictions, outperforming existing models. Interestingly, the interpretability analysis revealed that the variational representations can autonomously form event semantic information in an unsupervised manner, with the latent space representation measuring the similarity between DDI events. Case studies on antiplatelet activity reduction and non-small cell lung cancer drug synergy further validate the practical applicability and robustness of our model.
Beyond methodological advancements, RareDDIE has significant implications for clinical applications and drug development. By predicting rare DDIEs, our model enhances pharmacovigilance by identifying high-risk drug interactions that might otherwise evade detection, thereby improving drug safety. Moreover, its capacity to model drug synergy suggests potential applications in drug repurposing and rational design of combination therapies, particularly in fields such as oncology and infectious diseases. Future directions could explore the integration of patient-specific data, further extending its utility in personalized medicine.
RareDDIE provides a significant advancement in the early detection and prediction of rare DDIEs, contributing to safer drug development and improved patient care. However, our model has limitations that could be addressed in future research. Firstly, for few-shot prediction, the background graph we constructed currently uses only relatively important gene-drug relationships, which may bring one-sided information while reducing noise. Future work can address this by constructing larger-scale knowledge graphs with denoising technology. Secondly, for the transfer learning task of drug synergy, our model, designed for DDIE prediction, does not take cell line information into account. Although it has achieved competitive performance, this limitation affects the model’s generalization. Incorporating cell line-specific molecular and phenotypic features into the model could further enhance its predictive power for personalized drug combination strategies.
Methods
Dataset description
For the problem of drug-drug interaction events prediction, we mainly conduct experiments on the two public datasets: DrugBank54 and TWOSIDES55. The former is mainly used as the benchmark data set for comparison with SOTA, and the latter is utilized to construct an out-of-distribution data set to test the generalization ability of the model. Furthermore, according to the quantity of drug interaction events, we divided the set of drug pairs, \(D\), into three event disjoint sets, common event set of \({D}_{{{\rm{com}}}}\), fewer event set of \({D}_{{{\rm{few}}}}\) and rare event set of \({D}_{{{\rm{rare}}}}\), where the common event set has sample sizes greater than 50, fewer event set has sample sizes between 20 and 50, and rare event set has sample sizes fewer than 20. To construct the datasets for few-shot and zero-shot DDIEs prediction, we used the Dataset1 collected from Nyamabo et al.36, covering all three mentioned event types, including 1706 drugs with 191808 interactions under 86 events. In addition, due to the number of samples under all events exceeding 100, we use Dataset2, which contains 1258 drugs with 323539 interactions under 100 events, published by Lin et al.20, as the main source of common events. In order to compare the generalization ability of the model, the rare event samples in TWOSIDES are collected to build an independent test set, which includes 182 drugs with 346 interactions under 40 events. These datasets have similar characteristics of long-tail distribution, and we show the figures of drug interaction events distribution in Supplementary Figs. 1-2. In this study, the most common events are used to train the model, ensuring the generalization ability to learn how to predict rare and even unknown events. The remaining common events and all the fewer and rarer events are used to test the model performance. It is worth noting that, considering the different distribution of the data sets, the test tasks of common events and fewer/rare events are trained by common events under Dataset2 and Dataset1, respectively.
In order to investigate model performance in predicting drug synergy based on cross-domain knowledge transfer, we collected two drug combination datasets Dataset3 and Dataset4. The Dataset3 is a large-scale synergy dataset constructed by O’Neil et al.39, containing experiment results of 39 cell lines from 6 cancer tissue types and 583 diverse drug combinations among 38 drugs. In the specific experiment, all dosage regimens were repeated four times, and measured the cell growth rate relative to control group was after 48 hours. Preuer et al.41 integrated the dataset and calculated the synergy scores and generated 23052 samples of drug combinations under different cell lines, where samples with a score greater than 30 are considered to have a synergistic effect. Furthermore, aiming for lung cancer, Dataset4 is constructed as an independent test set from the experimental data of Nair et al.53 to verify the performance of the model in predicting the synergistic effect. Specifically, according to the three indicators of Nair’s work, we screen out the samples that meet the requirements of synergy_ratio less than 1, synergy_diff less than 0, and HAS less than 0 as the synergistic set. To screen high-quality negative samples, we chose samples that were unsatisfied with all three indicators to construct a negative sample set. After deleting the drugs without smiles, we got an independent test set with 2120 positive samples and 7242 negative samples among 77 drugs in 81 non-small cell lung cancer cell lines. The details of the four datasets are given in Table 4.
In addition, for the feature profile, we collected the structural information of all drugs and the knowledge graph DRKG56 is utilized for pre-training for the initialization of drugs. Notably, before the pre-training, we process the data of the knowledge graph and delete all DDIs to prevent information leakage, according to the previous work12.
Graphs construction and problem formulation
In this section, we first introduce the construction of drug structure graph, knowledge graph and background graph, and then give formal definitions of the DDIE task, few-shot DDIE predicting task, zero-shot DDIE predicting task, cross-domain DDIE predicting task, and drug synergy predicting task.
For all drugs in each dataset, we denote hydrogen-suppressed undirected molecular graph \({G}_{Mol}=({V}_{Mol},{E}_{Mol})\) through RDKit57 processing corresponding SMILES, where \({V}_{Mol}\) and \({E}_{Mol}\) are the node set and edge set representation of the atoms and the chemical bonds of a molecule, respectively. The pre-training representation of each drug is captured by self-supervised learning from the biomedical knowledge graph \({G}_{BKG}=({V}_{KG},{E}_{KG},{R}_{KG})\), where \({V}_{KG}=\{{v}_{1},{v}_{2},{v}_{3},{..}.,{{\mathrm{v}}}_{n}\}\) indicates biochemical entities such as drug, protein, biological process, etc., \({R}_{KG}=\{{r}_{1},{r}_{2},{r}_{3},...,{{\mathrm{r}}}_{m}\}\) stands for relation types including drug-enzyme, drug-disease, etc. and \({E}_{KG}=\{({v}_{i},r,{v}_{j})|{v}_{i},{v}_{j}\in {V}_{KG};\,r\in R\}\) represents \(r\) relations from entity \({v}_{i}\) to entity \({v}_{j}\). Furthermore, we extracted all the drug-target relations from \({G}_{BKG}\) and constructed a biological background graph \({G}_{B}=({V}_{B},{E}_{B},{R}_{B})\) to focus on the information on drug targets, which promotes our model to effectively infer rare or even unknown DDIE types. Intuitively, our background graph leverages information from biological entities associated with drugs as features, thereby constructing representations enriched with biological semantic relationships. This approach contrasts with models17,18,19 that directly compute drug relationships.
Definition 1: We define the DDIE task \({T}_{d}=\{{E}_{d},{M}_{d}\}\) for meta learning, where \(e\in {E}_{d}\) is a type of drug interaction event and \({M}_{d}^{e}=\{({d}_{i},{d}_{j})\}\) is the drug pair set of its corresponding event \(e\).
Definition 2: We define the few-shot DDIE predicting task inherited from DDIE task, which utilizes the event-specific model for prediction after learning a prototypical metric with samples of the type of event in meta-testing.
Definition 3: We define the zero-shot DDIE predicting task inherited from DDIE task, which utilizes the event-specific model for prediction without relearning a prototypical metric based on any samples.
Definition 4: We define drug synergy task \({T}_{c}=\{{E}_{c},{M}_{c}\}\) for meta learning, where \(c\in {E}_{c}\) is a type of cell line and \({M}_{c}^{e}\) is the drug pair set of its corresponding cell line \(c\).
Definition 5: We define the cross-domain drug synergy predicting task inherited from drug synergy task, which utilizes few-shot training strategy based on DDIE knowledge transferring.
Meta-learning settings
The meta-learning framework is designed to capture general knowledge from multiple related tasks, so that the model can use this general experience to adapt to novel tasks and improve performance23. Recent meta-learning approaches fall into two main types: (1) Metric-based methods: The matching network58 is a classic metric-based meta-learning algorithm that attends to learn generalizable metrics and matching functions for each category of training task. Once a good feature extractor is trained, the new category of samples can be determined by comparing the vector space of a small labeled support set; (2) Gradient-based methods: The classic gradient-based method MAML24 aims to learn the optimization of model parameters, summing up multiple task losses and updating the parameters across tasks with the gradients of few-shot examples. In this work, we apply metric-based methods matching networks as the training strategy. Besides, we follow GMatching31 to make some improvements to stabilize the training process of matching networks in biomedical graph scene.
In the details of RareDDIE, several techniques, including molecular chemical substructure information extraction (CSE), neighborhood adaptive integration with task guidance (NAI), pair variational representation (PVR), and biological semantic transferring (BST) are applied to improve the performance of RareDDIE. We first describe the few-shot learning setting for training and evaluation. According to different tasks, meta-train set \({{\rm T}}_{meta-train}=\{{E}_{dtr},{M}_{dtr}\}\) can be sampled from the distribution of the event task set \({T}_{d}\), and further be split as a support set \(S\) and query set \(Q\), where the former is used to build metric, and the latter is used to predict the matching score to calculate the loss. In the same way, the meta-test set \({{\rm T}}_{meta-test}=\{{E}_{dte},{M}_{dte}\}\) is constructed. For imitating the few-shot prediction, \({{\rm T}}_{meta-train}\) and \({{\rm T}}_{meta-test}\) have the same number of support samples. Notably, we construct negative samples through randomly replacing an entity of the sample pairs. Then we randomly initialize the parameters \(\theta\) of our metric model and sample \(t\) tasks from \({{\rm T}}_{meta-train}\) in each epoch to train the model. To learn a metric for new facts through a few examples, we repeatedly sample support samples for each task and calculate loss with the function:
which indicates how well RareDDIE works on \({Q}_{{e}_{k}}\) while giving few-shot data from \({S}_{{e}_{k}}\) under event \({e}_{k}\). After obtaining the loss of each task, RareDDIE applies the gradient descent with the average loss across all tasks. Based on the above learning process, our model has a good ability to predict drug-drug interactions under novel events with few support samples.
We further describe the zero-shot learning setting for training and evaluation. The meta-train and meta-test sets are nearly identical to those in the few-shot learning setting, but the configuration and function of the support set differ. To enable our model to be applied to zero-shot tasks, biological semantic transferring module is used to align the semantic information with the biological functional structure information. During training, we retain the support set to learn a function \(M(x)\) to align biological semantic space \({X}_{bs}\), which is directly utilized to build the metric. During testing, since any known samples are not required, the process includes only the query set. Similar to the few-shot learning setting, the loss can be calculated by the function:
where \({\ell }_{align}\) indicates alignment loss.
Architecture of base model in RareDDIE
As Fig. 1 shows, the architecture of the base model in RareDDIE mainly consists of five modules: an initialized module to obtain the embedding for each entity and relation in the background graph, a CSE module to obtain molecular chemical substructure information for each drug, a NAI module to adaptatively integrate biological neighborhood information with corresponding task guidance, a PVR module to effectively map the pairwise data to general relation metric space, and a comparator module to score the probability of DDI events.
To equip the model with knowledge of multi-domain biomedical relations, we first construct a feature initialization (FIN) module based on knowledge graph embedding to extract rich feature representations. It aims to learn biological node representations, providing essential initialization for the NAI module. After constructing a biomedical knowledge graph \({E}_{KG}=\{({v}_{i},r,{v}_{j})|{v}_{i},{v}_{j}\in {V}_{KG};\,r\in R\}\) for each drug in the meta-train set, the TransE59 is used to learn the embeddings of all entities and relations in the knowledge graph in a self-supervised manner. The loss is calculated using the following formula:
where \(h\), \(l\) and \(t\) represent the embeddings of drug \({v}_{i}\), \({v}_{j}\) and relation \(r\); \(h'\) and \(l'\) indicate negative samples; function \(d(\cdot,\cdot )\) calculates the distance between two vectors; \(\gamma\) means tolerance of the distance between positive and negative samples.
Chemical substructure information extraction in RareDDIE for each drug. Considering that the crucial substructure of a molecule can determine its function60, we apply the Chemical Substructure Extraction (CSE) module to learn the most critical information associated with the DDIE task and identify the most interpretable substructure, which can bind to the protein pockets related to the events. This module applies GNN with attention mechanisms to extract and highlight key molecular substructures. The module takes the drug \({G}_{Mol}=({V}_{Mol},{E}_{Mol})\) as input, and then, the embedding of all atoms and chemical bonds is initialized by corresponding chemical knowledge, which is reported in Supplementary Table 10-11. For an atom \(i\), the initial feature is constructed into a one-hot vector \({x}_{ato{m}_{i}}\) with 55 properties, and for each chemical bond between atoms \(i\) and \(j\), the initial feature is constructed into a one-hot vector \({x}_{bon{d}_{i,j}}\) with 17 properties. We further employ the Graph Transformer61 architecture to learn the molecular graph structure, enhancing the memorization capabilities of the GNN model. With the information aggregation from the neighborhood atoms \(N(i)\) and corresponding bonds in molecular graph \({G}_{Mol}\), the representation of the atom \(i\) can be updated by the function:
where \({x}_{ato{m}_{i}}^{l+1}\) indicates the embedding after aggregating the atom feature \({x}_{ato{m}_{j}}^{l}\) and bond feature \({x}_{bon{d}_{i,j}}^{l}\) in \(l-{{\rm{th}}}\) convolutional iteration. \({\sigma }_{1}(\cdot )\) represents the elu activation function. \({\alpha }_{i,j}\) is the attention coefficient for weighting neighborhood information, which is defined as:
where \({W}_{4}^{l}\) and \({W}_{5}^{l}\) denote query and key projections, respectively. \(d\) is the hidden size. The representation of the bond between atoms \(i\) and \(j\) can be updated by a multilayer perceptron (MLP):
After obtaining the features of all atoms and chemical bonds, we aggregate all information to calculate molecular representations \({x}_{Mol}\), which can be generalized to any molecule regardless of the number of atoms:
where \({{\rm{SAGPooling}}}(\cdot,\cdot )\) is an aggregation operator of self-attention graph pooling62. \({x}_{ato{m}_{all}}^{l}\) and \({x}_{bon{d}_{all}}^{l}\) indicate \(l-{{\rm{th}}}\) atomic and chemical bond aggregation information, respectively. \({\beta }_{l}\) is the contribution attenuation coefficient of each layer, and ⊙ denotes Hadamard product.
Neighborhood adaptive integration with task guidance explicitly captures structural patterns from the biological background graph. Through these patterns, we infer whether pairwise drugs will interact in novel events and identify the most crucial neighbor to capture event-related target protein. Considering that different entities may exhibit various roles in a task, we propose the Neighborhood Adaptive Integration (NAI) module, using task-guided weak relationships to adaptively capture the contributions of neighbors. Weak relationships are represented by the structural feature differences between paired drugs. Intuitively, novel drugs often lack extensive known information, yet their target proteins typically exist within biological networks. Therefore, this module leverages relevant entities within the biological network to indirectly characterize new drugs. To mitigate potential biases toward biological functional information, we incorporate weak relationships to refine the feature construction process.
Specifically, for each drug \({v}_{i}\in {V}_{B}\), we capture its first-order neighbors based on the biological background graph \({G}_{B}=({V}_{B},{E}_{B},{R}_{B})\) to maintain sufficient neighborhood information and the scalability of the graph. With the neighbor set \(N({v}_{i})=\{({r}_{j},{v}_{j})|({v}_{i},{r}_{j},{v}_{j})\in {E}_{B};{r}_{j}\in {R}_{B}\}\) of drug \({v}_{i}\), we use the following function to adaptively capture its neighborhood feature \({x}_{neig{h}_{i}}\):
where \({C}_{{r}_{j},{v}_{j}}\) indicates the feature of neighborhood pairwise relation-entity, which can be defined as the function:
where the relation feature \({x}_{{r}_{j}}\) and the entity feature \({x}_{{v}_{j}}\) are initialized by the FIN module and \(\oplus\) denote concatenation. We further defined \({\lambda }_{{v}_{i},{r}_{j},{v}_{j}}\) to learn the contribution of different entities:
where \({X}_{{v}_{i},{v}_{j}}\) is the weak relation representation, \(A\) and \({b}_{A}\) indicate the weight and bias of the bilinear transformation. \({x}_{Mo{l}_{i}}\) is the chemical structure feature of \({v}_{i}\), extracted by the CSE module. Finally, the single drug feature \({x}_{dru{g}_{i}}\), incorporating dual-granular structure information, can be calculated by the following function:
Construction of a general relation metric space of pairwise drugs. While extracting dual-granular structure information effectively describes each drug, it lacks the capability to directly represent drug pairs. Therefore, we employ the pair variational representation (PVR) module to learn an effective general relation metric space capable of adapting to diverse tasks through the mapping of universal representations. It employs a VAE to transform individual drug features into a general relation metric space, improving adaptability. Specifically, based on VAE63, the encoder is utilized to map pairwise features \({x}_{pair}={x}_{dru{g}_{i}}\oplus {x}_{dru{g}_{j}}\) to a metric space \({z}_{pair}\sim q({z}_{pair}|{x}_{pair})\), whose mean and variance are controlled by the output of the encoder parameters. Then, the decoder maps \({z}_{pair}\) back to the original data space to reconstruct \({x}_{pair}\sim p({x}_{pair}|{z}_{pair})\). Constructing a loss function by maximizing the lower bound of joint probability \(p({x}_{pair},{z}_{pair})\):
where \({{\rm{KL}}}(\cdot )\) means Kullback-Leibler divergence. The PVR module ensures the applicability of our model to different tasks and enhances its generalization ability when performing cross-domain predictions.
Constructing a comparator to make the final prediction. It determines whether the query sample and reference sample belong to the same category by computing their differences and leveraging a neural network-based architecture for classification. Utilizing the Comparator modules of RareDDIE, we calculate the general metric embeddings of the samples from the support set \(S\) and query set \(Q\) as \({z}_{pair}^{s}\) and \({z}_{pair}^{q}\), respectively. However, a crucial challenge is determining the similarity between \({z}_{pair}^{s}\) and \({z}_{pair}^{q}\). Prior work64 suggests simple Euclidean distance and other distance-based methods rely on the strict conditional assumption that each dimension of the vector is independent. Therefore, to accurately evaluate whether interactions exist between drugs in novel events, we construct a comparator and define a loss function to optimize the comparator module:
where \(scor{e}^{+}\) and \(scor{e}^{-}\) denote the scores of positive and negative samples. Hence, in the training process of few-shot learning, the complete loss function of RareDDIE primarily consists of two parts: the loss of the NAI module and the comparator module, which can be defined as the function:
where \(\mu\) is a weight coefficient.
Application of RareDDIE to zero-shot problem
Although the prediction analysis for new DDIEs can be defined as a few-shot prediction problem, since discovering DDI of a new event in clinical treatment often accompanies known drug usage, many undiscovered events might still exist without associated drug records. To address this zero-shot problem, the incorporation of semantic information serves as an effective approach11, and we developed the ZetaDDIE framework. ZetaDDIE extends its generalization to novel events by performing a distribution mapping of embedding vectors between pairwise drug and medical concepts. Building on the RareDDIE model architecture, we incorporated the Biological Semantic Transferring (BST) module and modified the model training/testing process to adapt to zero-shot tasks.
Intuitively, due to the lack of a known support set for constructing interaction event representations during testing, we aim to leverage a large-scale sentence embedding model to directly generate semantic feature vectors as inputs to Comparator. However, the discrepancy between the semantic space and the pair representation space prevents direct input. To address this, the BST module employs an adversarial learning-based mapping mechanism to transform the semantic vectors from the pretrained language model into the representation space, enabling their use for subsequent predictions. During training, we retained the support set to train the BST module, enabling it to align the clinical semantic distribution of events \({X}_{bs}\) with the general relation metric space constructed by the NAI. This alignment builds a semantic information metric for aleatoric events without any known examples. This constructed metric is used to predict the query set samples and optimize the BST module. Finally, during testing, the learned metric is directly used for query set samples without requiring the support set.
For constructing clinical semantic embeddings of events, we used the large-scale sentence embedding model BioSentVec26,27, which employs a Continuous Bag-of-Words model at the sentence level and extends the model by using n-grams of sentences. It is trained on various text genres in biomedical and clinical domains of PubMed65 and MIMIC-III Clinical Database66, containing 4,893,178,115 tokens. Details of data are provided in Supplementary Table 12. After training, the model parameters occupy 21GB and all DDI event description sentences can be embedded into 700-dimensional vectors \(\{{x}_{b{s}_{e}}|e\in {E}_{d}\}\).
Motivated by adversarial transfer learning, we next construct a superivised BST module, containing a Mapper Network \(M(x)\) and a Discriminator network \(D(x)\), to map the clinical semantic information onto our general relation metric space. Specifically, in each iteration of meta-learning task sampling, we learn mapping rules based on the support set. Given a task event \(e\) and the corresponding support samples \({S}_{e}\), we encode the task event and map it to the metric space, obtaining \(M({x}_{b{s}_{e}})\). Similarly, \({S}_{e}\) is mapped to the metric space via the first four modules of RareDDIE, resulting in \({z}_{pair}^{s}\). To assess the dissimilarity, we use a discriminator \(D(x)\) to distinguish whether \(M({x}_{b{s}_{e}})\) and \({z}_{pair}^{s}\) originate from the same distribution space. Ultimately, we train the Mapper Network and the Discriminator Network by optimizing the following equation:
Then, using BST, we obtain the embedding \(D(M({x}_{b{s}_{e}}))\) for the current task event, replacing \({z}_{pair}^{s}\) that must be computed during Rare DDIE training. Consequently, the loss function of the comparator is modified to the following equation:
Hence, in the training process of zero-shot learning, the complete loss function of ZetaDDIE primarily consists of three parts: the loss of the BST module, the loss of the NAI module and the comparator module, which can be defined as the function:
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the drug-drug interaction information was collected from DrugBank54 and TWOSIDES55. The knowledge graph and background graph were collected from DRKG. Drug synergy datasets were collected from O’Neil et al.39 and Preuer et al.41. We have compiled all data supporting key findings at https://github.com/MrPhil/RareDDIE. All accession codes used in this study,are listed below: Albumin (P02768 [https://www.uniprot.org/uniprotkb/P02768/entry#structure], PDB ID: 6YG9). Source data are provided with this paper.
Code availability
An open-source RareDDIE is available at the following GitHub repository (https://github.com/MrPhil/RareDDIE) with a DOI67 of https://doi.org/10.5281/zenodo.15068536.
References
Baragaña, B. et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 522, 315–320 (2015).
Jaaks, P. et al. Effective drug combinations in breast, colon and pancreatic cancer cells. Nature 603, 166–173 (2022).
Palmer, A. C. & Sorger, P. K. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell 171, 1678–1691.e1613 (2017).
Kommu, S., Carter, C. & Whitfield, P. Adverse Drug Reactions. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: https://www.ncbi.nlm.nih.gov/books/NBK599521/ (accessed on 10 December 2024).
Percha, B. & Altman, R. B. Informatics confronts drug–drug interactions. Trends Pharm. Sci. 34, 178–184 (2013).
Vandenbroucke, J. P. & Psaty, B. M. Benefits and risks of drug treatments: how to combine the best evidence on benefits with the best data about adverse effects. JAMA 300, 2417–2419 (2008).
Eichler, H.-G., Pignatti, F., Flamion, B., Leufkens, H. & Breckenridge, A. Balancing early market access to new drugs with the need for benefit/risk data: a mounting dilemma. Nat. Rev. Drug Discov. 7, 818–826 (2008).
Wu, H. & Huang, J. Drug-induced nephrotoxicity: pathogenic mechanisms, biomarkers and prevention strategies. Curr. Drug Metab. 19, 559–567 (2018).
Zhang, Y. et al. Emerging drug interaction prediction enabled by a flow-based graph neural network with biomedical network. Nat. Comput. Sci. 3, 1023–1033 (2023).
Karch, F. E. & Lasagna, L. Adverse drug reactions: a critical review. JAMA 234, 1236–1241 (1975).
Wang, Z., Xiong, Z., Huang, F., Liu, X. & Zhang, W. ZeroDDI: a zero-shot drug-drug interaction event prediction method with semantic enhanced learning and dual-modal uniform alignment. In Proc. Thirty-Third International Joint Conference on Artificial Intelligence, 6071–6079 (2024).
Ren, Z.-H. et al. A biomedical knowledge graph-based method for drug–drug interactions prediction through combining local and global features with deep neural networks. Brief. Bioinform. 23, bbac363 (2022).
Deng, Y. et al. A multimodal deep learning framework for predicting drug–drug interaction events. Bioinformatics 36, 4316–4322 (2020).
Harpaz, R. et al. Combing signals from spontaneous reports and electronic health records for detection of adverse drug reactions. J. Am. Med. Inf. Assoc. 20, 413–419 (2013).
Wang, Y., Zhai, Y., Ding, Y. & Zou, Q. SBSM-pro: support bio-sequence machine for proteins. Sci. China Inf. Sci. 67, 212106 (2024).
Ryu, J. Y., Kim, H. U. & Lee, S. Y. Deep learning improves prediction of drug–drug and drug–food interactions. Proc. Natl Acad. Sci. 115, E4304–E4311 (2018).
Nyamabo, A. K., Yu, H. & Shi, J.-Y. SSI–DDI: substructure–substructure interactions for drug–drug interaction prediction. Brief. Bioinform. 22, bbab133 (2021).
Shen, X., Li, Z., Liu, Y., Song, B. & Zeng, X. PEB-DDI: A task-specific dual-view substructural learning framework for drug–drug interaction prediction. IEEE J. Biomed. Health Inform. 28, 569–579 (2023).
Ren, Z.-H. et al. BioDKG–DDI: predicting drug–drug interactions based on drug knowledge graph fusing biochemical information. Brief. Funct. Genom. 21, 216–229 (2022).
Lin, S. et al. MDF-SA-DDI: predicting drug–drug interaction events based on multi-source drug fusion, multi-source feature fusion and transformer self-attention mechanism. Brief. Bioinform. 23, bbab421 (2022).
Yu, H., Dong, W. & Shi, J. Raneddi: relation-aware network embedding for drug-drug interaction prediction. Inf. Sci. 582, 167–180 (2022).
Wang, N.-N. et al. Comprehensive review of drug–drug interaction prediction based on machine learning: current status, challenges, and opportunities. J. Chem. Inf. Model 64, 96–109 (2023).
Hospedales, T., Antoniou, A., Micaelli, P. & Storkey, A. Meta-learning in neural networks: A survey. ITPAM 44, 5149–5169 (2021).
Finn C., Abbeel P. & Levine S. Model-agnostic meta-learning for fast adaptation of deep networks. In: International Conference on Machine Learning). PMLR, 1126–1135 (2017).
Yu, M. et al. Diverse few-shot text classification with multiple metrics. In Proc. 2018 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, vol. 1, 1206–1215 (2018).
Zhang, Y., Chen, Q., Yang, Z., Lin, H. & Lu, Z. BioWordVec, improving biomedical word embeddings with subword information and MeSH. Sci. Data 6, 52 (2019).
Chen Q., Peng Y. & Lu Z. BioSentVec: creating sentence embeddings for biomedical texts. In: 2019 IEEE International Conference on Healthcare Informatics (ICHI). IEEE, 1-5 (2019).
Cheng, F., Kovács, I. A. & Barabási, A.-L. Network-based prediction of drug combinations. Nat. Commun. 10, 1197 (2019).
Madushanka T. & Ichise R. Negative Sampling in Knowledge Graph Representation Learning: A Review. arXiv preprint arXiv:240219195 (2024).
Deng, Y. et al. META-DDIE: predicting drug–drug interaction events with few-shot learning. Brief. Bioinform. 23, bbab514 (2022).
Xiong, W., Yu, M., Chang, S., Guo, X. & Wang, W. Y. One-shot relational learning for knowledge graphs. In Proc. 2018 Conference on Empirical Methods in Natural Language Processing, 1980–1990 (2018).
Chen, M., Zhang, W., Zhang, W., Chen, Q. & Chen, H. Meta Relational Learning for Few-Shot Link Prediction in Knowledge Graphs. In Proc. 2019 Conference on Empirical Methods in Natural Language Processing and the 9th International Joint Conference on Natural Language Processing (EMNLP-IJCNLP), 4217–4226 (2019).
Li, Z. et al. DSN-DDI: an accurate and generalized framework for drug–drug interaction prediction by dual-view representation learning. Brief. Bioinform. 24, bbac597 (2023).
Xiong, Z. et al. Multi-relational contrastive learning graph neural network for drug-drug interaction event prediction. Proceedings of the AAAI Conference on Artificial Intelligence 37, 5339–5347 (2023).
Wang, Y., Yang, Z. & Yao, Q. Accurate and interpretable drug-drug interaction prediction enabled by knowledge subgraph learning. Commun. Med. 4, 59 (2024).
Nyamabo, A. K., Yu, H., Liu, Z. & Shi, J.-Y. Drug–drug interaction prediction with learnable size-adaptive molecular substructures. Brief. Bioinform. 23, bbab441 (2022).
Goodfellow I. et al. Generative adversarial networks. Stat. Mach. Learn. 63, 139-144 (2020).
Lee, J. et al. BioBERT: a pre-trained biomedical language representation model for biomedical text mining. Bioinformatics 36, 1234–1240 (2020).
O’Neil, J. et al. An unbiased oncology compound screen to identify novel combination strategies. Mol. Cancer Ther. 15, 1155–1162 (2016).
Wang, X. et al. PRODeepSyn: predicting anticancer synergistic drug combinations by embedding cell lines with protein–protein interaction network. Brief. Bioinform. 23, bbab587 (2022).
Preuer, K. et al. DeepSynergy: predicting anti-cancer drug synergy with Deep Learning. Bioinformatics 34, 1538–1546 (2018).
Zhang, T., Zhang, L., Payne, P. R. & Li F. Synergistic drug combination prediction by integrating multiomics data in deep learning models. Transl. Bioinform. Ther. Dev., 223–238 (2021).
Basak, D., Pal, S. & Patranabis, D. C. Support vector regression. Neural Inf. Process. -Lett. Rev. 11, 203–224 (2007).
Chen, T. & Guestrin, C. Xgboost: A scalable tree boosting system. In Proc. 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 785–794 (2016).
Chen, J. et al. Zero-shot and few-shot learning with knowledge graphs: A comprehensive survey. Proc. IEEE 111, 653–685 (2023).
Xiong, G. et al. DDInter: an online drug–drug interaction database towards improving clinical decision-making and patient safety. NAR 50, D1200–D1207 (2022).
Riniker, S. & Landrum, G. A. Similarity maps-a visualization strategy for molecular fingerprints and machine-learning methods. J. ChemInform. 5, 1–7 (2013).
Pinzi, L. & Rastelli, G. Molecular docking: shifting paradigms in drug discovery. Int J. Mol. Sci. 20, 4331 (2019).
Balius T. E., Tan Y. S. & Chakrabarti M. DOCK 6: Incorporating hierarchical traversal through precomputed ligand conformations to enable large‐scale docking. J. Comput. Chem. 45, 47–63 (2024).
Maaten, L. V. D. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
Wu Z. et al. MoleculeNet: a benchmark for molecular machine learning. Chem. Sci. 9, 513–530 (2018).
Kalyanasundaram, A. & Lincoff, A. M. Managing adverse effects and drug–drug interactions of antiplatelet agents. Nat. Rev. Cardiol. 8, 592–600 (2011).
Nair, N. U. et al. A landscape of response to drug combinations in non-small cell lung cancer. Nat. Commun. 14, 3830 (2023).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. NAR 46, D1074–D1082 (2018).
Tatonetti, N. P., Ye, P. P., Daneshjou, R. & Altman, R. B. Data-driven prediction of drug effects and interactions. Sci. Transl. Med 4, 125ra131–125ra131 (2012).
Ma, T., Lin, X., Song, B., Philip, S. Y. & Zeng, X. Kg-mtl: knowledge graph enhanced multi-task learning for molecular interaction. IEEE Trans. Knowl. Data Eng. 35, 7068–7081 (2022).
Landrum, G. RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling. Greg. Landrum 8, 5281 (2013).
Vinyals, O., Blundell, C., Lillicrap, T. & Wierstra, D. Matching networks for one shot learning. Adv. Neural Inf. Process Syst. 29, 3630–3638 (2016).
Bordes, A., Usunier, N., Garcia-Duran, A., Weston, J. & Yakhnenko, O. Translating embeddings for modeling multi-relational data. Adv. Neural Inf. Process Syst. 26, 2787–2795 (2013).
Ma, M. & Lei, X. A dual graph neural network for drug–drug interactions prediction based on molecular structure and interactions. PLoS Comp. Biol. 19, e1010812 (2023).
Shi, Y. et al. Masked label prediction: Unified message passing model for semi-supervised classification. In Proc. Thirtieth International Joint Conference on Artificial Intelligence, 1548–1554 (2021).
Lee J., Lee I. & Kang J. Self-attention graph pooling. In: International Conference on Machine Learning. PMLR, 3734-3743 (2019).
Kingma, D. P. & Welling, M. Auto-encoding variational Bayes. In International Conference on Learning Representations (2014).
Sung F. et al. Learning to compare: Relation network for few-shot learning. In Proc. IEEE Conference on Computer Vision and Pattern Recogn., 1199-1208 (2018).
Lu, Z. PubMed and beyond: a survey of web tools for searching biomedical literature. Database 2011, baq036 (2011).
Johnson, A. E. et al. MIMIC-III, a freely accessible critical care database. Sci. Data 3, 1–9 (2016).
Ren, Z. Predicting rare drug-drug interaction events with dual-granular structure-adaptive and pair variational representation. Zenodo. https://doi.org/10.5281/zenodo.15068536 (2025).
Zhang, Y.-J., Li, M.-P., Tang, J. & Chen, X.-P. Pharmacokinetic and pharmacodynamic responses to clopidogrel: evidences and perspectives. Int. J. Environ. Res. Public Health 14, 301 (2017).
Jeddi, R. Antineoplastics/idarubicin. Reactions 1208, 28 (2008).
Wang C.-L. et al. Risk of major bleeding associated with concomitant use of anticancer drugs and direct oral anticoagulant in patients with cancer and atrial fibrillation. J. Thromb Thrombolysis, 1–13 (2022).
Majfrand, E. P. J., Picard-Fraire, C., Vallée, E. & Roncucci, J. B. R. Ticlopidine: a promise for the prevention and treatment of thrombosis and its complications. Haemostasis 13, 1–54 (1983).
Chang, Y. et al. A novel role for tamoxifen in the inhibition of human platelets. Transl. Res. 157, 81–91 (2011).
Mazzucconi, M. G. et al. Danazol therapy in refractory chronic immune thrombocytopenic purpura. Acta Haematol. 77, 45–47 (1987).
Crooke, S. T. et al. The effects of 2′-O-methoxyethyl containing antisense oligonucleotides on platelets in human clinical trials. Nucleic Acid Ther. 27, 121–129 (2017).
Reis, E. T. D. et al. Revisiting hydroxychloroquine and chloroquine for patients with chronic immunity-mediated inflammatory rheumatic diseases. Adv. Rheumatol. 60, 32 (2020).
Miescher, P. A. & Pola, W. Haematological effects of non-narcotic analgesics. Drugs 32, 90–108 (1986).
Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 62425107 to Q.Z., No. 62450002 to Q.Z., No. 62432011 to C.L., No. 62425204 to X.Z. and No. U22A2037 to X.Z.), Zhejiang Provincial Natural Science Foundation of China (No. LD24F020004 to Q.Z.), the Municipal Government of Quzhou (No.2023D036 to Q.Z.), and Zhongguancun Academy (No. 20240310 to C.L.).
Author information
Authors and Affiliations
Contributions
Z.R., X.Z., and C.L. wrote the first draft of the manuscript. C.L., Q.Z., and Y.L. revised the manuscript to the submitted version. Z.R., Q.Z., X.Z., and Z.Y. conceived the study. Z.R. designed all the experiments and wrote the codebase of RareDDIE. Z.R., Q.Z., and Y.S. conduct the benchmarks and run all of the analysis. Z.R. and Y.S. collected and preprocessed all datasets. Z.R., X.Z., Y.L., and C.L. contributed to data analysis and model discussion. Z.R., Q.Z., and Z.Y. conducted the figure design for the overall framework. Z.R., X.Z., and Y.L. completed the visualizations. Q.Z. and C.L. supervised the research. All of the authors reviewed the manuscript and approved it for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Tunca Dogan and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ren, Z., Zeng, X., Lao, Y. et al. Predicting rare drug-drug interaction events with dual-granular structure-adaptive and pair variational representation. Nat Commun 16, 3997 (2025). https://doi.org/10.1038/s41467-025-59431-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-59431-9
