
Abstract
The accumulation of α-synuclein within Lewy bodies is a critical factor in the pathogenesis of Parkinson’s disease, with potential implications for axonal transport deficits. Activated asparagine endopeptidase enzymatically cleaves α-synuclein and tau, resulting in the formation of α-SynN103 and tauN368, which are markedly elevated in the brains with Parkinson’s disease. In this study, rats received intrastriatal injections of 15 µg of preformed α-SynN103 and tauN368 fibrils, and their behaviors were evaluated after a 2-month period. Subsequent analyses investigated alterations in axonal transport and the underlying molecular mechanisms. Our findings indicated that preformed fibrils reduced kinesin levels and excessively activated the AMPK and p38 MAPK, thereby compromising the function of kinesin and dynein in axonal transport. Pharmacological inhibition of AMPK and p38 MAPK ameliorated these dysfunctions in rat models, which identified Compound C and SB203580 as potent inhibitors, offering evidence for early interventions of Parkinson’s disease.

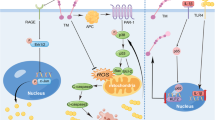
Mechanisms by which PFFs caused axonal transport defects of dopamine neurons in PD-like models. (A) Shows normal axonal transport. (B) Demonstrates how PFFs increase ?-Syn accumulation, reducing PIKE expression and triggering AMPK/p38 MAPK over-activation, which lowers kinesin levels and motor-cargo interaction. (C) AMPK activity inhibition with C.C significantly improves these deficits. (D) The p38 inhibitor enhances kinesin transport by preventing p38 MAPK over-activation, reducing its inhibition of kinesin-cargo binding.
Similar content being viewed by others

Introduction
Parkinson’s disease (PD) is a prevalent neurodegenerative disease characterized by the selective degeneration of nigrostriatal dopamine neurons and the presence of Lewy bodies (LBs) in cell bodies1. The pathological accumulation of α-synuclein (α-Syn*) in LBs is a key feature of PD, with high levels observed in affected brain regions. α-Syn* has been shown to trigger various pathophysiological processes in neurons, such as synaptic dysfunction, mitochondrial impairment, and lysosomal maturation defects, ultimately leading to neurodegeneration2.
Axonal transport is the primary mechanism by which neurons facilitate long-distance communication between the cytosol and synaptic terminals. Kinesin, located on microtubules, is responsible for the transportation of membrane organelles, cytoskeletal proteins, and other cargoes from the cell body to the synaptic terminal in a process known as anterograde transport. Conversely, dynein typically binds to endosomes, autophagosomes, and other intracellular vesicles, facilitating their retrograde transport back to the cytosol. The transportation of different cargoes is conducted via disparate modes along the axon3. Numerous studies have established that impaired axonal transport is a critical contributing factor to various neurodegenerative diseases, such as Alzheimer’s disease4, amyotrophic lateral sclerosis5 and Huntington’s disease6. Conversely, the correlation between axonal transport deficits and PD has been less extensively investigated. Furthermore, Lewy neurites (LNs) are predominantly located within axons, forming earlier and exhibiting a wider distribution than LBs7. However, the relationship between axonal transport defects and α-Syn* remains to be elucidated.
Researchers investigating neurons impacted by α-Syn* have discovered that α-synuclein (α-Syn) inclusions form within axons prior to neuronal degeneration and death. Additionally, the velocity and mobility of various cargoes, such as Rab7+ endosomes, TrkB, and LC3+ autophagic vesicles, were markedly diminished, and mitochondrial transport was similarly compromised8. However, electron microscopy analysis revealed that α-Syn inclusions do not obstruct the axonal transport process by physically occupying the axonal space. What’s more, kinesin levels were found to be significantly lower and more pronounced in dopamine neurons containing α-Syn*. These changes preceded the altered tyrosine hydroxylase (TH) phenotype of the neurons, as documented in the referenced study9. In contrast, alterations in dynein levels have been observed exclusively in advanced PD patients. Furthermore, these manifestations were also observed in A30P/A53T transgenic mice10. The findings suggest that α-Syn* may play a regulatory role in the modulation of motor protein levels, potentially disrupting axonal transport.
It Is Important to recognize that kinesin and dynein exhibit a competitive dynamic when transporting identical cargo. This competition is predominantly evident in their quest for microtubule-binding targets11 and surface-binding sites on the transported cargo12. Furthermore, dynein requires binding to dynactin and the corresponding adapter protein to form a functional transport unit13. Observations have demonstrated the co-localization of LBs and LNs with dynactin in the brains of patients with PD and Lewy body dementia14. This has led to the hypothesis that α-Syn* may influence retrograde transport through interactions with the transport unit. Additionally, snapin has been identified as one of the selective adapters for dynein. Upon binding to dynein, snapin facilitates the retrograde transport of internalized TrkB following signal reception at the synaptic terminal, thereby transmitting the signal to the cytosol for a subsequent response. Recent studies have demonstrated that the retrograde transport of TrkB is affected by α-Syn* in vitro15. This dysfunction primarily manifests as a deceleration in the movement rate of dynein-loaded TrkB along microtubules and a reduction in the overall migration rate16. Furthermore, certain pathological proteins, including APP and SOD1, have been demonstrated to disrupt the assembly of the snapin-dynein unit, resulting in aberrant retrograde transport17. These studies suggest that α-Syn* may obstruct dynein transport during the early stages of PD, even when changes in dynein protein levels are not yet detectable, through mechanisms distinct from those influencing anterograde kinesin transport. Consequently, the inhibition of retrograde transporter unit formation presents itself as a promising area for further research.
Asparagine endopeptidase (AEP) exhibits significant activation in the brains of individuals with PD, particularly within the substantia nigra (SN)18. Empirical evidence indicates that activated AEP cleaves α-Syn at the N103 residue, resulting in the formation of α-SynN10319, and similarly cleaves tau protein at the N368 residue to produce tauN36820. These cleavage events enhance the biotoxicity of the respective proteins and facilitate each aggregation. Recent investigation has demonstrated that these monomeric fragments can aggregate and interact to form preformed fibrils complexes (PFFs). Notably, the presence of α-SynN103 and tauN368 complexes is markedly elevated in the brains of PD patients, underscoring their potential pathological relevance21. Furthermore, our findings indicate that PFFs exhibit significant neurotoxicity and transmissibility, leading to endogenous α-Syn aggregation and dopaminergic neuronal degeneration in the rat brain22. These results suggest that PFFs may constitute a substantial advancement in the investigation of the pathogenic mechanisms underlying α-Syn* in axonal transport disorders of PD.
In the present study, rats received intra-striatal injections of 15 µg PFFs, and their behaviors were evaluated at the conclusion of the second month post-surgery. Subsequently, we assessed the pathological effects and distribution of PFFs on nigrostriatal dopaminergic neurons through Western blot analysis and immunostaining techniques. Further investigations were conducted to evaluate the alterations in kinesin-mediated anterograde axonal transport and dynein-mediated retrograde axonal transport in dopamine neurons induced by PFFs, as well as to elucidate the potential molecular mechanisms involved. We also implemented pharmacological interventions for mechanism validation, with the aim of exploring potential therapeutic targets in the early stages of PD and providing a theoretical basis for the research and development of novel interventional drugs.
Results
PFFs induced motor behavioral alterations and dopaminergic neurodegeneration in rats
Pathological features of Parkinson’s disease include the accumulation of misfolded α-Syn into insoluble LBs23 and elevated levels of phospho-S129 α-Syn (pS129)24. We investigated the effects of injecting the PFFs into the striatal region of rats for 2 months on Parkinson’s disease-related behaviors and neuropathological changes. We conducted four tests to evaluate motor disorders in rats. Rats treated with PFFs showed more incoordination on the affected side in the adjusting steps test (Fig. 1A), more slips and longer crossing times in the balance beam test (Fig. 1B), less use of the contralateral forelimb in the cylinder test (Fig. 1C), and reduced exploration of the central area in the open field experiment compared to the sham group (Fig. 1D, E). In other words, rats treated with PFFs exhibited motor retardation, impaired coordination, reduced activity, and decreased muscle strength, flexibility, and coordination in one limb.

A Two-month post-injection, rats showed increased bilateral limb incoordination. B PFFs resulted in more hindlimb slips and increased balance beam time. C PFFs rats had fewer contralateral forelimb wall touches in the cylinder test. D, E In the open field, PFFs rats entered the central area less frequently and briefly and showed reduced autonomous behavior. G PFFs increased α-Syn aggregation in the SNpc (left). Dopaminergic neurons displayed varying loss of TH phenotype (right). F, H, I Relevant statistical analyses followed. J Lamp1 co-staining indicated insoluble inclusions. Scale bar = 50 μm in (G, middle) and 200 μm in other images. n = 12 in (A–E) and 3 in (F, H and I). *p < 0.05, **p < 0.01, ***p < 0.001.
We stained the SN and striatal regions of different rat groups to examine pathological aggregates. Notable pS129 signals appeared in the ipsilateral SNpc of PFFs rats (Fig. 1G, H). In the nervous system, Lamp1 is usually present in autophagy- and lysosome-related organelles, many of which lack detectable lysosomal hydrolases25,26. Co-immunofluorescence with pS129 and Lamp1 showed increased insoluble inclusion bodies in the PFFs group (Fig. 1J). Sporadic PD pathology involves loss of neuronal TH phenotypes in the SNpc and striatum, and LB accumulation22. Brain staining revealed reduced TH immunofluorescence in the ipsilateral striatum and fewer TH+ cells in the SNpc of PFFs rats (Fig. 1F–G, I). Overall, the data indicated that PFFs induced abnormal α-Syn phosphorylation and accumulation in the rat brain, resulting in LB deposition, dopamine neuron degeneration, and motor disorders similar to sporadic PD.
PFFs disrupted anterograde transport by interfering with kinesin expression and binding to cargoes
Previous studies show that cytoskeleton-associated proteins change significantly before dopamine neuron loss in rats22. We hypothesize that PFFs-induced α-Syn* affects axonal transport in rat dopamine neurons, as evidenced by a significant shift in transport track. Kinesin, composed of two heavy chains (KHC) and two light chains (KLC), uses ATP to move cargoes along microtubules by binding to the light chain (see Graphical abstract A)27. Mitochondria provide energy for anterograde transport from the soma to distal synapses, facilitated by MTX1, MTX2, and Miro128,29. Western blot analysis showed decreased kinesin and mitochondrial proteins in the ipsilateral striatum of rats (Fig. 2A, B). The findings showed abnormal protein distribution in the striatum, particularly in nigral nerve endings. Immunofluorescence co-staining for TH and KLC1 in the SNpc revealed reduced fluorescence intensity in the PFFs-treated group (Fig. 2E), indicating impaired anterograde transport in TH-positive neurons in the SN. To confirm anterograde transport impairment, we transfected primary cortical neurons with Miro1-overexpressing lentiviruses and tracked Miro1 using mCherry fluorescence. After 2 days of PFF or PBS treatment, we recorded Miro1 transport every 5 s for 120 s, displaying images at the 5th, 60th, and 120th seconds (Fig. 2F, G). Analysis with Image J showed slowed mitochondrial movement in PFFs-treated group, confirming abnormal distribution of mitochondrial proteins and anterograde transport deficit (Fig. 2D).

A, B The images indicate a significant reduction in kinesin and mitochondrial proteins in the striatum. C–E Kinesin reduction mainly occurred in dopaminergic neurons, as suggested by KLC1 and TH co-staining. D PFFs restricted mitochondrial anterograde transport. F, G Images of Miro1-mCherry within cortical neuron axons are also presented. Scale bar = 20 μm. n = 3 in (B, C), n = 10 in (D). *p < 0.05, **p < 0.01, ***p < 0.001.
MTX1/2 and Miro1 bind to KLC1 to facilitate mitochondrial transport in axons29. Co-IP was conducted on SN tissues to examine PFFs’ effects on protein binding in transport complexes. Results indicated reduced KLC1 binding to Miro1 and MTX1/2 in the SNpc of PFFs-treated rats compared to the sham group (Fig. 3A). KLC2 binding showed a similar trend (Fig. 3C). Co-staining of KLC1 and Miro1 in the SNpc confirmed consistent co-localization and decreased fluorescence intensity in the PFFs group (Fig. 3D, E). KHC not only binds to cargo for transport but also interacts with α-Tubulin on microtubules to aid anterograde transport. Our prior research indicated that the PFFs affects cytoskeletal proteins, leading us to explore its impact on KHC and α-Tubulin binding. Co-staining revealed that KHC and α-Tubulin co-localized in the SNpc but showed reduced fluorescence intensity in the PFFs group (Fig. 3F, G). Our findings revealed reduced expression and impaired binding of kinesin and mitochondrial proteins, causing abnormal distribution and mitochondrial dysfunction.

A Immunoprecipitation of KLC1 revealed reduced binding to mitochondrial transport proteins. B Illustrates kinesin’s attachment to mitochondria. C KLC2 showed decreased binding to these proteins. D, E Immunofluorescence indicates a drop in kinesin co-localization with Miro1. F, G Kinesin’s co-localization with the cytoskeleton also decreased. Scale bar = 200 μm in (D) and (G). n = 3, **p < 0.01, ***p < 0.001 in (E, F).
PFFs impeded retrograde transport by affecting the binding of dynein to cargoes
Dynein, made up of heavy, intermediate, light-intermediate (DIC), and light chains, enables retrograde axonal transport in axon terminals30. Dynactin and snapin are also involved (see Graphical abstract A)31,32. In the rat striatum, increased levels of DIC, dynactin, and snapin suggest impaired retrograde transport due to PFFs, with abnormal transport protein distribution observed (Fig. 4D, E) and confirmed by DIC immunofluorescence staining (Fig. 4F, G).

This is illustrated by images of Erk1/2-GFP in cortical neuron axons (A–C) and the accumulation of motor proteins and cargoes in the striatum (D, E). PFFs increased DIC in the striatum of treated rats. F–H Additionally, PFFs caused TrkB disorganization in axons and increased its presence at the cell membrane following BDNF stimulation. Scale bar = 20 μm in (B), and 200 μm (top) and 50 μm (bottom) in (G). n = 10 in (C), n = 3 in (E–F, H). *p < 0.05, **p < 0.01, ***p < 0.001.
The TrkB receptor on the synaptic membrane binds to BDNF, triggering internalization and retrograde transport to the soma33, which increased in the ipsilateral striatum of PFFs rats (Fig. 4D, E). After adding PFFs to primary neurons for 24 h without using Triton-100X, more TrkB remained on the neuronal membrane (Fig. 4G, H), suggesting impaired internalization and signaling transport. Rab7 is essential for TrkB signaling and endosomal transport34. Studies show that pErk is co-transported with TrkB to the soma, transmitting extracellular signals35. In PFFs rats, Rab7 and pErk accumulated in the striatum, aligning with TrkB expression, which suggests PFFs affect retrograde transport in neuronal axons. Additionally, we examined impaired retrograde transport in living cells by overexpressing Erk in primary neurons. This caused GFP fluorescence in neuronal Erk molecules, confirming that Erk movement was slower in the PFFs-treated group, indicating restricted retrograde axonal transport (Fig. 4A–C).
We hypothesized that PFFs might inhibit the binding of retrograde transport proteins and cargoes. Using Co-IP experiments on rat striatum, we found that in rats exposed to PFFs, dynein’s binding to dynactin, snapin, TrkB, and pErk1/2 was reduced (Fig. 5A). This suggests that PFFs disrupt dynein’s interactions, impairing retrograde transport. Research indicates that α-Syn inclusion bodies appear in axons before neurons degenerate and die, slowing the retrograde transport of cargoes like Rab7+ endosomes and TrkB receptors8. Dynein inhibition raises Rab7 levels, implying delayed endosome maturation and reduced lysosomal degradation36. The PFFs rats showed higher TrkB and Rab7 co-localization in SNpc, indicating TrkB might be trapped in the nuclear endosome and not transported to the lysosome for degradation (Fig. 5C, D). Our results show increased expression of dynein, dynactin, and snapin, and restricted interactions between these proteins and retrograde transport cargoes.

A Immunoprecipitation of DIC indicated reduced binding with its adapters and cargoes. B A model of retrograde transport dynamin complex binding to cargoes, such as TrkB. C, D Enhanced co-localization of TrkB and Rab7 indicated limited retrograde transport to lysosomes in the cytosol for degradation. Scale bar = 200 μm. E–H Lower PIKE expression led to over-activation of the downstream AMPK/p38 MAPK. n = 3 in (C, F–H), *p < 0.05, **p < 0.01, ***p < 0.001.
PFFs downregulated the expression of PIKE and activate the AMPK/p38 MAPK signaling pathway
Our earlier findings indicate that PFFs reduce motor protein expression and disrupt binding, though the mechanism remains unclear. The PFFs rats showed reduced PIKE expression and elevated p-AMPK/AMPK and p-p38 MAPK/p38 MAPK levels in both the striatum and SN compared to the sham group (Fig. 5E–G, Supplemental Fig. 3A–D). And the intensity of DAT fluorescence decreased, while the intensity of pS129 fluorescence increased and co-localized with TH+ dopamine neurons (Supplemental Fig. 3E, F). We chose KLC1 and DIC to study protein binding changes in neurons for anterograde and retrograde transport. Co-IP results showed that PFFs increased AMPK/p38 MAPK binding to DIC and KLC1 in cortical neurons (Supplemental Fig. 3G, H). These findings indicate that PFFs can abnormally phosphorylate α-Syn, lowering PIKE protein levels and activity. This diminishes PIKE’s inhibition of AMPK, causing abnormal activation of the AMPK/p38 MAPK pathway, which may affect axonal transport in the striatum and SN.
PFFs affected axonal transport in dopamine neurons by upregulating AMPK activity
We investigated how AMPK affects axonal transport in dopamine neurons by manipulating its activity with AICAR and Compound C (C.C) in rats divided into four groups: PBS/DMSO, PFFs/DMSO, PFFs/AICAR, and PFFs/C.C. After 14 days of drug injections, brain tissue was tested using western blot assay. Both AICAR and C.C were found to cross the blood-brain barrier to regulate AMPK in the brain tissue. AICAR inhibited the expression of kinesin and mitochondrial proteins in comparison to controls, while C.C. attenuated the inhibitory effect of AMPK (Fig. 6A–D). As shown in Fig. 7F, C.C. promoted kinesin transcription, while AICAR further inhibited it. Treating PFFs-exposed primary neurons with C.C for 24 h enhanced anterograde transport of Miro1 (Fig. 6E–G) and retrograde transport of Erk1/2 (Fig. 7A–C). These indicated that PFFs-induced AMPK activation impairs axonal transport, but this impairment can be partially mitigated by C.C.

A–D Compound C boosted kinesin and mitochondrial transport proteins in the ipsilateral striatum, while AICAR did the opposite. Images (F, G) depicted Miro1-mCherry in cortical neuron axons. Scale bar = 20 μm. The statistical analysis of these images is shown in (E). n = 4 in (B–D) and 10 in (E). *p < 0.05, **p < 0.01, ***p < 0.001.

A–C The images depict GFP-ERK1/2 within cortical neuron axons. AMPK inhibitor increased the binding of KLC1 and DIC to their cargoes (D), as well as kinesin transcription (F). In contrast, AICAR inhibits this (E). A, B Scale bar = 20 μm. n = 10 in (C) and 3 in (F) *p < 0.05, **p < 0.01, ****p < 0.0001.
We performed Co-IP to examine changes in transport-related protein interactions in the ipsilateral striatum and SN of rats’ post-drug administration. In the ipsilateral SN, KLC1 binding to Miro1 and MTX2 increased, and p-p38 MAPK decreased after C.C treatment (Fig. 7D). In the striatum, binding of DIC, dynactin, snapin, TrkB, and pErk1/2 was enhanced (Fig. 7E). Inhibiting AMPK activation enhanced KLC1 and DIC’s cargo-binding ability, indicating that PFFs disrupts axonal transport by altering the phosphorylation of the motor-cargo binding site in dopamine neurons, and improved the levels of kinesin as well (Fig. 7F). Our research showed that PFFs caused abnormal AMPK activation, leading to reduced expression, altered distribution, and impaired binding of motor proteins, disrupting axonal transport and worsening dopamine neuron degeneration (see Graphical abstract B). Inhibiting AMPK activity with C.C significantly improved these deficits (see Graphical abstract C).
AMPK inhibitors can be neuroprotective on dopaminergic neurons
BDNF/TrkB signaling endosomes in axons activate CREB and protein synthesis, supporting learning and memory37. Enhancing ERK, CREB, and BDNF expression could protect against cerebral ischemia-reperfusion injury38. Treatment with C.C. improved retrograde transport and dopamine neuron function in rats, with Western blot showing increased Erk and CREB phosphorylation, and higher BDNF and DAT levels (Supplemental Fig. 4A–C). Our earlier research showed that PFFs reduced DA, DOPAC, and HVA in dopamine neurons22. Treatment with C.C increased dopamine and HVA in the SN of PFFs-treated rats as expected (Supplemental Fig. 4D–F). These findings suggest that C.C. may protect neurons by improving endosomal signaling transport and increasing the production of neurotrophic factors and dopamine in dopamine neurons.
PFFs impaired anterograde axonal transport in dopamine neurons by increasing p38 MAPK activity
Previous experiments indicated that abnormal AMPK activation disrupts axonal transport and boosts p38 MAPK activation. We hypothesized that increased p38 MAPK activation impairs axonal transport as well. To test this, rats were divided into three groups: PBS/DMSO, PFFs/DMSO, and PFFs/SB203580, following flowchart C. Western blot analysis revealed that SB203580 increased DAT and TH expression in rat brain tissues (Fig. 8A, B), and significantly elevated Miro1 expression beyond PBS control levels (Fig. 8A, D). Additionally, SB203580 did not inhibit p38 MAPK but significantly reduced MK2 phosphorylation (Fig. 8A, C). Moreover, SB203580 mildly increased kinesin protein levels in the striatum (Fig. 8A, B) by inhibiting p38 MAPK activation, indicating more kinesin transported from the SN.

A–D SB203580 lowered MK2 activation levels, increased kinesin and mitochondrial transport proteins in the striatum. n = 3. E–G Mitochondrial transport was partially restored. Scale bar = 20 μm. n = 10, *p < 0.05, **p < 0.01.
Moreover, SB203580 partially enhanced the anterograde transport of Miro1 (Fig. 8F–H), aligning with Western blot results, but did not impact the retrograde transport of Erk1/2 (Supplemental Fig. 5A–C). Interestingly, there were no differences in fluorescence intensity changes for either motor protein (Supplemental Fig. 5D, G), suggesting that the improved axonal transport with SB203580 was likely not due to changes in protein expression levels but via other ways. To clarify p38 MAPK’s role in axonal transport, Co-IP experiments on motor and cargo proteins were performed. SB203580 increased KLC1-cargo binding and reduced KLC1-p-p38 MAPK interaction, with no effect on DIC-cargo binding (Supplemental Fig. 5E–G). Immunofluorescence co-staining showed that the p38 MAPK inhibitor enhanced motor-cargo co-localization in anterograde but not retrograde transport (Supplemental Fig. 6A–E). In summary, abnormal p38 MAPK activation by PFFs hinders anterograde transport by disrupting kinesin-cargo binding, while its effect on retrograde transport remains uncertain.
The administration of p38 MAPK inhibitor partially ameliorated dopaminergic synapses dysfunction
Previous experiments demonstrated that the p38 MAPK inhibitor enhances axonal transport and prevents cell death in vivo39. To assess its impact on dopamine neurons, we examined fresh dorsolateral striatal tissue from rats using TEM. Treatment with SB203580 partially restored vesicle numbers in the striatum (Supplemental Fig. 6H–I). Normal mitochondrial transport along axons is essential for synaptic terminal function. Enhanced kinesin-based anterograde transport of mitochondria may correlate with restored synaptic function seen in TEM results. SB203580 partially mitigated the loss of DAT and TH due to PFFs but did not completely restore them to control levels (Supplemental Fig. 6F–G). TH fluorescence in the striatum significantly increased, aligning with Western blot findings (Supplemental Fig. 6H, J).
Above all, the PFFs caused abnormal phosphorylation of α-Syn, leading to p38 MAPK overactivation. SB203580 inhibited p38 MAPK activation of downstream, improving transport of mitochondria to the synapse and slowing dopaminergic neuron degeneration (see Graphical abstract D).
Discussion
Our current investigation illustrates that the levels of proteins associated with axonal transport are disrupted by α-Syn* aggregates at an early stage of PD, preceding the significant degeneration of dopamine neurons characteristic of the disease.
Axonal transport plays a crucial role in the maturation and physiological activities of neurons, and its dysfunction is a prominent factor in various neurodegenerative diseases27. The findings of the current investigation demonstrate a notable decrease in the levels of kinesin in the striatum of rats subjected to PFFs. Simultaneously, the mRNA transcript levels of the proteins associated with these three proteins were notably decreased. These results implies that PFFs influence the expression of kinesin, might leading to a decline in kinesin-mediated axonal transport and a substantial build-up in the cytosolic compartment. Kinesin mainly transports organelles like mitochondria in neurons40. Recent studies highlight a complex involving MTX1, MTX2, and Miro1, where MTX1/2 are central and Miro1 is supplementary, aiding kinesin in mitochondrial transport29. Our findings show that MTX1/2 expression remains largely unchanged in neurons. Changes in kinesin levels can reduce axonal transport efficiency, causing decreased mitochondrial transport and abnormal protein localization (Fig. 2). This limited transport may lead to mitochondrial buildup in the cytosol, affecting energy supply to axons and accelerating synaptic degeneration. The reduced affinity of KLC1/2 for Miro1 and MTX2 (Fig. 2) further suggests kinesin’s impaired role in mitochondrial transport.
Our study observed an increase in levels of dynein, along with its activator dynactin and adapter snapin, in the striatum on the focal side of PFFs rats. This finding highlights the competitive nature of transport functions between kinesin and dynein, which primarily occurs through competition for microtubule binding targets12 and tug-of-war dynamics with transported cargoes11. Furthermore, the suppression of kinesin transport by the PFFs may lead to an increase in the expression of dynein, potentially contributing to the improved efficacy of retrograde transport. Previous research suggested that snapin binds to DIC41, aiding in the retrograde transport of TrkB signaling endosomes42. Our study found elevated TrkB levels in the striatum of PFFs rats versus controls. Additionally, PFFs-treated neurons showed increased TrkB retention on neuronal terminal and axon membranes, indicating disrupted internalization and signaling transport. Rab7 is involved in TrkB-mediated endosomal transport and signaling34. A study shows that pErk is often co-transported with TrkB along axons to the soma, aiding extracellular signal transmission35. The accumulation of Rab7 and pErk in the striatum of PFFs rats suggests restricted retrograde transport of pErk in healthy cells. These findings imply that PFFs might affect the retrograde transport of cargoes like TrkB/Erk signaling in neurons. Furthermore, reduced BDNF levels in PD patients may be related to these observations43.
The hypothesis of TrkB/Erk signaling transport defects is challenged by changes in dynein and its associated proteins in the striatum. Immunoprecipitation revealed decreased binding of dynein to dynactin, snapin, TrkB, and pErk in rats exposed to PFFs, along with increased α-Syn* binding to DIC, which may reduce dynein-snapin binding to TrkB/Erk. In summary, PFFs might indirectly influence TrkB/Erk signaling by causing abnormal α-Syn phosphorylation. Increased dynein levels could also lead to α-Syn* buildup and protein degradation in the cytosol.
The aforementioned results showed that PFFs influenced kinesin and dynein transcription and expression, as well as α-Syn* binding to motor proteins, hindering motor-cargo binding. Future research aims to understand the mechanism behind this phenomenon, which is currently unknown. PIKE plays a crucial role in various intracellular processes in the brain44, which is mainly found in nerve endings45. Previous research indicates that α-Syn* inhibits PIKE activity, leading to overactivation of downstream molecules and neuronal death15. AMPK is crucial in the PIKE signaling pathway, inhibiting kinesin transcription in breast cancer46. And the p38 MAPK is important downstream of AMPK47, with α-Syn* activating it and causing mitochondrial dysfunction48. In our investigation, we observed a decrease in PIKE levels and an increase in AMPK and p38 MAPK phosphorylation in response to PFFs. These findings are consistent with prior research and support our initial hypotheses. Furthermore, our Co-IP analysis indicated that PFFs enhanced the interaction between AMPK/p38 MAPK and DIC and KLC1 in cortical neurons. Collectively, our results suggest that the PIKE/AMPK/p38 MAPK pathway may play a crucial role in mediating the effects of PFFs on axonal transport proteins.
Our study showed that PFFs rats had significantly reduced kinesin mRNA, likely due to AMPK activation. Administering the AICAR and C.C. separately to these rats produced opposite effects, confirming this hypothesis. Additionally, C.C. improved dynein cargo binding by reducing AMPK hyperphosphorylation, partially restored TrkB/Erk signaling transport, decreased p38 MAPK phosphorylation, and stabilized kinesin-based mitochondrial transport. The AMPK inhibitor C.C. restored axonal transport in neurons and provided neuroprotection for dopaminergic neurons. Pre-treating with heparin before cerebral ischemia-reperfusion injury increased ERK, CREB, and BDNF expression, contributing to neuroprotection38. In rats, C.C. improved TrkB endosomal signaling and dopamine neuron function. Western blot analysis revealed that C.C. enhanced phosphorylation of Erk and CREB, and increased BDNF and DAT levels (Supplemental Fig. 3). Our earlier study found that PFFs reduced DA, DOPAC, and HVA levels in dopamine neurons22. Treatment with C.C increased DA and HVA levels in the ipsilateral SN of PFFs rats. Moreover, C.C activated CREB, which boosted BDNF expression.
Previous experiments indicated that abnormal AMPK activation impairs axonal transport and increases p38 MAPK activation by PFFs. SB203580 intervention did not significantly alter p38 MAPK phosphorylation, likely due to its mechanism of action, which involves inhibiting MK2 activation rather than directly binding to p38 MAPK’s phosphorylation site49. Western blot analysis showed reduced p-p38 MAPK activation on MK2 in the striatum of PFFs rats treated with SB203580. SB203580 reduced the interaction between KLC1 and p38 MAPK, while increasing the binding of Miro1 and MTX2 to KLC1, enhancing mitochondrial transport in neurons. This improved kinesin transport efficiency and slowed degeneration in dopamine neurons in PFFs rats, likely via better mitochondrial transport. Mitochondria at the presynaptic terminal boost ATP production in response to intracellular Ca2+ changes from action potentials50, facilitating neurotransmitter release and synaptic vesicle recycling. In our experiment, SB203580 partially restored kinesin transport, increasing mitochondria and vesicles in synaptic terminals. This suggests mitochondria might be key for TH-catalyzed dopamine synthesis51. SB203580 also boosted synaptic mitochondria, enhancing DA synthesis and improving rat behavior by raising TH and DAT levels in the striatum.
SB203580 has been shown to inhibit p38 MAPK activity and reduce COX2 protein levels52. Evidence suggests that neuroinflammation and COX-2 upregulation are crucial in degenerative brain diseases53. PFFs can cause astrocyte hyperactivation and neuroinflammation22, so SB203580 may aid kinesin transport and provide a protective effect by inhibiting p38 MAPK.
Currently, there is not enough data to strongly support the therapeutic use of SB203580 in treating PD. Studies have shown some promising effects in preventing cell death39, but its use is limited due to potential side effects and low efficacy in clinical trials. Further research is recommended to study the therapeutic effects of SB203580 on PD, including its specificity on PD pathology, potential cross-reactivity with other cellular components, and any underlying factors affecting drug metabolism.
In summary, we first demonstrated that α-Syn* led to impaired bidirectional axonal transport in neuronal cells, as indicated by decreased binding capacity of kinesin and dynein to cargo and plumbing. We subsequently demonstrated that AMPK/p38 MAPK was the key signaling pathway through which α-Syn* affected axoplasmic transport. Finally, we selected the appropriate AMPK inhibitor and p38 MAPK inhibitor to achieve stable improvement in PFFs rats, which provided a new proof and direction for the etiology and treatment of PD.
Methods
Animals
All animal experiments followed ethical guidelines and were approved by the Institutional Animal Care and Use Committee at Huazhong University of Science and Technology, China (IACUC number: 3859). The 8-week-old male SD rats weighing 220–250 g were obtained from Vital River Laboratory Animal Technology Company for animal experiments. They were housed in a SPF animal laboratory at Huazhong University of Science and Technology with regulated temperature, light cycle, and provided with clean food and water. Bedding and water bottles were replaced weekly. The experiment in vivo process is shown in the Supplemental Fig. 1.
Isolation and culture of primary cortical neurons
Before isolating primary cortical neurons, cell well plates were coated with 0.1 mg/mL poly-L-lysine (Sigma-Aldrich, P1399) for at least 30 min, washed twice with sterile water, and dried. C57BL/6 embryos aged 16–18 days were used54. Fetal mouse cortices were isolated in a biosafety cabinet using sterile tubes, mechanically separated, and centrifuged at 300 × g for 3 min to reduce enzymatic digestion damage. The sediments were resuspended in a medium containing 39% DMEM/F12 (Gibco, 11330032), 10% FBS (Gibco, 10099141), and 1% penicillin/streptomycin (Gibco, 15140122) and incubated at 37 °C with 5% CO2. After 4 h, the medium was replaced with a neuronal growth medium consisting of 96% Neurobasal (Gibco, 10888022), 2% B-27 Supplement (Gibco, 17504044), 1% penicillin/streptomycin, and 1% GlutaMAX (Gibco, 35050061). The cell culture medium was changed at least every 3 days, with daily axon growth assessments via optical microscope. The in vitro experiment process is illustrated in the Supplemental Fig. 2.
PFFs purification and preparation
To facilitate the expression of protein monomers in Escherichia coli BL21, plasmids were individually constructed utilizing human-derived α-SynN103 cDNA and tauN368 cDNA and verified via enzyme digestion. IPTG induced the expression of α-SynN103 and tauN368 monomer, which were purified using nickel column chromatography. The monomers were stored at 3 mg/mL in PBS, then incubated at 37 °C and 1000 rpm for 7 days to form fibrils before being stored at −80 °C for later use22.
Use of in vivo and in vitro models for investigation
Modeling was conducted utilizing Sprague-Dawley rats selected by the researchers as outlined above. Following anesthesia with sodium pentobarbital (supplied by the affiliated hospital experimental platform), the rats were secured on a digital brain stereotaxic apparatus to verify the correct striatal position at the coordinates: anteroposterior +1.2 mm, dorsoventral −4.0 mm, and mediolateral −2.4 mm. Consistent with previous experimental findings, the rats were injected with 15 μg of PFFs or phosphate buffered saline (PBS) and divided into two groups, PFFs and sham22. Following the modeling procedure, the rats were placed on a thermostatic pad set at 37 °C to mitigate the risk of hypothermia. Subsequently, they were housed in the SPF animal laboratory for a period of 2 months and subsequently underwent a series of behavioral and biochemical assessments. At 5 days in vitro (DIV), discernible axons were observed in primary cortical neurons. Following this observation, PFFs (2 μg/mL) were introduced into the medium for a duration of 48 h, after which biochemical tests were conducted55.
Imaging of axonal transport in living neurons
Cultured primary cortical neurons were individually transfected with lentiviruses overexpressing miro1-cherry and ERK-GFP, obtained from Shanghai Genechem Company, two days prior to the introduction of PFFs. Following a 48-h incubation period with PFFs, live imaging of the neurons was conducted at 5-s intervals for a total of 120 s using a confocal laser scanning microscope (Olympus, FV3000). Subsequently, the stacks of fluorescence images were resliced using ImageJ software to generate gray-scale images depicting the motion of particles within the axon for trajectory analysis. The speed of motion of fluorescent molecules can be evaluated by measuring the angle between the vertical line and the diagonal line of molecule motion on the gray-scale image.
Treatment in both in vitro and in vivo settings
After a 2-month period of feeding in the SPF animal laboratory, the rats were allocated into three groups for experimentation: a negative control group (PBS + DMSO), a positive control group (15 μg PFFs+DMSO), and a drug-treated group (15 μg PFFs+drugs). In vitro experiments were organized in a similar manner. The treatment drugs utilized in this study, in accordance with our experimental design, included AICAR (AMPK activator, MedChemExpress, HY-13417), Compound C (AMPK inhibitor, MedChemExpress, HY-13418A), and SB203580 (p38 MAPK inhibitor, MedChemExpress, HY-10256), with DMSO serving as a solvent for drug dissolution. In vivo experiments involved the daily intraperitoneal administration of the three drugs individually within an equivalent volume of solvent over a period of 14 days. The injection concentrations utilized were as follows: AICAR (1 mg/mL)56, Compound C (0.2 mg/mL)57, and SB203580 (1 mg/mL)58. In vitro experiments included the addition of two inhibitors, Compound C (1 μM)59 and SB 203580 (5 μM)60, were added to the medium separately and treated for 24 h.
Behavioral tests
The four behavioral tests are briefly described. Adjusting Steps Test: Rats were held by their tails and hind limbs were pinched to move them away from the table while keeping their upper limbs touching the table. Rats were then moved forward at a steady rate, recording the number of adjusted steps taken by both affected and healthy limbs61. Balance Beam Test: Rats are placed on a wooden beam and their motor coordination and balance are assessed by measuring the time taken to reach a cage and the number of times they slide off. Cylinder Test: Rats explore the walls of an upright transparent cylinder with their forelimbs. Rats were observed in an open field for 5 min to track their movements. The number of times each rat’s affected forelimb, healthy forelimb, and both forelimbs touched the wall were counted separately. The total moving distance, time spent in the central area, and time spent motionless were recorded. Tests should be done in a quiet space, and any waste should be cleaned up after each recording62. The behavioral tests for each rat were recorded on video, followed by a detailed count and analysis conducted through slow-motion playback.
Western blot
Fresh brain tissues were lysed with pre-cooled RIPA lysate (Servicebio, G2003) containing PMSF (Servicebio, G2008), phosphorylated protease inhibitor (Servicebio, G2007), and cocktail protease inhibitor (Servicebio, G2006), then centrifuged to extract the protein supernatant. Equal amounts of protein were analyzed using PAGE Gel Rapid Preparation Kit (Epizyme Biotech, PG111-113), transferred to PVDF membranes (Millipore), blocked with quick blocking buffer (Newcells Biotech, P30500), and incubated with primary and secondary antibodies. Finally, the blot was developed using chemiluminescent substrate (Abbkine, BMU102) and exposed using GeneGnome XRQ-NPC (SYNGENE, UK). Images were captured with GeneSys intelligent control image acquisition software and analyzed with Image J. The antibodies used in western blot were as follows: KIF5B (Abcam, ab5629), KLC1 (Abcam, ab174273), KLC2 (Proteintech, 17668), Miro1 (ABclonal, A5838), MTX1 (Proteintech, 11529), MTX2 (Proteintech, 11610), Dynactin (ABclonal, A1783), DIC (Abcam, ab171964), snapin (Proteintech, 10055), TrkB (CST, Q16620), BDNF (Proteintech, 66292), Erk1/2 (Proteintech, 66192), pErk1/2 (ABclonal, AP0974), Rab7 (ABclonal, A12784), PIKE (Proteintech, 14690), AMPK (CST, 5831S), p-AMPK (CST, 2535S), p38 (CST, 8690S), p-p38 (CST, 4511S), α-synuclein (Abcam, ab212184), α-synuclein pSer129 (Abcam, ab51253), TH (Abcam, ab75875), DAT (Proteintech, 22525), MK2 (ABclonal, A10668), CREB (ABclonal, A11989), p-CREB (ABclonal, AP0903). The above primary antibodies dilution is 1:1000. Internal reference antibody including GAPDH (Abbkine, A01020) and β-tubulin (Abbkine, A01030) with dilution 1:2000. The secondary antibodies are the corresponding-specie HRP-conjugated antibodies with dilution 1:10,000.
Immunohistochemistry
Brain tissues were cut into 4-μm sections and preserved using paraffin. The sections were rehydrated, treated with citrate solution (pH = 6.0) for antigen recovery, washed with PBS, blocked with BSA, and incubated with primary and secondary antibodies. They were stained with DAB for 5 min, followed by Mayer’s hematoxylin (Absin, abs9215). Sections were washed with PBS three times after each step, imaged with an Olympus VS120 digital scanner, and analyzed with Image J. Three rats were randomly selected from each group for analysis. For each rat, five consecutive tissue sections were examined, and data were averaged from five identical fields of view per section to ensure consistency in the calculations. Antibodies used in western blotting were α-synuclein pSer129 (Abcam, ab51253) and TH (Abcam, ab75875), both diluted at 1:1000, with corresponding-species HRP-conjugated secondary antibodies also diluted at 1:1000.
Immunofluorescence
Brain tissues were sliced into 4-μm sections and preserved via the Frozen-section method. These sections were kept at room temperature for 20 min before blocking. Primary neurons were cultured in 24-well plates with cell slides and directly blocked. Secondary antibody incubation was done in a light-protected environment at room temperature for 1 h. The sections were then incubated with DAPI (Solarbio, 28718) for 5 min and mounted using antifade medium (Solarbio, S2100). Brain sections were also scanned with VS120. Cell slides were photographed with FV3000. Finally, the data is analyzed with Image J. The antibodies utilized in the western blot analysis included α-synuclein pSer129 (Abcam, ab51253, 1:1000), TH (Proteintech, 66334, 1:200), Lamp1 (CST, 9091, 1:400), KLC1 (Abcam, ab174273, 1:200), Miro1 (ABclonal, 1:400), KIF5B (Abcam, ab5629, 1:500), α-Tubulin (SANTA CRUZ, sc-8035, 1:500), DIC (Abcam, ab171964, 1:100), TrkB (Proteintech, 13129, 1:100), Rab7 (ABclonal, A12784, 1:200), DAT (Proteintech, 22525, 1:200), p-Erk1/2 (ABclonal, 1:200). Fluorescent secondary antibodies used were Alexa-fluor488, Alexa-fluor594, or cy3 (Abbkine) with dilution 1:250.
Transmission electron microscopy
Each 20 µl sample was pipetted onto a carbon-coated copper grid that had been discharged for a duration of 3 min, after which 2% phosphotungstic acid was added for 2 min. Any remaining liquid was absorbed using filter paper. It is important to ensure the grid remains dry prior to imaging. Subsequently, images were obtained and digitized utilizing a transmission electron microscope (HITACHI, HT7800/HT7700).
Real-time quantitative PCR analysis
Total RNA was isolated using the FastPure Cell/Tissue Total RNA Isolation Kit (Vazyme, RC112-01), and its concentration was measured with a Nanodrop One UV spectrophotometer (ThermoFisher, 840-317500). Subsequently, the RNA was reverse transcribed into cDNA using a cDNA synthesis kit (Vazyme, R223-01), and all amplifications were conducted on a StepOnePlus™ Real-Time Fluorescent Quantitative PCR System (ThermoFisher, 4376600). Relative mRNA expression levels were determined utilizing the comparative Ct method (2-∆∆Ct). The primer sequence is shown in the Supplementary raw data.
High performance liquid chromatography with electrochemical detection
Each 20 mg of fresh striatal tissues was homogenized in ice-cold precipitation solution containing 0.1 M perchloric acid and 0.1% L-cysteine. The supernatant was collected after centrifugation, and mixed standards of the targets were prepared. Samples were separated on a Spherisorb ODS1 column (5 μm, 4.6 mm × 250 mm, Waters, USA), and the biogenic amines tested included Norepinephrine (NE), 3,4-dihydroxyphenylacetic acid (DOPAC), dopamine (DA), Hom vanillic acid (HVA), and 5-hydroxytryptamine (5-HT)22. The biogenic amines were bought from Sigma-Aldrich and detected using an electrochemical detector from Hewlett-Packard. The mobile phase consisted of methanol and a buffer solution. Results were reported in ng/mg wet weight.
Co-immunoprecipitation
Fresh rat brain tissues from the striatum and substantia nigra were lysed using a preconfigured buffer with various inhibitors (same as western blot did) and centrifuged to extract the protein supernatant. For the precipitating antibody group, 1 μg of antibody was added per 2000 μg of protein, while the IgG control group received 1 μg of IgG antibody (Proteintech, B900610 and B900620). Both groups were incubated on a shaker at 4 °C for over 16 h. Input group proteins were denatured with 5X protein uploading buffer (Epizyme, LT101S) and stored at −20 °C. Protein L agarose beads (Millipore, P3351) were washed three times with cold PBS and resuspended in PBS to a 50% volume ratio. Each 40 μl of protein L solution was added to the primary antibody incubation solution and incubated at 4 °C for 4 h. The precipitates were collected by centrifugation at 500 × g, 4 °C for 30 s, washed three times with cold IP lysate, and then denatured in a protein metal bath with 2X protein uploading buffer. The subsequent experimental procedure is identical to western blot.
Statistics analysis
Data analysis was conducted using GraphPad Prism 9.0 software. Normality tests were performed with D’Agostino and Pearson tests. Student’s t-test compared two groups, while one-way ANOVA compared three groups. Brown-Forsythe and Tukey tests were used for further analysis between two groups. A p-value < 0.05 was considered significant.
Data availability
Data is provided within the supplementary information files. Requests for further information and resources should be directed to and will be fulfilled by the lead contact, Xuebing Cao (caoxuebing@126.com).
Abbreviations
- 3,4-dihydroxyphenylacetic acid:
-
DOPAC
- 5-Aminoimidazole-4-carboxamide1-β-D-ribofuranoside:
-
AICAR
- 5-hydroxytryptamine:
-
5-HT
- Adenosine 5‘-monophosphate-activated protein kinase:
-
AMPK
- Amyloid Precursor Protein:
-
APP
- Asparagine endopeptidase:
-
AEP
- brain-derived neurotrophic factor:
-
BDNF
- cAMP-response element binding protein:
-
CREB
- Compound C:
-
C.C
- days in vitro:
-
DIV
- dopamine:
-
DA
- Dopamine Transporter:
-
DAT
- dynein intermediate chain:
-
DIC
- extracellular regulated protein kinases:
-
Erk
- Hom vanillic acid:
-
HVA
- kinesin heavy chain:
-
KHC
- kinesin light chain 1/2:
-
KLC1/2
- Lewy bodies:
-
LBs
- Lewy neurites:
-
LNs
- Metaxin 1/2:
-
MTX1/2
- microtubule-associated proteins light chain 3:
-
LC3
- Mitochondrial Rho GTPase 1:
-
Miro1
- mitogen-activated protein kinase-activated protein kinase 2:
-
MK2
- Norepinephrine:
-
NE
- p38 mitogen-activated protein kinase:
-
p38 MAPK
- Parkinson’s disease:
-
PD
- Pathological α-synuclein:
-
α-Syn*
- phosphate buffered saline:
-
PBS
- phosphoinositide-3 kinase enhancer:
-
PIKE
- phospho-S129 α-Syn:
-
pS129
- preformed fibrils complexes of α-SynN103 and tauN368:
-
PFFs
- Ras-related GTP binding protein 7:
-
Rab7
- substantia nigra:
-
SN
- substantia nigra pars compacta:
-
SNpc
- Superoxide Dismutase 1:
-
SOD1
- tyrosine hydroxylase:
-
TH
- Tyrosine Kinase receptor B:
-
TrkB
- α-synuclein:
-
α-Syn
References
Belur, N. R., Bustos, B. I., Lubbe, S. J. & Mazzulli, J. R. Nuclear aggregates of NONO/SFPQ and A-to-I-edited RNA in Parkinson’s disease and dementia with Lewy bodies. Neuron 112, 2558–2580 (2024).
Wong, Y. C. & Krainc, D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat. Med. 23, 1–13 (2017).
Berth, S. H. & Lloyd, T. E. Disruption of axonal transport in neurodegeneration. J. Clin. Invest 133, e168554 (2023).
Wang, W., Zhao, F., Ma, X., Perry, G. & Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol. Neurodegener. 15, 30 (2020).
Baron, D. M. et al. ALS-associated KIF5A mutations abolish autoinhibition resulting in a toxic gain of function. Cell Rep. 39, 110598 (2022).
Sawant, N., Morton, H., Kshirsagar, S., Reddy, A. P. & Reddy, P. H. Mitochondrial abnormalities and synaptic damage in Huntington’s Disease: a focus on defective mitophagy and mitochondria-targeted therapeutics. Mol. Neurobiol. 58, 6350–6377 (2021).
Prots, I. et al. α-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J. Biol. Chem. 288, 21742–21754 (2013).
Volpicelli-Daley, L. A. et al. Formation of α-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol. Biol. Cell 25, 4010–4023 (2014).
Chu, Y. et al. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain 135, 2058–2073 (2012).
Chung, C. Y., Koprich, J. B., Siddiqi, H. & Isacson, O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J. Neurosci. 29, 3365–3373 (2009).
Mizuno, N. et al. Dynein and kinesin share an overlapping microtubule-binding site. EMBO J. 23, 2459–2467 (2004).
Belyy, V. et al. The mammalian dynein-dynactin complex is a strong opponent to kinesin in a tug-of-war competition. Nat. Cell Biol. 18, 1018–1024 (2016).
Canty, J. T. & Yildiz, A. Activation and regulation of cytoplasmic dynein. Trends Biochem Sci. 45, 440–453 (2020).
Shen, C. et al. Dynactin is involved in Lewy body pathology. Neuropathology 38, 583–590 (2018).
Kang, S. S. et al. α-Synuclein binds and sequesters PIKE-L into Lewy bodies, triggering dopaminergic cell death via AMPK hyperactivation. Proc. Natl Acad. Sci. USA 114, 1183–1188 (2017).
Kang, S. S. et al. TrkB neurotrophic activities are blocked by α-synuclein, triggering dopaminergic cell death in Parkinson’s disease. Proc. Natl Acad. Sci. USA 114, 10773–10778 (2017).
Lie, P. P. Y. & Nixon, R. A. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis. 122, 94–105 (2019).
Kang, S. S. et al. α-Synuclein stimulation of monoamine oxidase-B and legumain protease mediates the pathology of Parkinson’s disease. EMBO J. 37, e98878 (2018).
Wang, H. et al. C/EBPβ/AEP is age-dependently activated in Parkinson’s disease and mediates α-synuclein in the gut and brain. NPJ Parkinsons Dis. 9, 1 (2023).
Xiang, J. et al. Gut-induced alpha-Synuclein and Tau propagation initiate Parkinson’s and Alzheimer’s disease co-pathology and behavior impairments. Neuron 112, 3585–3601.e5 (2024).
Ahn, E. H. et al. Initiation of Parkinson’s disease from gut to brain by δ-secretase. Cell Res. 30, 70–87 (2020).
Yang, X. et al. Time-dependent alterations in the rat nigrostriatal system after intrastriatal injection of fibrils formed by α-Syn and tau fragments. Front Aging Neurosci. 14, 1049418 (2022).
Praschberger, R. et al. Neuronal identity defines α-synuclein and tau toxicity. Neuron 111, 1577–1590 (2023).
Ramalingam, N. et al. Dynamic physiological α-synuclein S129 phosphorylation is driven by neuronal activity. NPJ Parkinsons Dis. 9, 4 (2023).
Cheng, X. T. et al. Revisiting LAMP1 as a marker for degradative autophagy-lysosomal organelles in the nervous system. Autophagy 14, 1472–1474 (2018).
Cheng, X. T. et al. Characterization of LAMP1-labeled nondegradative lysosomal and endocytic compartments in neurons. J. Cell Biol. 217, 3127–3139 (2018).
Yang, X., Ma, Z., Lian, P., Xu, Y. & Cao, X. Common mechanisms underlying axonal transport deficits in neurodegenerative diseases: a mini review. Front Mol. Neurosci. 16, 1172197 (2023).
Panchal, K. & Tiwari, A. K. Miro (Mitochondrial Rho GTPase), a key player of mitochondrial axonal transport and mitochondrial dynamics in neurodegenerative diseases. Mitochondrion 56, 118–135 (2021).
Zhao, Y. et al. Metaxins are core components of mitochondrial transport adaptor complexes. Nat. Commun. 12, 83 (2021).
Canty, J. T., Tan, R., Kusakci, E., Fernandes, J. & Yildiz, A. Structure and mechanics of dynein motors. Annu Rev. Biophys. 50, 549–574 (2021).
Chaaban, S. & Carter, A. P. Publisher Correction: Structure of dynein-dynactin on microtubules shows tandem adaptor binding. Nature 611, E8 (2022).
Lee, Y. H. et al. DYRK3 phosphorylates SNAPIN to regulate axonal retrograde transport and neurotransmitter release. Cell Death Discov. 8, 503 (2022).
Barford, K., Deppmann, C. & Winckler, B. The neurotrophin receptor signaling endosome: where trafficking meets signaling. Dev. Neurobiol. 77, 405–418 (2017).
Deinhardt, K. et al. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 52, 293–305 (2006).
Valdez, G. et al. Pincher-mediated macroendocytosis underlies retrograde signaling by neurotrophin receptors. J. Neurosci. 25, 5236–5247 (2005).
Yap, C. C., Digilio, L., McMahon, L. P., Wang, T. & Winckler, B. Dynein is required for Rab7-dependent endosome maturation, retrograde dendritic transport, and degradation. J. Neurosci. 42, 4415–4434 (2022).
Moya-Alvarado, G. et al. BDNF/TrkB signaling endosomes in axons coordinate CREB/mTOR activation and protein synthesis in the cell body to induce dendritic growth in cortical neurons. Elife 12, e77455 (2023).
Liu, W. et al. The ERK/CREB/PTN/syndecan-3 pathway involves in heparin-mediated neuro-protection and neuro-regeneration against cerebral ischemia-reperfusion injury following cardiac arrest. Int Immunopharmacol. 98, 107689 (2021).
Kheiri, G., Dolatshahi, M., Rahmani, F. & Rezaei, N. Role of p38/MAPKs in Alzheimer’s disease: implications for amyloid beta toxicity targeted therapy. Rev. Neurosci. 30, 9–30 (2018).
Guedes-Dias, P. & Holzbaur, E. L. F. Axonal transport: driving synaptic function. Science 366, eaaw9997 (2019).
Cai, Q. et al. Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron 68, 73–86 (2010).
Mitchell, D. J. et al. Trk activation of the ERK1/2 kinase pathway stimulates intermediate chain phosphorylation and recruits cytoplasmic dynein to signaling endosomes for retrograde axonal transport. J. Neurosci. 32, 15495–15510 (2012).
Li, R. et al. Serum BDNF levels are involved in the diagnosis and treatment response in patients with PD. J. Affect Disord. 327, 31–37 (2023).
Chan, C. B. & Ye, K. Multiple functions of phosphoinositide-3 kinase enhancer (PIKE). ScientificWorldJournal 10, 613–623 (2010).
Ye, K. et al. Phospholipase C gamma 1 is a physiological guanine nucleotide exchange factor for the nuclear GTPase PIKE. Nature 415, 541–544 (2002).
Zhang, S. et al. Fyn-phosphorylated PIKE-A binds and inhibits AMPK signaling, blocking its tumor suppressive activity. Cell Death Differ. 23, 52–63 (2016).
Gu, X. et al. AMPK/SIRT1/p38 MAPK signaling pathway regulates alcohol‑induced neurodegeneration by resveratrol. Mol. Med. Rep. 17, 5402–5408 (2018).
Wiesen, T. & Atlas, D. Novel anti-apoptotic L-DOPA precursors SuperDopa and SuperDopamide as potential neuroprotective agents for halting/delaying progression of Parkinson’s disease. Cell Death Dis. 13, 227 (2022).
Alimbetov, D. et al. Small molecule targeting of the p38/Mk2 stress signaling pathways to improve cancer treatment. BMC Cancer 23, 895 (2023).
Plotegher, N. Physiological roles of organelles at the pre-synapse in neurons. Int J. Biochem Cell Biol. 154, 106345 (2023).
Wang, J., Lou, H., Pedersen, C. J., Smith, A. D. & Perez, R. G. 14-3-3zeta contributes to tyrosine hydroxylase activity in MN9D cells: localization of dopamine regulatory proteins to mitochondria. J. Biol. Chem. 284, 14011–14019 (2009).
Zhou, Y. et al. Reduced nicotinamide adenine dinucleotide phosphate inhibits MPTP-induced neuroinflammation and neurotoxicity. Neuroscience 391, 140–153 (2018).
Prabhakaran, J., Molotkov, A., Mintz, A. & Mann, J. J. Progress in PET imaging of neuroinflammation targeting COX-2 enzyme. Molecules 26, 3208 (2021).
Watters, O., Connolly, N. M. C., König, H. G., Düssmann, H. & Prehn, J. H. M. AMPK Preferentially depresses retrograde transport of axonal mitochondria during localized nutrient deprivation. J. Neurosci. 40, 4798–4812 (2020).
Mao, X. et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353, aah3374 (2016).
Yang, H. et al. AMPK activation attenuates cancer-induced bone pain by reducing mitochondrial dysfunction-mediated neuroinflammation. Acta Biochim. Biophys. Sin.55, 460–471 (2023).
Guo, Y. et al. AMPK inhibition blocks ROS-NFκB signaling and attenuates endotoxemia-induced liver injury. PLoS ONE 9, e86881 (2014).
Yan, W. et al. SB203580 inhibits epithelial-mesenchymal transition and pulmonary fibrosis in a rat silicosis model. Toxicol. Lett. 259, 28–34 (2016).
Liu, M. L., et al. Small molecules enable neurogenin 2 to efficiently convert human fibroblasts into cholinergic neurons. Nat. Commun. 4, 2183 (2013).
Davies, S. P., Reddy, H., Caivano, M. & Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105 (2000).
Salaramoli, S., Joshaghani, H. R., Hosseini, M. & Hashemy, S. I. Therapeutic effects of selenium on alpha-synuclein accumulation in substantia Nigra pars compacta in a rat model of Parkinson’s disease: behavioral and biochemical outcomes. Biol. Trace Elem. Res 202, 1115–1125 (2024).
Miyanishi, K. et al. Behavioral tests predicting striatal dopamine level in a rat hemi-Parkinson’s disease model. Neurochem. Int. 122, 38–46 (2019).
Acknowledgements
We sincerely thank all participants for their participation in this study. We thank the Medical sub-center of Huazhong University of Science & Technology Analytical & Testing Center for technical support. This work was supported by the National Natural Science Foundation of China (No. 81974200 and No. 82371270).
Author information
Authors and Affiliations
Contributions
Conceptualization, X.C., Y.X., and X.Y; methodology, X.Y. and Z.M.; investigation, X.Y., P.L., and Z.M.; formal analysis, Y.W. and X.Y.; resources, Z.M., P.L., Y.W., K.L., Z.Z., and Z.T.; writing—original draft, X.Y. and Z.M.; writing—review & editing, Z.M. and X.Y.; visualization, X.C., X.Y., and Y.X.; supervision, X.Y. and X.C.; funding acquisition, X.C. and Y.X. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, X., Ma, Z., Lian, P. et al. Disruption of axonal transport in Parkinson’s disease: the role of pathological α-Syn and AMPK/p38 MAPK signaling. npj Parkinsons Dis. 11, 114 (2025). https://doi.org/10.1038/s41531-025-00926-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-025-00926-z
