
Abstract
The long-standing challenges facing Pt-based alloy catalysts in oxygen reduction reactions (ORRs) are rapid oxidation and loss of transition metal/Pt in proton exchange membrane fuel cells (PEMFCs). In this work, we report a concept of “covalentization” in intermetallic L10-PtMM’ (M = Fe, Co, Ni and M’ = one of the 4th-period elements (from Ti to Ge)) alloys to enhance their electrochemical stability. Specifically, the formation of a quasi-covalent bond network in L10-PtMM’ due to the less occupied antibonding states induced by high d-band positions of M’ elements (e.g., Ti, V, Cr) enhances atomic bond order and strength, diminishing Co anodic dissolution via strengthened Pt/Co-M’ bonds and reducing Co cathodic corrosion by inhibiting Pt oxidation through an electron buffering effect. The developed L10-PtCoCr/C catalysts show a high mass activity (MA = 1.27 A mgPt−1) and rated power (16.5 W mgPt−1) in PEMFCs at a low total Pt loading of 0.075 mgPt cm−2. The catalysts also exhibit high electrochemical stability with ~3% and 5% loss of MA and rated power after 30,000 accelerated durability testing cycles and projects a lifetime of about 42,000 hours.
Similar content being viewed by others


Introduction
Proton exchange membrane fuel cells (PEMFCs) have emerged as a critical technology for the widespread application of hydrogen energy to reduce the dependence on fossil fuels1,2,3,4,5. However, limited by the sluggish kinetics of the cathode oxygen reduction reaction (ORR), the overall efficiency of PEMFC is unsatisfactory. Previous research has demonstrated that alloying Pt with other transition metals M is an effective method to improve the intrinsic activity of Pt-based catalysts6,7,8,9. The interaction between valence orbitals of Pt and M dominates the electronic structure modulation of the active site (e.g., ligand effect and strain effect). On the other hand, the harsh electrochemical working conditions in fuel cells (0.6–1.5 V, pH < 1, and 60–80 °C) are extremely challenging for the long-term performance and durability of Pt-M alloy catalysts. Specifically, the rapid etching of M and oxidation corrosion of Pt may cause the degradation of cathode catalysts, leading to performance decay during the long-term operation.
Some early theoretical research on the thermodynamic stability of metal alloys noted that the variation in the cohesive energy of transition metals is mainly driven by their d-band contribution10,11,12. Also, Norskov et al. found that some typical Pt-based alloys with approximately half-filled metal-metal d bonds (e.g., Pt3Y and Pt3Sc) exhibit large negative enthalpies of formation, implying highly stable structures13. In particular, recent studies have demonstrated that chemically ordered L10-PtM intermetallic alloys show impressive advantages in stabilizing transition metal elements8,14,15. The sequential arrangement of Pt and M atomic layers along the c-axis of the lattice leads to a considerable overlap between their d orbitals, inducing a stronger 3d (M)−5d (Pt) interaction that can prevent the transition metal atoms from etching out of the ordered lattice16,17. For instance, Sun et al. utilized such an interaction to construct the structurally ordered L10-PtFe and L10-PtCo, effectively reducing M leaching in PEMFC18,19. Another effective method is to strengthen d-p interaction between Pt and some p-block elements (e.g., B, N) to improve the electrochemical stability of catalysts20,21. All these results highlight the critical role of orbital modulation in improving the structural stability of metal alloys. However, most regulation methods are confined to enhancing a particular type of bond/interaction. The intricate atomic coordination and inherent weakness of metal bonds in PtM alloys make the dissolution of metal M more susceptible to the influence of less tightly bonded atoms, akin to the bucket effect, which leads to the dissolution of M in these M-rich Pt alloys, especially for the anodic M dissolution. On the other hand, the weakly bonded M atoms could be more easily bonded with subsurface oxygen atoms formed due to the place-exchange oxidation of surface Pt at a high potential window, thus accelerating the cathode M corrosion process and resulting in a fast decay of PtM alloy catalysts. In this regard, L10 intermetallic alloys exhibit passable advantages in anti-metal dissolution as the strong internal 3d (M)−5d (Pt) orbital coupling. However, the more detailed mechanism concerning the electrochemical stability of L10-PtM alloys and how it can be further enhanced remain unexplored.
Herein, we develop an orbital interaction enhancement strategy via constructing a quasi-covalent bond network within Pt crystallines to improve the catalytic stability of intermetallic L10-PtMM’ (M = Fe, Co, Ni; M’ = 4th period element from Ti to Ge) nanocrystals (NC) ORR catalysts for PEMFCs. Density functional theory (DFT) calculations demonstrate that through increasing bonding-antibonding energy level splitting of Pt/M-M’ in L10-PtMM’ (especially when M’ = Ti, V, and Cr with high d-band position), the conversion of metallic to covalent interaction can be selectively promoted with enhanced atomic bond order, thereby improving the atomic bond strength and forming a highly stable thermodynamic state of L10-PtMM’. Comprehensive operando characterizations indicate that the quasi-covalent bond network in L10-PtCoCr diminishes the direct anodic dissolution of Co by enhancing the Pt/Co-Cr bonding strength and alleviating the cathodic Co corrosion by suppressing surface Pt oxidation, effectively reducing the attenuation of catalyst. The best-performing L10-PtCoCr/C reveals high performance in PEMFC tests under light-duty vehicle (LDV) conditions, with the mass activity (MA = 1.27 A mgPt−1) and rated power (16.5 W mgPt−1) in MEAs at a low total Pt loading (0.075 mgPt cm−2). In addition, the L10-PtCoCr/C catalyst demonstrates high durability with 3% MA loss after 30,000 accelarated potential cycles and a long projected lifetime of 42,000 h.
Results
Bonding characteristics of L10-PtCoM’
The joint influence of electronic structures and lattice types restricts understanding electronic regulation for atomic bonding strength. In particular, the order of atomic arrangement in the lattice has a significant impact on the catalytic performance of the catalyst. To minimize the lattice effect, the L10-PtCo with a space group of P4/mmm is chosen as the model catalyst for understanding the orbital interaction effect on the structural stability (Fig. 1a). The strictly aligned Pt/Co layers along the c-axis of L10-PtCo can result in a precise orbital overlap between the interlayer Pt and Co, leading to the 5d−3d interaction16. This could induce the splitting of bonding and antibonding orbitals between Pt and Co (Fig. 1b). To investigate such an orbital coupling effect on the metal-metal bonding strength, doped metal atoms M’ (4th period element from Ti to Ge) were introduced into L10-PtCo. Thus, the structure of L10-PtCoM’ catalysts can be interpreted as a network consisting of the interlayer Pt-Co/M’ bond and intralayer Pt-Pt and Co-Co/M’ bonds. The metal-metal bonding strength, therefore, could be a crucial indicator of structural stability. To this end, the Crystal Orbital Hamilton Population (COHP) for the metal-metal interactions in the intermetallic structure has been calculated. Here, the bond order (BO), defined as half the difference between the number of bonding orbital electrons and the number of antibonding orbital electrons, is used to describe the bonding tendency between interacting atoms in L10-PtCoM’. Specifically, a high bond order signifies a high degree of atomic covalency. For all of the studied L10-PtCoM’ systems, the introduction of M’ atoms can greatly influence the strength of Pt-M’ and Co-M’ bonds compared to the Pt-Co and Co-Co bonds in the undoped L10-PtCo, while that of Pt-Co bond is less affected (Fig. 1c). In particular, the BOs of the Pt-M’ and Co-M’ bonds decrease as the atomic number of the doping metal increases from Ti to Zn. Among them, Pt-M’ and Co-M’ containing early transition metal (ETM, i.e., Ti, V and Cr) atoms show the highest BOs, which is increased by about 123% (Pt-Ti) and 57% (Co-Ti) compared with those of Pt-Co and Co-Co in L10-PtCo, respectively. In contrast, the BOs of Pt-M’ and Co-M’ bonds in L10-PtCoM’ containing post-transition metals (PTM, i.e., Ni, Cu and Zn) exhibit obvious decreases to varying extents. Our further calculations show that the interatomic bonding strength between Pt/Co and M’ is primarily determined by the d-band position of the M’ metals referencing the Fermi level according to the projected density of states (PDOS) of the d-band of the Pt, Co, and M’ atoms in L10-PtCoM’ (Supplementary Fig. 1, 2). Importantly, the d-band of ETM atom in L10-PtCoM’ is generally higher in energy than the d-band of Co in L10-PtCo, resulting in a less-occupied antibonding state of Pt-M’ bond in L10-PtCoM’ and a higher BO. The enhanced BO indicates stronger covalency and strength of the Pt/Co-ETM bonds in L10-PtCoM’ than that of Pt-Co in L10-PtCo (Fig. 1b). Interestingly, alloying L10-PtCo with main group metal (MGM) Ga and Ge can also result in strong Pt-Co-Ga and Pt-Co-Ge networks, as suggested by the prominent Pt-Ga, Co-Ga, and Go-Ge BOs (Fig. 1c), which was identified as the p-d interaction effect in some previous works7,22. It is notable that the BO trends for transition metals and main group metals are slightly different and the trend of main group metals changes faster, which may be caused by the changes in the interacting valence band structures. The electron localization function analysis of the studied L10-PtCoM’ systems reveals that the electrons are more localized between Pt and ETM/MGM atoms compared to that of PTM, suggesting the formation of quasi-covalent bonds (QCBs) in these systems (Fig. 1d). For ETM/MGM-doped L10-PtCo intermetallic alloy, the strong orbital coupling between the interlayer Pt-M’ and the intralayer Pt/Co-M’ leads to the formation of a spatial QCB network, which could exhibit a significant positive effect on improving the overall electrochemical stability of the alloy (Fig. 1e).

a Structural model of the L10-PtCo. b Schematic representation of the DOS showing the orbital coupling of the ETM/PTM 3d band with the Pt 5d band. c Bond order of the Pt-M’, Pt-Co, and Co-M’ bonds in L10-PtCoM’. d Two-dimensional display of the electron localization function analysis of the corresponding L10-PtCoM’ systems. The iso-values of 0 and 0.5 imply low and high electron localization, respectively. e Schematic diagram of interlayer and intralyer QCB network in the L10-PtCoM’ nanoparticle (NP).
Synthesis and structural characterizations
To study the relationship between electronic structures and electrochemical durability in L10-PtCo alloys, we first prepared PtCoM’ (atomic ratio: Pt/Co/M’ = 50/35/15) NPs through a facile wet-chemical approach and then these NPs were anchored on a carbon support (C). After a series of annealing (700 °C), acid-treatment and a followed reannealing (400 °C), all the samples can achieve a phase transformation from A1 to L10 accompanied by forming a thin Pt shell23. The X-ray diffraction (XRD) patterns indicated that the annealed PtCo/C sample matched well with the standard powder diffraction file (PDF) card of L10-PtCo (PDF #65-8968) according to the superlattice peaks located at around 24° and 33° (Fig. 2a). When Co is partially replaced by M’ in the ternary alloys, the intermetallic L10 structure could still be formed. However, a minor shift in peaks occurs due to different atomic radius of M’ in these L10-PtCoM’ alloys. The ordering degree of these samples was also quantitatively analyzed to minimize its impact on subsequent measurements and characterizations (Supplementary Table 1)24. Transmission electron microscopy (TEM) images show that the annealed alloy NPs disperse uniformly with an average diameter of 5.0 ± 0.5 nm (Supplementary Fig. 3). The compositions of L10-PtCoM’ NPs were measured by X-ray fluorescence (XRF) and inductively coupled plasma-mass spectrometry (ICP-MS) analysis.

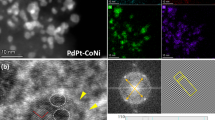
a XRD patterns of L10-PtCoM’ (M = Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga and Ge) catalysts. b HAADF-STEM image of a L10-PtCoCr NP. Inset: corresponding FFT pattern. c Local HAADF-STEM image and corresponding atomic stacking schematic of the L10-PtCoCr NP. d EDS elemental mapping images of Pt, Co, and Cr. e Lattice strain distribution of L10-PtCoCr NP. f Pt L3-edge XANES spectra of Pt foil, L10-PtCo/C and L10-PtCoCr/C. Wavelet transforms of Pt foil (g) and L10-PtCoCr/C (h) for the Pt L3 edge k3-weighted XAFS signals in the L10-PtCoCr/C.
Spherical aberration (Cs)-corrected high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive spectroscopy (EDS) elemental mapping were performed to analyze the structural properties of an individual NP. The ordered intermetallic structure can be viewed along the [−110] zone axis. The distinct alternating layers of Co/Cr and Pt atoms along the <001> direction can be clearly observed, proving the formation of L10 structure. Inset of Fig. 2b reveals the corresponding fast Fourier transformation (FFT) pattern of this NP, showing the presence of (110) and (001) superlattice spots6. Furthermore, alternating intensity profiles further confirm the ordered structures. The edge intensity distribution of NP proved the formation of a thin Pt shell with 2–3 atomic layers (Fig. 2c). EDS elemental mappings confirm that the three elements distributed uniformly within the NP, and a clear Pt shell could be observed by the EDS line scan profile (Fig. 2d and Supplementary Fig. 4). In addition, a compressive surface strain (~−2.2 %) can be clearly seen on the Pt shell due to biaxial compression inside the core (Fig. 2e). Strain analysis also demonstrates that the shell strain resulted from the strain relaxation, proved by the lattice difference between the core and shell (Supplementary Fig. 5). X-ray absorption near-edge structure (XANES) spectra have proven the decrease in Pt valence state of L10-PtCoCr/C compared to that of L10-PtCo/C according to the white line peak intensity, indicating the electronic modulation effect of Cr (Fig. 2f). The continuous Cauchy wavelet transforms of Pt L-edge extended X-ray absorption fine structure (EXAFS) results exhibit a contoured feature with maximum intensities of metallic Pt in L10-PtCoCr/C with the slightly shortened Pt coordination bond lengths (Fig. 2g, h, Supplementary Table 2).
Electrochemical measurements
To investigate the effectiveness of the covalentization strategy to the structural stability of L10-PtCoM’, the ORR performance of the obtained L10-PtCoM’/C catalysts and commercial Pt/C were evaluated through cyclic voltammetry (CV) and linear scanning voltammetry (LSV) technique. CV curves reveal that the electrochemically active surface areas (ECSAs) of L10-PtCoM’/C ranged from 60–70 m2 gPt−1, which is comparable to that of Pt/C (72.2 m2 gPt−1) (Supplementary Figs. 6–16). Notably, the SA of L10-PtCoM’/C reveals a volcano-type relationship with the atomic radius of M’ (Supplementary Fig. 17), which suggests the ORR activity is governed by lattice strain25. DFT calculations based on the compressive strain on L10-PtCoM’ surface demonstrate that L10-PtCoCr exhibits the theoretically most optimized oxygen adsorption energy among all these transition metal-doped samples (Supplementary Fig. 17). Notably, the incorporation of Cr can alleviate the excessive compressive strain in L10-PtCo lattice, leading to a slight rise of d band center of surface Pt layer for better ORR activity. Experimentally, L10-PtCoCr/C demonstrates the best ORR activity with the highest mass activity (MA) of 2.56 A mgPt−1 and specific activity (SA) of 5.03 mA cm−2 (Fig. 3a, b), as proved by the DFT calculations. After a standard ADT protocol of 30,000 potential cycles, nearly all the L10-PtCoM’/C catalysts exhibit varying degrees of activity decline. Besides, the SA retention results of L10-PtCoM’/C show a strong correlation with the BOs of Pt/Co-M’ (Fig. 3c, Supplementary Fig. 18). Specifically, a high interatomic BO indicates a high SA retention. Among these catalysts, L10-PtCoCr/C and L10-PtCoTi/C demonstrate the best ORR stability with 99.0% and 99.4% SA retention after ADT, respectively.

a ORR polarization curves (without iR correction, R = 24.8 ± 0.4 Ω.) of L10-PtCoCr/C, L10-PtCo/C, and Pt/C in O2-saturated 0.1 M HClO4 (rotating speed, 1600 rpm; scan rate, 10 mV s−1). b ECSA and MA (@0.90 V vs. RHE) of the studied catalysts. c SA and SA retentions of the studied catalysts before and after ADT. d SA and SA retentions of L10-Pt-M-ETM catalysts. The potentials reported here are calibrated in RHE scale. The error bars are the s.d. from three separate measurements.
The ETM elements with similar electronic structures in the 5th and 6th period (Zr, Nb, Hf and Ta) were also introduced in L10-PtCo for the rotating disk electrode (RDE) measurements (Supplementary Figs. 19–23). In addition, the typical ETM elements (Ti, V and Cr) were also introduced into PtFe and PtNi intermetallic catalysts to verify their generality in improving electrochemical stability (Supplementary Figs. 24–30). As shown in Fig. 3d, nearly all the catalysts can achieve a high SA retention of over 95% after 30,000 ADT potential cycles, indicating the universal applicability of the covalentization strategy. These results suggest that building such metal-metal QCB networks, which can be manipulated by introducing different alloy elements, could be an effective strategy to improve the structural stability and catalytic activity of intermetallic alloys.
Dissolution mechanism of L10-PtCoM’
To shed light on the characteristic electrochemical degradation behavior of L10-PtCoM’ with enhanced QCB networks, the component differences of L10-PtCoM’/C catalysts were analyzed after the long-term ADT through XRF. As shown in Supplementary Fig. 31, after the acid-treatment and annealing process, all the as-prepared L10-PtCoM’/C catalysts show apparent transition metal loss, corresponding to the dissolution of surface transition metals and the formation of surface Pt-skin shell. Then, a standard ADT process was performed to study the specific component change. Notably, the total loss of transition metals exhibits a similar trend to the change in SA, consistant with the DFT calculation results (Supplementary Figs. 32 and S33). A clear relationship can also be observed between Pt/Co-M’ BOs and SA retention/metal dissolution rates. Such a result and the unchanged particle size distributions of post-ADT samples, demonstrate that the component change of catalysts dominates the activity loss during the electrochemical cycles (Supplementary Fig. 34). The loss ratios of Co and M’ were then separated to understand the role of doped elements (Fig. 4a). For the ETM and MGM doped samples, the Co loss ratios are significantly smaller than those of other doped samples, especially for typical post-transition metals (i.e., Fe, Ni, Cu, and Zn). Moreover, the relative loss ratios of M’ of ETM doped catalysts are lower than those of PTM doped samples, implying that the much stronger interaction between ETM and Co/Pt can stabilize both Co and ETM simultaneously, consistent with the DFT calculation results.

a The content change of Co and M’ in L10-PtCoM’/C catalysts. b Real-time Co dissolution rates for A1-PtCo/C, L10-PtCo/C and L10-PtCoCr/C. c Co dissolution amounts for A1-PtCo/C, L10-PtCo/C and L10-PtCoCr/C under a RDE and GDE conditions. d CNPt-Pt, CNPt-Pt/Co/ Cr and CNPt-O of L10-PtCoCr/C and L10-PtCo/C as a function of the operando potential-dependent process. e XANES of Pt L3-edge of L10-PtCo/C and L10-PtCoCr/C before and after ADT. The inset shows the enlarged post-edge region f Atomic pair distribution function spectra of L10-PtCo/C and L10-PtCoCr/C before and after ADT. The potentials reported here are calibrated in RHE scale. The error bars in (c) are the s.d. from three separate measurements.
The leaching profiles of the L10-PtCoM’/C were further investigated using an online ICP-MS26. Figures S35 and S36 exhibited the potential-dependent Co and Pt dissolution behaviors of A1-PtCo/C, L10-PtCo/C and L10-PtCoCr/C under a RDE measurement condition. The dissolution of transition metal Co was first considered (Fig. 4b). All the catalysts show varying onset potentials of Co dissolution, which were defined as the intersection point of the tangent of the dissolution curve in the starting region with the horizontal baseline (Supplementary Fig. 35). Under two typical operating potential windows (0.05–1.0 V and 0.05–1.2 V), the dissolution of Co in A1-PtCo/C and L10-PtCo/C occurs almost simultaneously with the applied potential. In contrast, the onset potential of Co dissolution in L10-PtCoCr/C is delayed to about 0.2 V, proving the inhibitory effect of Cr on the electrochemical dissolution behaviors of Co. We noticed the characteristic peak difference on the Co transient dissolution curves of these catalysts27,28. In A1-PtCo/C and L10-PtCo/C, there are three typical Co dissolution peaks, those are, two anodic dissolution peaks (A1 and A2) and one dissolution cathodic peak (C1). Notably, the relative intensities of anodic and cathodic dissolution peaks in L10-PtCo/C are smaller than those in A1-PtCo/C, especially for the first anodic dissolution peak, indicating the critical role of atomic ordering in suppressing transition metal dissolution. Moreover, the first anodic dissolution peak (A1) in L10-PtCoCr/C seems to disappear. The subsequent second anodic dissolution peak (A2) also exhibits a rather low peak intensity, which illustrates that the Co oxidation process is suppressed during the potential scanning process, that is, the introduction of Cr further reduces the dissolution tendency of Co in the intermetallic alloy. Especially in the typical operating potential window of PEMFC (<1.0 V), such dissolution behavior of transition metal Co can be more obvious (Supplementary Fig. 35). The dissolution of Pt shows very different etching behavior compared with that of Co in these catalysts. As the dissolution almost occurs exclusively above 1.0 V, none of these samples exhibit obvious Pt dissolution peaks, even under a gas diffusion electrode (GDE) measurement condition (Supplementary Figs. 37, 38)29. Interestingly, we also notice almost no significant dissolution of Cr in L10-PtCoCr/C (Supplementary Fig. 39). Thus, we can confirm that the most activity degradation comes from the etching of transition metal Co in PtCo alloy.
Further, the amounts of Co dissolution during a complete potential cycle were also quantified through the loading-normalization, which varied significantly among these catalysts. Generally, the total transition metal Co dissolution amount follows the order of L10-PtCoCr/C<L10-PtCo/C<A1-PtCo/C (Fig. 4c). After normalizing different dissolution peaks, the contribution of different dissolution processes in these three catalysts can be further distinguished. The absolute anodic and cathodic dissolution values still show the same order as above (Supplementary Fig. 40-41). The reduced anodic dissolution of L10-PtCoCr/C compared to L10-PtCo/C can be attributed to the strong Pt/Co-Cr interaction preventing the migration of Co atom towards the surface as the anodic dissolution process is directly related to the oxidation and dealloying of Co in the lattice. Moreover, it is noteworthy that the Co anodic dissolution values of these catalysts are maintained while the cathodic dissolution rises significantly and gradually dominates the loss of the dissolution process when the upper potential limit (UPL) increases from 1.0 to 1.2 V. Considering the place-exchange oxidation (PEO) process of Pt (>1.0 V), the subsurface O may directly participate in the dissolution of Co in the lattice, leading to an increased cathodic dissolution. This is a consequence of the reduction of Pt-oxide triggering the removal of transition metal Co in the process30. Trace dissolution of Pt could be detected in all the samples and was much slower than that of Co when UPL is set at 1.2 V. Especially, the dissolution rate of Pt in L10-PtCoCr/C is significantly less than that in L10-PtCo/C (Supplementary Fig. 42). Combining previous research, we can confirm that introducing Cr with variable valence helps weaken the possible surface polarization and reduce the formation of Pt oxides. On the other hand, the much-enhanced interatomic interaction helps to weaken the Co-O bonding during the reduction process to inhibit the detachment of Co. These results imply that the increased Pt/Co-M’ bond strength has a similar inhibitory effect on both anodic and cathodic processes.
The local structures of studied catalysts during the electrochemical process were investigated by operando X-ray absorption spectroscopy (XAS) analysis to further uncover the mechanism of Cr-facilitated anti-dissolution process. Supplementary Fig. 43-44 show the potential-dependent Pt-L3-edge XANES of L10-PtCo/C and L10-PtCoCr/C during the ORR-relevant potential region (0.54, 0.70, and 0.90 V vs RHE). The white-line peak intensity (μnorm) of both samples shows a raised trend with the increased applied potential, indicating an increased polarization of Pt in catalysts (Supplementary Fig. 45). It is noticed that the relative white-line peak intensity (Δμnorm) of L10-PtCoCr/C increased with potential is much lower than that of L10-PtCo/C, indicating a much lower valence state change of Pt in L10-PtCoCr/C than that in L10-PtCo/C during the whole potential region. Furthermore, the FT-EXAFS analysis was performed to analyze the structural changes of the studied catalysts during the operation. Both catalysts exhibit similar changes in bond length in response to the applied potentials, which mainly originates from the strain change induced by the surface charge distribution at the catalyst-electrolyte interface. The L10-PtCoCr/C catalyst shows a much lower increased trend of Pt-Pt bond length (dPt-Pt) and a decreased trend of Pt coordination number (CN) than that of L10-PtCo/C with the applied potential (Fig. 4d, (Supplementary Table 3). According to the coordination parameter analysis, such a difference can be attributed to the change of Pt-O and Pt-Co/Pt coordinated structure induced by the Co dissolution and Pt oxidation species generation during the electrochemical process (Supplementary Fig. 45). As the potential increases from open circuit potential (OCP) to 0.54 V, a noticeable decrease of CNPt-Co in L10-PtCo/C can be observed, indicating that Co dissolution dominates the structural evolution at the initial electrochemical stage. L10-PtCoCr/C shows a much smaller reduction in CNPt-Co/Cr, which corresponds to the online ICP-MS results. It is noticeable that a sustained decrease of CNPt-Co can be observed in the subsequent stage when the applied potential jumps to 0.70 V. And the increase of dPt-Pt and CNPt-O in L10-PtCo/C can be clearly captured, mostly due to the lattice expansion induced by the surface Pt oxidation species generation. The similar changes in dPt-Pt and CNPt-Co/O are not obvious in L10-PtCoCr/C, suggesting the significant role of Cr in mitigating surface Pt oxidation and inhibiting Co dissolution (Fig. 4d, Supplementary Fig. 45)6,31. Even when the potential is further increased to 0.90 V, the change of dPt-Pt and CNPt-O in L10-PtCoCr/C are much smaller than those in L10-PtCo/C.
The XAS analysis of post-ADT catalysts was also performed (Fig. 4e, Supplementary Figs. 46, 47, Supplementary Table 4). The much higher white line intensity (HA) of L10-PtCo/C indicated a severe oxidation of Pt than that in L10-PtCoCr/C. The features in the post edge of XANES, corresponding to the interference of photoelectrons with local atoms, can serve as an indicator of the extent of local structural ordering surrounding the target atom32. Compared with L10-PtCoCr/C, A significantly attenuated intensity of the oscillation hump (Hp) of L10-PtCo/C indicates the increased local structural disordering around Pt, which is caused by the loss of Co around Pt and the formation of Pt oxides during ADT. Such a result proves that the introduction of Cr improves the structural stability of L10-PtCo even in the electrochemical environment. According to the post-ADT CN analysis of these two samples, L10-PtCoCr/C exhibits much more CNPt-Co/Cr retention and less CNPt-O increase than those of L10-PtCo/C, demonstrating a remarkably resistance to Pt oxidation and dissolution (Supplementary Fig. 48). Moreover, the significant peaks located at ~2.7 Å can be identified in the atomic pair distribution function spectra, indicating that the first coordination distance of L10-PtCo/C increased significantly compared to that of L10-PtCoCr/C after ADT, proving the severe loss of Co in L10-PtCo/C during the long-term electrochemical cycles (Fig. 4f, Supplementary Fig. 49). The post-ADT structure of L10-PtCoCr/C was further investigated through HAADF-STEM and elemental mapping (Supplementary Figs. 50, 51). The well-preserved core-shell structure and the virtually unchanged lattice parameters, strain and element distribution, confirm the high electrochemical stability of L10-PtCoCr/C.
DFT calculations were performed to provide more insights into the enhanced ORR stability. of L10-PtCoCr. As shown in Fig. 5a, the 5d-3d interaction in Pt-Co and Pt-Cr bonds gives rise to bonding and antibonding states around the Fermi level. The Pt-M bonding strength is primarily determined by the d-band position of the M metals referencing the Fermi level. The d-band of Cr in L10-PtCoCr is higher in energy than the d-band of Co in L10-PtCo, resulting in a less-occupied Pt-Cr antibonding state in the Pt-Cr bond of the Pt-Cr-Co triangle and, thus a higher BO as compared with that of the Pt-Co bond in the Pt-Co-Co triangle. A similar principle also leads to the higher bonding strength of Co-Cr than Co-Co. Thus, introducing Cr atoms can greatly improve the Pt-Cr and Cr-Co bond strength, while the Pt-Co bond is almost unaffected (Fig. 1c). In particular, the bond order of the Pt-Cr and Cr-Co bonds is ~44% larger than that of the Pt-Co and Co-Co bonds, suggesting an enhanced structural network and thus improved stability of the L10-PtCoCr. Consequently, the interlayer/intralayer diffusion energy barrier of Co atom in L10-PtCoCr exhibits significant increase of 68%/24% compared to those of the ordinary L10-PtCo structure due to the increased Pt/Co-Cr bonding strength (Fig. 5b, Supplementary Fig 52). This explains our post-ADT samples’ low Co loss ratio in Cr-doped L10-PtCo.

a Projected density of states (DOS) for the d-band (spin up) of Pt, Co, and Cr atoms and crystal orbital Hamilton population (COHP) for the Pt-Co and Pt-Cr interactions (spin up) in L10-PtCo and L10-PtCoCr. b The interlayer/intralayer diffusion energy barrier of Co atom in L10-PtCo and L10-PtCoCr. c Schematic diagram of the step-by-step electrochemical processes occurring during a whole potential cycle (top: L10-PtCo, buttom: L10-PtCoCr).
Here, a schematic representation of possible surface processes is given to elucidate the electrochemical dissolution behavior of studied catalysts during a whole potential cycle (Fig. 5c). During the anodic sweep process, L10-PtCo would undergo significant Co dissolution due to the weak intralayer/interlayer bonding. In stark contrast, the presence of a spatial QCB network could significantly impede the migration of Co out of the L10-PtCoCr lattice. And the Pt layers of L10-PtCoCr also exhibit much weaker surface polarization and lower surface tensile strain compared with those of L10-PtCo due to the electron buffering effect of the strongly ionized Cr. When the applied potential enters a high potential window (>1.0 V), the PEO process of Pt layers promotes oxygen atom to enter the subsurface. The formed Pt-O could neutralize part of the enriched positive charge on the surface during the anodic sweep process, effectively weakening the Coulomb repulsion of the surface and thus leading to a partial shrinkage of surface Pt layers. On the one hand, due to the flow of ionized electrons from the inner lattice Cr to the surface Pt layers, the surface polarization could be significantly suppressed. As a consequence, L10-PtCoCr shows a feebler oxidation of Pt layers compared with that of L10-PtCo. On the other hand, the near-surface Co in L10-PtCo presents a strong bonding with the subsurface oxygen atom owing to the inherent oxygen affinity of Co, while in L10-PtCoCr, the bonding between Co and oxygen is significantly weakened on account of the restraint of the internal QCB network. During the reduction process of cathodic sweep, the subsurface oxygen atoms depart from the lattice, which could simultaneously trigger the escape of near-surface Co in L10-PtCo due to its weak bonding with the internal lattice and strong oxophilicity. Because of a lower surface polarization and the protection of the strong QCB network, the near-surface Co in L10-PtCoCr can be well preserved. After the scanning potential back to OCP, the reduced surface polarization in L10-PtCoCr results in a considerably weaker surface tensile strain. The Pt layers of L10-PtCoCr displays a lower oxidation degree, indicating that the corrosion dissolution of Pt can also be suppressed.
MEA performance
The MEA performances of studied catalysts were evaluated in a practical single fuel cell to ascertain their viability in real vehicles. According to the standard DOE 2025 protocols for light-duty vehicles (LDVs), the MEA was prepared with the L10-PtCoCr/C catalyst as the cathode with a total Pt loading of 0.075 mgPt cm−2 (anode/cathode loading: 0.025/0.05 mgPt cm−2) (Fig. 6a). Impressively, the L10-PtCoCr/C cathode achieves a high begin-of-life (BOL) MA of 1.27 A mgPt−1 at 0.9 ViR-free, about 1.3 times and 5.3 times those of L10-PtCo/C (0.98 A mgPt−1) and commercial Pt/C (JM 20%, 0.24 A mgPt−1), respectively (Supplementary Fig. 53). Even at a higher cathode loading (0.1 mgPt cm−2), the L10-PtCoCr/C cathode can still exhibit a high initial MA of 1.04 A mgPt−1 (Supplementary Fig. 54). The L10-PtCoCr/C exhibits an impressive current density distribution during the typical operating voltage range (>0.7 V) of H2-air LDV condition (Fig. 6a, Supplementary Fig. 55). The L10-PtCoCr/C achieves a high current density of 1.34 A cm−2 at 0.7 V and a peak power density of 1.40 W cm−2, much higher than those of L10-PtCo/C (1.06 A cm−2, 1.01 W cm−2), Pt/C (0.62 A cm−2, 0.85 W cm−2) and most of reported Pt-based catalysts (Supplementary Fig. 56, Supplementary Table 5)33,34,35,36,37. To satisfy the heat rejection target of DOE, the practical BOL rated power of L10-PtCoCr/C was also measured, that is, the area-normalized and mass-normalized rated power of 1.24 W cm−2 and 16.5 W mgPt−1, respectively, outperforming L10-PtCo/C (1.09 W cm−2, 14.5 W mgPt−1) and Pt/C (0.73 W cm−2, 5.8 W mgPt−1) and representing one of the most promising Pt-based catalysts for practical PEMFCs.

a H2–air fuel cell polarization curves of Pt/C, L10-PtCo/C, and L10-PtCoCr/C under LDV conditions (anode/cathode Pt loading: 0.025/0.05 mgPt cm−2, back pressure: 150 kPaabs, 80 °C, 100% RH, and H2/air). b H2-air fuel cell polarization curves of L10-PtCoCr/C before and after 30,000 and 50,000 voltage cycles. c MA retention and voltage loss at 0.8 A cm−2 of the studied catalysts before and after ADT. d Comparison of rated power (normalized by the total PGM loading) and current density at 0.8 V of the studied catalysts. e Comparison of peak power density and current density at 0.7 V of the studied catalysts. f Comparison of critical metrics for studied catalysts and reference catalysts34. The voltages reported here are non-iR corrected.
A standard ADT protocol of 30,000 square-wave potential cycles was applied to evaluate the long-term service feasibility of studied catalysts (Fig. 6b, Supplementary Fig. 56). Noteworthy, the prepared L10-PtCoCr/C exhibits negligible performance loss. Specifically, L10-PtCoCr/C revealed only 3% MA loss after ADT, much better than commercial L10-PtCo/C (52% MA loss) and Pt/C (67% MA loss) and surpassing the DOE 2025 targets and most of the reported electrocatalysts (Fig. 6c and Supplementary Table 5). Moreover, the current density and voltage losses of L10-PtCoCr/C are only 8.4% (@0.8V), 4.3% (@0.7V) and 5 mV (@0.8 A cm−2), respectively, exceeding L10-PtCo/C, Pt/C and DOE 2025 targets (Fig. 6c, e and Supplementary Table 5). In addition, the end-of-life (EOL) rated power of L10-PtCoCr/C can maintain over 90% of initial performance (15.6 W mgPt−1). Even after extra 20,000 potential cycles, the aforementioned indicators can still maintain (Fig. 6b, Supplementary Fig. 57). L10-PtCoCr/C emerges as one of few catalysts that meets the DOE benchmarks for both MA and rated power durability (Fig. 6f). Especially, based on the accelerated failure time conversion of voltage loss, the catalyst is estimated to have a long lifetime of 42,000 h, which is 8.4 times than the DOE target.The almost unchanged high-frequency resistance (HFR) of MEA with the L10-PtCoCr/C cathode before and after ADT demonstrates no significant metal dissolution/migration to the membrane (Supplementary Fig. 55). Suppressed Fenton reaction damage to proton exchange membrane can also be confirmed by the UV-vis absorption spectra results (Supplementary Fig. 58). The detailed characterizations of studied catalysts after stability tests in MEA prove that the particle sizes, compositions, element valence states and intermetallic structures of L10-PtCoCr/C catalysts exhibit insignificant change in contrast to those of L10-PtCo/C, demonstrating the intrinsic stability of L10-PtCoCr/C and the feasibility of the covalentization strategy (Supplementary Figs. 59, 60). Overall, all the activity and stability metrics of L10-PtCoCr/C can achieve or exceed the DOE 2025 targets, representing one of the most promising ORR catalysts (Fig. 6f, Supplementary Table 5).
In summary, we report a concept of constructing a quasi-covalent bond network strategy to bolster the electrochemical stability of L10-PtMM’ alloy ORR catalysts in PEMFCs. We theoretically demonstrate that the increased Pt/M-M’ bond order induced by a less occupied antibonding orbital between Pt/Co and M’ (i.e., ETM) with a high d-band position promotes the conversion from tranditianl metal bond to quasi-covalent bond in L10-PtMM’, which enables the improvement of electrochemical stability. The online ICP-MS and in situ X-ray-based spectroscopy results uncover that the mechanism of the Cr-facilitated anti-dissolution process of L10-PtCo includes directly mitigated anodic dissolution suppressed by the strong QCB network and curtailed cathodic dissolution-driven by the diminished surface Pt oxidation. In MEA tests, the developed L10-PtCoCr/C catalyst achieves high activity and stability, with a high initial MA of 1.27 A mgPt−1 and rated power of 16.5 W mgPt−1 at the critical heat rejection limit of 0.67 V, and ca. 3% MA loss and 5% power loss after 30000 ADT cycles, surpassing U.S. DOE 2025 targets. In particular, the assembled MEA with L10-PtCoCr/C catalyst is highly promising for long-term applications by achieving a projected lifetime of 42,000 h. The developed strategy is applicable to other materials serving in similar harsh electrochemical environments and can be extended to broader energy conversion technologies.
Methods
Chemicals and materials
Pt(acac)2 (98%), Fe(acac)3 (97%), TiO(acac)2 (98%), VO(acac)2 (99%), Cr(acac)3 (98%), Mn(acac)2 (97%), Fe(acac)3 (98%), Co(acac)2 (98%), Ni(acac)2 (95%), Cu(acac)2 (97%), Zn(acac)2 (97%), Ga(acac)3 (99.99%), C2H5Cl3Ge (97%), Zr(acac)4 (98%), NbCl5 (99%), Hf(acac)4 (97%), and TaCl5 (95%) (acac = aceylacetonate), oleylamine (OAm, 70%) were purchased from Aladdin. Tert-butylamine borane (BTB, 97%) was purchased from Sigma-Aldrich. Isopropanol (AR), hexane (AR), ethanol (AR) were purchased from SINOPHARM GROUP CO.LTD. Nafion (5% in a mixture of lower aliphatic alcohols and water) were purchased from Alfa Aesar. Commercial Pt/C was purchased from Johnson Matthey (HiSPEC4000, 40 wt%). All chemicals were used without further purification.
Synthesis of PtCoM’ nanoparticles
The synthesis of PtCoM’ nanoparticles was based on previous reports6,17. The synthesis of PtCoM’ nanoparticles was initiated by combining 0.025 mmol of Pt(acac)2, 0.018 mmol of Co(acac)2, and 0.007 mmol of various M’ precursors (including TiO(acac)2, VO(acac)2, Cr(acac)3, Mn(acac)2, Fe(acac)3, Co(acac)2, Ni(acac)2, Cu(acac)2, Zn(acac)2, Ga(acac)3, C2H5Cl3Ge, Zr(acac)4, NbCl5, Hf(acac)4, and TaCl5) with 5 mL of OAm in a four-necked flask. The mixture was first heated to 110 °C under N2 protection for 30 min. Subsequently, 0.36 mmol of BTB was introduced into the reaction mixture. The temperature was then gradually increased to 280 °C at a rate of 5 °C min−1 and maintained at this temperature for 60 min. After cooling to room temperature, the resulting nanoparticles were precipitated using ethanol and collected via centrifugation (7800 g, 20 min). The product was then redispersed in hexane, reprecipitated with ethanol, and collected again by centrifugation (7800 g, 20 min). Finally, the purified nanoparticles were dispersed in hexane for further use.
Synthesis of carbon-supported intermetallic L10-PtCoM’ (L10- PtCoM’/C)
PtCoM’ NPs dispersion was gradually added to XC-72 carbon that was dispersed in hexane via 10 min sonication, followed by 1 h sonication. The mass percentage of Pt was kept at 20–25 wt%. The suspension was then centrifuged to obtain a solid. The solid was then nitrogen-dried. To obtain carbon-supported intermetallic L10-PtCoM’/C, the product was annealed at 700 °C for 2 h in an Ar/H2 (95/5) gas flow. Next, the sample was dispersed in 50 mL of 0.1 M HClO4, sonicated for 4 h, and then centrifuged and washed three times with DI water and ethanol. Finally, after drying in ambient conditions, the sample was annealed at 400°C for 2 h in an Ar/H2 (95/5) gas flow to produce the final catalyst.
Material characterizations
Material characterization operations refered to our previous work6,7,38.TEM and HRTEM images were collected from a FEI Tecani G2 20 (200 kV) and a FEI Tecani G2 F30 (300 kV), respectiviely. Scanning transmission electron microscopy (STEM) analyses were performed on a JEOL JEMARM200F STEM/TEM with a guaranteed resolution of 0.08 nm. XRD were collected from Rigaku MiniFlex 600 diffractometer with a Cu radiation source (λ = 0.15406 nm). The ordering degree is calculated according to the following equations:
Where Shkl is the peak area of the obtained samples and Ihkl is the peak intensity obtained from the standard JCPDF card. ICP-MS result was carried out from ELAN DRC-e. X-Ray Fluorescence (XRF) results were obtained from EAGLE III operated at 40 kV.
Electrochemical measurements
Electrochemical measurements were performed in a five-port glass electrochemical cell (PINE, 150 mL) through CHI760e electrochemical station (Shanghai Chenhua Instrument Corporation, China). A glassy carbon rotating disk (diameter: 5 mm, area: 0.196 cm2), a graphite rod (diameter: 5 mm) and a Ag/AgCl (saturated KCl) electrode are used as the working electrode, the counter electrode, and the reference electrode, respectively. To prevent Cl− ion contamination, the Ag/AgCl reference electrode was isolated from the working and counter electrodes using a salt bridge. All the potentials were experimentally converted to values with reference to an RHE. A catalyst ink was formulated by blending the catalysts with deionized water (490 μL), isopropanol (1.5 mL), and Nafion® (10 μL, 5 wt%) to achieve a concentration of 2 mg mL−1. Following 60 min of sonication to ensure thorough mixing, 10 μL of the ink was deposited onto a glassy carbon electrode (geometric area: 0.196 cm2) and dried at room temperature. ICP-MS analysis indicated that the final catalyst loading for the Pt alloy electrocatalysts was ~20 µg cm−2. The practical Pt loading on the electrode is calculated according to the following equations:
Where \({{\mbox{w}}}_{{\mbox{Pt}}}\) is the Pt loading of catalyst calculated from ICP-MS results. The CV measurements (0.05−1.2 V vs. RHE, 50 mV s−1) were performed in 100 mL N2-saturated 0.1 M HClO4 (pH = 1 ± 0.1, made from 70.0–72.0% HClO4, Sigma-Aldrich, Merck) for the calculation of ECSA. The ECSA value is calculated from the equation:
Where \({{\mbox{Q}}}_{{\mbox{H}}-{\mbox{UD}}}\) is the hydrogen desorption charge obtained by integrating the area in CVs, \({{\mbox{M}}}_{{\mbox{Pt}}}\) is the actual Pt loading amount. The ORR measurements (roation rate: 1600 rpm, anodic sweep rate: 10 mV s−1) were carried out in 100 mL O2-saturated 0.1 M HClO4. Accelerated durability tests (25 °C, 0.6−1.0 vs. RHE, 100 mV s−1) were performed in 100 mL O2-saturated 0.1 M HClO4. All the electrode potentials were converted to reference the reversible hydrogen electrode (RHE) using:
The resistance of the electrochemical cell was measured by iR compensation on a CHI760e electrochemical station (Shanghai Chenhua Instrument Corporation, China). The specific resistance values (0.1 M HClO4) in CV and LSV techniques are 24.8 ± 0.4 Ω.
Online ICP-MS characterization
Online ICP-MS characterization operation was based on our previous work26. In the flow cell, a glassy carbon electrode (diameter: 5 mm, area: 0.196 cm2), an Au wire and a saturated calomel electrode (SCE) were used as the working electrode, the counter electrode and the reference electrode, respectively. In the ICP-MS experiment, the flowing electrolyte consisted of a N2-saturated 0.1 M HClO4 solution (pH = 1 ± 0.1, flowing rate: 13 mL min−1). The metal dissolution was recorded by performing a CV sweeping operation (0.05–1.0/1.2 V, 2 mV s−1). All the standard curves for Pt, Co, and Cr were calibrated to quantify their dissolution amounts. For GDE-based ICP-MS measurements (80 °C), the anode/cathode consisted of commercial Pt/C (0.4 mg cm−2) and prepared catalysts (1 mg cm−2). A Nafion 117 membrane (Dupont®, 183 μm) was used to separate the anode and cathode. An SCE reference electrode was used. The electrochemical tests are the same as the aqueous electrode testing.
Membrane electrode assembly (MEA) preparation
MEA preparation process was based on our previous work6. Cathode catalysts, including L10-PtCoCr/C, L10-PtCo/C, and commercial Pt/C (Johnson Matthey, HiSPEC4000, 40 wt%), were mixed with D520® perfluorosulfonic acid (PFSA) ionomer in an isopropanol-to-water ratio of 1:1. The ionomer-to-carbon (I/C) ratio was maintained at 0.6, and the mixture was ultrasonicated for 60 min to form a homogeneous catalyst ink. The catalyst concentration in the ink was controlled to be between 2 and 3 mg mL−1. The catalyst-coated membrane (CCM) with an active area of 5 cm2 was prepared on a Gore membrane (Gore-select®, 12 μm, used as received without further treatment). An ultrasonic spray technique was employed for deposition, with a temperature of 70 °C, a spraying rate of 40 μL min−1, and a spraying distance of 2–4 cm. For the anode, a commercial Pt/C catalyst (Johnson Matthey, HiSPEC4000, 40 wt%) was used, and the anode catalyst ink was prepared using the same method and sprayed onto the opposite side of the membrane to form the anode catalyst layer. The Pt loadings for the anode and cathode were confirmed by drying the layers completely and weighing them. The anode had a Pt loading of 0.025 mgPt cm−2, while the cathode had a Pt loading of 0.05 mgPt cm−2. Gas diffusion layers (GDLs) with an integrated microporous layer (SIGRACET®, GDL 22BB, thickness: 215 μm) were used. The GDLs, gaskets, and prepared CCM were hot-pressed together to form MEA. The actual catalyst loadings were verified using ICP-MS analysis.
Fuel cell testing
Fuel cell testing was carried out in a single cell (Scribner 850e, Hephas Energy Corporation)6,38. The MEA was assembled between two graphite flow plates with 14 parallel flow channels to reduce the pressure gradients between the inlet and outlet. Break-in process was performed by cycling between 0.75 V and 0.35 V under H2-air condition until a stable current was obtained. The mass activity (H2/O2) of catalyst was tested at 80 °C, 100% relative humidity (RH), 150 kPaabs and with a gas flow rate of 200 sccm (anode) /500 sccm (cathode). The H2–air fule cell polarization curves was recorded at 80 °C, 100% RH, 150 kPaabs and with a gas flow rate of 500 sccm (anode)/2000 sccm (cathode). The ADT was conducted by applying square-wave protocol (0.6 V for 3 s, 0.95 V for 3 s, 80 °C, 100% RH, atmospheric pressure, H2/N2 200/200 sccm). The projection lifetime of fuel cell is equal to the operation time until a 10% loss is reached in cell voltage at the rated power. According to the previous report,34,39 the lifetime can be calculated through the equation:
The lifetime of the tested MEA is predicted from the ratio between 10% of the initial voltage (V1.5 A cm-2) at 1.5 A cm−2 and the voltage loss rate at 1.5 A cm−2.
XAS characterizations
All the Pt L3-edge XAS spectra were recorded under ambient conditions at the BL11B beamline (Shanghai Synchrotron Radiation Facility)6,38. Potential-dependent in situ XAFS spectra were performed by using a homemade electrochemical cell in N2-saturated 0.1 M HClO4 solution. A carbon paper loaded with catalysts (2 mg cm−2), an Au wire and a SCE electrode were used as the working electrode, the counter electrode and the reference electrode, respectively (Supplementary Fig. 61). All the in situ X-ray absorption signals were collected after the electrodes reach a steady state at the set potential. The Athena and Artemis software codes were utilized for the XAS analysis. The Fourier transformation of the EXAFS data in R space was performed, and the resulting data were interpreted using models of metallic Pt and L10-PtCo to account for the contributions from Pt-Pt and Pt-Co/Cr interactions.
DFT computational details
All DFT calculations were performed using the Vienna ab initio Simulation Package, with projector-augmented-wave pseudopotentials to describe the valence electrons-core ions interactions40,41. The exchange-correlation effects were modeled using the Perdew-Burke-Ernzerhof functional within the generalized gradient approximation42,43. The plane-wave energy cutoff was set as 400 eV and the Brillouin zone was sampled with a Monkhorst-Pack 3 × 3 × 1 k-point mesh44. The slab model for the (111) surface of L10-PtCoM’ was created within a 3 × 3 in-plane supercell. A vacuum spacing of 15 Å was maintained between adjacent slabs in the normal direction. The COHP analysis was performed by using the LOBSTER software45,46. The bond order (BO) for a metal-metal bond is defined as one-half of the difference between the number of bonding and antibonding electrons. The minimum energy pathways for Co diffusion were calculated using the climbing image nudged elastic band method47.
Data availability
The data that support the findings of this study are available from the corresponding authors upon request. Source data is provided with this paper.
References
Fan, J. et al. Bridging the gap between highly active oxygen reduction reaction catalysts and effective catalyst layers for proton exchange membrane fuel cells. Nat. Energy 6, 475–486 (2021).
Cullen, D. A. et al. New roads and challenges for fuel cells in heavy-duty transportation. Nat. Energy 6, 462–474 (2021).
Kodama, K., Nagai, T., Kuwaki, A., Jinnouchi, R. & Morimoto, Y. Challenges in applying highly active Pt-based nanostructured catalysts for oxygen reduction reactions to fuel cell vehicles. Nat. Nanotechnol. 16, 140–147 (2021).
Liang, J. et al. Metal bond strength regulation enables large-scale synthesis of intermetallic nanocrystals for practical fuel cells. Nat. Mater. 23, 1259–1267 (2024).
Liang, J. et al. Gas-balancing adsorption strategy towards noble-metal-based nanowire electrocatalysts. Nat. Catal. 7, 719–732 (2024).
Liu, X. et al. Introducing electron buffers into intermetallic Pt alloys against surface polarization for high-performing fuel cells. J. Am. Chem. Soc. 146, 2033–2042 (2024).
Liu, X. et al. Inducing covalent atomic interaction in intermetallic Pt alloy nanocatalysts for high-performance fuel cells. Angew. Chem. Int. Ed. 62, e202302134 (2023).
Lin, F., Li, M., Zeng, L., Luo, M. & Guo, S. Intermetallic nanocrystals for fuel-cells-based electrocatalysis. Chem. Rev. 123, 12507–12593 (2023).
Liu, X., Wu, G. & Li, Q. Orbital modulation in platinum-group-metal (PGM) electrocatalysts: an effective approach to boost catalytic performance. eScience 5, 100270 (2025).
Pettifor, D. G. A physicist’s view of the energetics of transition metals. Calphad 1, 305–324 (1977).
Pettifor, D. G. Theory of the heats of formation of transition-metal alloys. Phys. Rev. Lett. 42, 846–850 (1979).
Pettifor, D. G. Theoretical predictions of structure and related properties of intermetallics. Mater. Sci. Technol. 8, 345–349 (1992).
Greeley, J. et al. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nature Chemistry 1, 552–556 (2009).
Nakaya, Y. & Furukawa, S. Catalysis of alloys: classification, principles, and design for a variety of materials and reactions. Chem. Rev. 123, 5859–5947 (2023).
Zeng, Y. et al. Regulating catalytic properties and thermal stability of Pt and PtCo intermetallic fuel-cell catalysts via strong coupling effects between single-metal site-rich carbon and Pt. J. Am. Chem. Soc. 145, 17643–17655 (2023).
Li, J. & Sun, S. Intermetallic nanoparticles: synthetic control and their enhanced electrocatalysis. Acc. Chem. Res. 52, 2015–2025 (2019).
Li, J. et al. Anisotropic strain tuning of L10 ternary nanoparticles for oxygen reduction. J. Am. Chem. Soc. 142, 19209–19216 (2020).
Li, J. et al. Hard-Magnet L10-CoPT nanoparticles advance fuel cell catalysis. Joule 3, 124–135 (2019).
Li, J. et al. Fe stabilization by intermetallic L10-FePt and Pt catalysis enhancement in L10-FePt/Pt nanoparticles for efficient oxygen reduction reaction in fuel cells. J. Am. Chem. Soc. 140, 2926–2932 (2018).
Mao, Z. et al. Interstitial B-doping in Pt lattice to upgrade oxygen electroreduction performance. ACS Catal 12, 8848–8856 (2022).
Zhao, X. et al. High-performance nitrogen-doped intermetallic PtNi catalyst for the oxygen reduction reaction. ACS Catal 10, 10637–10645 (2020).
Gao, L. et al. Unconventional p–d hybridization interaction in PtGa ultrathin nanowires boosts oxygen reduction electrocatalysis. J. Am. Chem. Soc. 141, 18083–18090 (2019).
Stamenkovic, V. R., Mun, B. S., Mayrhofer, K. J. J., Ross, P. N. & Markovic, N. M. Effect of surface composition on electronic structure, stability, and electrocatalytic properties of Pt-transition metal alloys: Pt-skin versus Pt-skeleton surfaces. J. Am. Chem. Soc. 128, 8813–8819 (2006).
Yang, C.-L. et al. Sulfur-anchoring synthesis of platinum intermetallic nanoparticle catalysts for fuel cells. Science 374, 459–464 (2021).
Luo, M. & Guo, S. Strain-controlled electrocatalysis on multimetallic nanomaterials. Nat. Rev. Mater. 2, 17059 (2017).
Gao, X. B. et al. Mechanism of particle-mediated inhibition of demetalation for single-atom catalytic sites in acidic electrochemical environments. J. Am. Chem. Soc. 145, 15528–15537 (2023).
Đukić, T. et al. Adjusting the operational potential window as a tool for prolonging the durability of carbon-supported pt-alloy nanoparticles as oxygen reduction reaction electrocatalysts. ACS Catal 14, 4303–4317 (2024).
Moriau, L. et al. Towards improved online dissolution evaluation of Pt-alloy PEMFC electrocatalysts via electrochemical flow cell - ICP-MS setup upgrades. Electrochim. Acta 487, 144200 (2024).
Topalov, A. A. et al. Dissolution of platinum: limits for the deployment of electrochemical energy conversion? Angew. Chem. Int. Ed. 51, 12613–12615 (2012).
Gatalo, M. et al. Comparison of Pt–Cu/C with benchmark Pt–Co/C: metal dissolution and their surface interactions. ACS Appl. Energy Mater. 2, 3131–3141 (2019).
Sheyfer, D. et al. Operando nanoscale imaging of electrochemically induced strain in a locally polarized Pt grain. Nano Lett 23, 1–7 (2023).
Dai, S. et al. Platinum-trimer decorated cobalt-palladium core-shell nanocatalyst with promising performance for oxygen reduction reaction. Nat. Commun. 10, 440 (2019).
Zhao, Z. et al. Graphene-nanopocket-encaged PtCo nanocatalysts for highly durable fuel cell operation under demanding ultralow-Pt-loading conditions. Nat. Nanotechnol. 17, 968–975 (2022).
Peng, B. et al. Embedded oxide clusters stabilize sub-2 nm Pt nanoparticles for highly durable fuel cells. Nat. Catal. 7, 818–828 (2024).
Song, T.-W. et al. Small molecule-assisted synthesis of carbon supported platinum intermetallic fuel cell catalysts. Nat. Commun. 13, 6521 (2022).
Zhao, Z. et al. Tailoring a three-phase microenvironment for high-performance oxygen reduction reaction in proton exchange membrane fuel cells. Matter 3, 1774–1790 (2020).
Qiao, Z. et al. Atomically dispersed single iron sites for promoting Pt and Pt3Co fuel cell catalysts: performance and durability improvements. Energy Environ. Sci. 14, 4948–4960 (2021).
Li, Y. et al. Introducing covalent metal-phosphorus bonds into intermetallic platinum-based catalysts for high-performance fuel cells. Renewables 2, 223–232 (2024).
Stariha, S. et al. Recent advances in catalyst accelerated stress tests for polymer electrolyte membrane fuel cells. J. Electrochem. Soc. 165, F492 (2018).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Hammer, B., Hansen, L. B. & Nørskov, J. K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 59, 7413–7421 (1999).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Deringer, V. L., Tchougréeff, A. L. & Dronskowski, R. Crystal Orbital Hamilton Population (COHP) analysis as projected from plane-wave basis sets. J. Phys. Chem. A 115, 5461–5466 (2011).
Maintz, S., Deringer, V. L., Tchougréeff, A. L. & Dronskowski, R. LOBSTER: a tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 37, 1030–1035 (2016).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Acknowledgements
This work is supported by the National Key Research and Development Program of China (grant no. 2021YFA1501001 to Q. Li), National Nature Science Foundation of China (grant no. U24A20499, no. 22479056 and no. 22122202 to Q. Li, grant no. 22102077 and no. 22462022 to Z. Zhao), the Fundamental Research Funds for the Central Universities (grant no. 5003110132 to Q. Li), and the HUST-TROOWIN hydrogen fuel cell technology center. The work at California State University Northridge was supported by NSF-PREM program (grant no. DMR−1828019 to G. Lu). We thank the Analytical and Testing Center of Huazhong University of Science and Technology for carrying out the TEM, XRF and XRD measurements. The XAS and atomic pair distribution function spectra experiments were performed at BL11B and BL02U2 beamline, Shanghai Synchrotron Radiation Facility.
Author information
Authors and Affiliations
Contributions
Q. Li and X. Liu conceived the idea and designed the experiments. X. Liu, and S. Zhang carried out the sample synthesis, characterization, and electrochemical measurements. Y. Wang and D. Su carried out HAADF-STEM characterizations. X. Liu and H. He performed XAS measurements. Y. Wang and X. Luo performed online ICP-MS measurements. Z. Zhao and G. Lu provided theoretical calculations. X. Liu, G. Lu, Y. Huang, and Q. Li wrote and revised the manuscript. All the authors contributed to the overall scientific discussion and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jong-Sung Yu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, X., Wang, Y., He, H. et al. Regulating orbital interaction to construct quasi-covalent bond networks in Pt intermetallic alloys for high-performance fuel cells. Nat Commun 16, 4895 (2025). https://doi.org/10.1038/s41467-025-60171-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-60171-z
