
Abstract
Although respiratory failure and hypoxaemia are the main manifestations of COVID-19, kidney involvement is also common. Available evidence supports a number of potential pathophysiological pathways through which acute kidney injury (AKI) can develop in the context of SARS-CoV-2 infection. Histopathological findings have highlighted both similarities and differences between AKI in patients with COVID-19 and in those with AKI in non-COVID-related sepsis. Acute tubular injury is common, although it is often mild, despite markedly reduced kidney function. Systemic haemodynamic instability very likely contributes to tubular injury. Despite descriptions of COVID-19 as a cytokine storm syndrome, levels of circulating cytokines are often lower in patients with COVID-19 than in patients with acute respiratory distress syndrome with causes other than COVID-19. Tissue inflammation and local immune cell infiltration have been repeatedly observed and might have a critical role in kidney injury, as might endothelial injury and microvascular thrombi. Findings of high viral load in patients who have died with AKI suggest a contribution of viral invasion in the kidneys, although the issue of renal tropism remains controversial. An impaired type I interferon response has also been reported in patients with severe COVID-19. In light of these observations, the potential pathophysiological mechanisms of COVID-19-associated AKI may provide insights into therapeutic strategies.
Key points
-
Over a quarter of patients hospitalized with coronavirus disease 2019 (COVID-19) have been reported to develop acute kidney injury (AKI).
-
Low molecular weight proteinuria, Fanconi syndrome and histological findings point towards tubular injury.
-
Analyses of kidney biopsy samples from patients with COVID-19 and AKI have inconsistently reported viral infection of kidney cells.
-
Collapsing glomerulopathy has been identified in patients with high-risk APOL1 genotypes, mostly in those without severe respiratory symptoms.
-
Regional inflammation, endothelial injury and renal microthrombi have been reported but their implication in the pathogenesis of COVID-associated AKI remains uncertain.
-
Anti-inflammatory drugs (for example, steroids and IL-6 receptor blockers) seem to limit the development of severe AKI in patients with COVID-19.
Similar content being viewed by others

Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first described in December 2019 and is responsible for coronavirus disease 2019 (COVID-19) and the current global pandemic. The pulmonary manifestations of COVID-19 are most prominent, but acute kidney injury (AKI) is also now recognized as a common complication of the disease, and is often evident at hospital admission. Although initial reports from China suggested relatively low rates of kidney involvement1,2,3,4, subsequent reports from the USA and Europe indicate much higher rates of AKI, particularly in the intensive care setting, with up to 45% of patients in the intensive care unit (ICU) requiring kidney replacement therapy (KRT)5,6,7,8. Mortality among hospitalized patients with COVID-19-associated AKI (COVID-19 AKI) is higher than for those without kidney involvement8,9. As with all instances of AKI in the context of multi-organ failure requiring ICU admission, mortality among patients admitted to the ICU with COVID-19 AKI requiring KRT is especially high10. Anecdotal reports of a lack of renal recovery in those who survive relative to that reported for other forms of AKI is of particular concern7,9,10. However, long-term patient outcomes are not yet fully understood as they are complicated by prolonged hospital admissions and lack of reported follow-up. Ascertaining the true epidemiology of COVID-19 AKI is difficult owing to differences in the underlying comorbidities of the populations examined, as well as possible variations in the practice and methods of AKI diagnosis and reporting. Age, history of hypertension and diabetes mellitus have been repeatedly associated with a higher risk of AKI in patients with COVID-19. Chronic kidney disease (CKD) is a well identified risk factor for AKI in hospitalized patients, and was indicated to be the most relevant risk factor for AKI requiring KRT in 3,099 critically ill patients with COVID-19 (ref.9). Indeed, several epidemiological studies have clearly demonstrated that CKD represents a relevant and independent risk factor for worse outcomes in COVID-19. A 2021 case–control study that compared patients with COVID-19 with the Danish general population matched for age, gender and comorbidities identified an association between lower estimated glomerular filtration rate (eGFR) and rate of hospital-diagnosed COVID-19 and death10. An OpenSAFELY analysis of variables associated with COVID-19-associated death in ~17 million patients identified CKD as one of the most prevalent comorbidities associated with mortality (HR 2.52 for patients with eGFR <30 ml/min/1.73 m2)11. Moreover, CKD is often associated with other comorbidities such as diabetes mellitus, hypertension and obesity, which have also been linked to mortality in patients with COVID-19 (ref.3). In this clinical scenario, the high mortality observed in comorbid and elderly patients may be related to a reduction in renal functional reserve (RFR), an impaired capacity of the kidney to increase GFR in response to stress and reduced functioning nephron mass12.
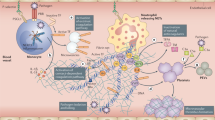
Decreased GFR and RFR levels may also support the development of AKI, as suggested by epidemiological studies. In a study of 4,020 consecutively hospitalized patients with COVID-19 in Wuhan, China, 285 (7.09%) were identified as having AKI. Both early and late forms of AKI (that is, AKI at presentation and AKI developing after presentation) were associated with an increased risk of in-hospital mortality. Moreover, CKD, older age and levels of inflammatory biomarkers were associated with an increased risk of late AKI13. In another study of 1,603 patients consecutively admitted to a university reference hospital in Spain, 21.0% of patients demonstrated elevated serum creatinine levels at admission, of whom 43.5% had previous CKD; 11.4% of patients with normal serum creatinine level at admission developed AKI14. In yet another study of 777 patients hospitalized in Genoa, Italy, 176 (22.6%) developed AKI; of these, 79 (45%) showed an acute worsening of pre-existing CKD, and 21 (12%) required KRT. Independent variables for AKI development were the presence of CKD, C-reactive protein level and requirement for ventilatory support15. Nevertheless, it is clear that the pathophysiology is multifactorial and different sub-phenotypes of COVID-19 AKI exist. In this Review, we discuss current understanding of the pathophysiology of COVID-19 AKI, examining potential mechanisms by which SARS-CoV-2 infection might induce direct and indirect effects on the kidney, and factors that are not specific to COVID-19 but may influence kidney function through haemodynamic changes and/or organ crosstalk (Fig. 1).

Coronavirus disease 2019 (COVID-19)-associated acute respiratory distress syndrome involves regional inflammation with the recruitment of immune cells, including macrophages, effector T cells and polymorphonuclear neutrophils. Cytokines are released locally within the lung in response to damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) and contribute to the further recruitment of inflammatory cells and tissue damage. Secretion of interferon (IFN) from immune cells contributes to viral clearance. Neutrophil extracellular traps (NETs), released by activated neutrophils, may also contribute to the local inflammatory response, pathogen clearance and thrombosis. Acute respiratory distress syndrome likely contributes to the development of acute kidney injury through systemic processes (for example, venous congestion and decreased cardiac output as a consequence of right-sided heart failure, high levels of intrathoracic pressure and hypoxia). Increased renal interstitial pressure due to tissue oedema is also likely to contribute to tubular injury. Release of DAMPs and PAMPs into the circulation contributes to regional inflammation within the kidney, the immune response and immune-mediated thrombosis. Direct infection of kidney cells has been observed in some patients and may also contribute to local inflammation and kidney damage. Conversely, acute kidney injury in other settings has been shown to contribute to promoting lung injury by stimulating regional inflammation, lung capillary permeability and fluid overload. DC, dendritic cell; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Features of COVID-19 AKI
Epidemiology
The reported incidence and severity of AKI in the setting of COVID-19 depends on the clinical setting and definitions used. Most studies have used the Kidney Disease Improving Global Outcomes (KDIGO) consensus definition of AKI and several studies that have used this definition have reported that upwards of 30–50% of hospitalized patients with COVID-19 develop some form of AKI, with the proportion increasing in those requiring intensive care3,7,9,12,13. According to one meta-analysis from 2020, the pooled incidence of AKI among hospitalized patients with COVID-19 was 28.6% (95% CI 19.8–39.5) in the USA and Europe and 5.5% (95% CI 4.1–7.4) in China16. Worldwide, among patients admitted to the ICU, an estimated 29% have AKI; this proportion is up to 78% in those requiring intubation17. Other studies have reported that up to 20% of patients in the ICU required KRT18,19,20,21,22. In a large-scale retrospective observational cohort of a New York City health-care system, 46% of 3,993 inpatients developed AKI with 39%, 19% and 42% having KDIGO stage 1, 2 and 3 AKI, respectively19. These data are mirrored in a separate large cohort of New York-based patients, of whom 3,854 (39.9%) had inpatient COVID-19 AKI, with 42.7%, 21.8% and 35.5% having stages 1, 2 and 3 AKI, respectively8. In this second cohort, which also included both on-ward and ICU patients, 638 of the 1,370 patients with stage 3 AKI (46.5%, or 16.6% of the total) received KRT. Importantly, both of these cohort studies are limited in that they only used the serum creatinine criteria of the KDIGO consensus definitions to identify those with AKI. Of note, wide geographical disparities in the incidence of AKI among US veteran patients hospitalized with COVID-19 have been reported, ranging from 10% to 56%23. This finding, combined with evidence that rates of COVID-19 AKI have declined over time (from 40% in March 2020 to 27% in July 2020)23 with similar findings reported in a New York study24, suggests that changes in patient management have had a positive impact on kidney outcomes and the incidence of AKI among patients with COVID-19.
Clinical features
Early reports of COVID-19 AKI noted the presence of haematuria and/or proteinuria1,18. In one cohort study of 701 patients with COVID-19, 44% and 26% of patients presented with proteinuria and haematuria, respectively2; severity of haematuria or proteinuria (2–3+ on dipstick) was associated with the risk of hospital mortality in a step-wise manner2,18. A more recent cohort study demonstrated much higher rates of proteinuria (defined as a protein-to-creatinine ratio of >0.5, 1+ or higher on dipstick or > 30 mg/dl on urinalysis) and haematuria (defined as 1+ or higher on dipstick or urinalysis), in 80% of patients with COVID-19 AKI19. Furthermore, >50% of patients without AKI as defined by KDIGO serum creatinine criteria had haematuria and over 70% presented with proteinuria. The presence of urinalysis abnormalities in those not meeting the definition of AKI suggests the existence of kidney injury without notable acute changes in kidney function. Fanconi syndrome (characterized by proteinuria, renal phosphate leak, hyperuricosuria and normo-glycaemic glycosuria) has been reported to precede episodes of AKI25 (Fig. 2). This presentation is in keeping with stage 1S of new recommendations for AKI staging, when evidence of kidney injury exists that is not detected by creatinine and urine output criteria26.

Proteinuria and/or haematuria is indicative of kidney injury, even in the absence of a rise in serum creatinine (SCr) level or a drop in glomerular filtration rate (GFR). Further injury is associated with a drop in GFR and rise in SCr. Underlying chronic kidney disease or factors such as ageing limits the baseline functional reserve and can precipitate the development of acute kidney injury (AKI). Kidney replacement therapy (KRT) is required for severe cases of AKI. COVID-19, coronavirus disease 2019.
The proteinuria detected in patients with COVID-19 AKI is of low molecular weight rather than albuminuria, suggesting a tubular origin rather than glomerular injury, and can be used to identify patients with early-stage AKI27. The contribution of underlying CKD is unknown, but the proportion of patients with COVID-19 and proteinuria far exceeds the prevalence of stage 1 and 2 CKD in the general population28. Future studies are required to determine the association of biomarkers of glomerular and tubular function with serum creatinine-based AKI in the context of COVID-19 AKI. In addition, these clinical data should be integrated with pathological findings from analyses of kidney biopsy samples29,30,31,32,33 to gain greater insights into the pathophysiology of COVID-19 AKI. Evidence from a study of biopsy data from 47 patients in France demonstrated that kidney injury seen in cases with the most severe respiratory disease in the ICU is predominantly tubular (66.7%); by contrast, collapsing glomerulopathy and focal segmental glomerulosclerosis were not seen in critically ill patients but were observed in 70.6% of cases not in the ICU34. More than two coexisting comorbidities were seen in over 60% of cases and the occurrence of collapsing glomerulopathy correlated highly with the expression of high-risk APOL1 genotypes.
Pathophysiology of COVID-19 AKI
The pathophysiology of COVID-19 AKI is thought to involve local and systemic inflammatory and immune responses, endothelial injury and activation of coagulation pathways and the renin–angiotensin system31,35. Direct viral infection with renal tropism of the virus has also been proposed but remains controversial36. Non-specific factors that are common in critically ill patients, such as mechanical ventilation, hypoxia, hypotension, low cardiac output and nephrotoxic agents, might also contribute to kidney injury and/or functional decline in the most severely affected patients (Box 1).
Insights from renal histology
Autopsy studies demonstrate that acute tubular injury is by far the most common finding in kidneys of patients with COVID-19 AKI (Supplementary Table 1). Of note, tubular autolysis is a confounding factor in post-mortem histological analyses of acute tubular injury31,37. Analyses of post-mortem kidney samples from patients with stage 2 or 3 AKI and COVID-19 have revealed acute tubular injury characterized by mostly mild focal acute tubular necrosis29,33,35,38, illustrating an apparent uncoupling between the extent of histological injury and decline of kidney function — a finding previously reported in patients with non-COVID sepsis39. In an autopsy series of 9 patients in the UK, evidence of acute tubular injury was noted in all patients; viral load quantified by the use of quantitative real-time PCR targeting the viral E gene was observed in the kidneys of 3 patients and detection of subgenomic viral RNA in only 1 (11%) kidney sample38,40.
Another analysis of kidney biopsy samples from 17 patients with SARS-CoV-2 infection and mostly mild COVID-19 symptoms identified AKI and proteinuria in 15 and 11 patients, respectively. Acute tubular injury (n = 14; 82%), collapsing glomerulopathy (n = 7; 41%) and endothelial injury or thrombotic microangiopathy (n = 6; 35%) were the most common histological findings41 (Supplementary Table 1). Virus detection (using immunohistochemistry for SARS-CoV-2 nucleocapsid and RNA in situ hybridization) were negative in patient samples on which it was performed. Another series from France demonstrated tubular injury in the most severely ill cohort whereas glomerular pathology was restricted to the non-ICU patients34. Of note, most biopsies were performed several weeks after the onset of COVID-19 symptoms, and most have failed to show notable SARS-CoV-2 infection of the kidney. Despite initial concerns about the methodology and interpretation of some early studies that reported direct viral tropism of the kidney36,42,43,44, one study that identified and isolated SARS-CoV-2 from post-mortem kidney tissue demonstrated that the virus could replicate in non-human primate kidney tubular epithelial cells, showing its ability to infect kidney cells45. The researchers further identified that 23 of 32 patients with AKI (72%) showed viral RNA in kidney tissue, whereas viral RNA was identified in only 3 of 7 (43%) patients without AKI. Another autopsy study that performed microdissection of kidneys from 6 patients with COVID-19 identified SARS-CoV-2 in different kidney compartments, particularly in the glomerulus43. Viral RNA and protein were also detected in kidney by in situ hybridization with confocal microscopy. In addition, SARS-CoV-2 particles have been observed in urine samples33,46,47 — a finding that either reflects the release of virus from infected, damaged tubule epithelial cells or the filtration of viral fragments, as the high molecular weight of SARS-CoV-2 (600 kDa) should prevent it from being filtered through the intact glomerular filtration barrier48. Thus, a substantial body of evidence now suggests that SARS-CoV-2 can infect kidney tissue; however, a direct role of the virus in the development of AKI remains to be confirmed.
Collapsing glomerulopathy
Collapsing glomerulopathy has been reported in several patients with COVID-19 (Supplementary Table 1). This entity has been described as COVID-19-associated nephropathy (COVAN), and seems to occur mostly in patients with non-severe respiratory symptoms of COVID-19 and isolated AKI or in those presenting with glomerular proteinuria30,32,34. Of note, collapsing glomerulopathy has previously been described in the context of other viral infections, including HIV parvovirus B19, cytomegalovirus and Epstein–Barr virus infections. COVAN is associated with high-risk APOL1 genotypes, and has been observed mostly in Black patients. The true incidence of collapsing glomerulopathy and its contribution to kidney failure in the context of COVID-19 compared with the effects of other underlying conditions (for example, hypertension or CKD) is unknown. Although the exact pathophysiology of COVAN remains unknown, it may share common mechanisms with HIV-associated nephropathy, with podocyte injury through disruption of autophagy and mitochondrial homeostasis31.
Endothelial dysfunction and coagulation
Biomarkers of coagulation and fibrinolysis activation (for example, fibrinogen and D-dimer) have been repeatedly associated with an increased risk of death in patients with COVID-19. Autopsy studies have reported a ninefold higher incidence of observed microvascular and macrovascular thrombosis in lungs of patients with COVID-19 than that of patients with influenza pneumonia49. Systemic microvascular and macrovascular thrombosis in organs, including the kidneys, have also been repeatedly reported in the context of COVID-19 (refs50,51,52). Many critical illnesses are associated with microvascular and endothelial injury but SARS-CoV-2 is believed to specifically affect the endothelium. Post-mortem studies have reported vascular endotheliitis in patients with COVID-19 (refs49,53). Moreover, findings from at least one report indicate viral infection of kidney endothelial cells53; however, that report used electronic microscopy to identify viral elements, which is insufficiently specific and thus firm evidence of direct viral infection of kidney endothelial cells is lacking. Nonetheless, increased levels of plasma biomarkers of endothelial injury (for example, soluble (s) E-selectin, sP-selectin, ANG2, sICAM1 and von Willebrand factor antigen) and platelet activation (soluble thrombomodulin) are associated with poor prognosis54,55,56. Microvascular inflammation can trigger endothelial activation, leading to vasodilation, increased vascular permeability and pro-thrombotic conditions57,58,59. Complement activation — evidenced by increased circulating levels of soluble complement components C5b–9 and C5a and by tissue deposition of C5b–9 and C4d in lung and kidney tissues60,61,62 — may further promote inflammation and coagulation pathways in COVID-19. The release of damage-associated molecular patterns from cells undergoing necrosis might further contribute to endothelial injury in COVID-19 (ref.63). SARS-CoV-2 has furthermore been shown to bind to platelets via ACE2, leading to platelet activation and immunothrombosis64,65,66. Thus, platelet activation may represent a potential player in the pathophysiology of COVID-19 AKI67,68. Circulating prothrombotic autoantibodies that target phospholipids and phospholipid-binding proteins have also been reported69. In a cohort of 172 hospitalized patients with COVID-19, higher titres of prothrombotic antibodies were associated with lower eGFR. In vitro studies confirmed the autoantibodies to be drivers of endothelial cell activation, potentially contributing to the thrombo-inflammatory effects observed in severe COVID-19 (ref.70).
However, macrothrombi and microthrombi have been inconsistently observed in kidneys of patients who have died with COVID-19, or have involved only a small proportion of renal capillaries. A small autopsy study from New York, USA, observed thrombotic microangiopathy within glomeruli in only 1 of 7 cases51. Another series of kidney biopsy samples from 17 patients with mild COVID-19 symptoms identified evidence of acute glomerular endothelial cell injury in 6 patients, most of whom demonstrated laboratory features of thrombotic microangiopathy41. Of note, no evidence of peritubular vascular injury was observed in that study. Neutrophils and neutrophil extracellular traps — frequently aggregating with platelets — have been observed in many organs including the kidneys, despite the sporadic presence of virus on histology, suggesting a role of inflammation in the development of intravascular thrombi71. Cases of renal artery thrombosis have also been anecdotally reported72,73. Finally, patients with severe COVID-19 often present with complications associated with chronic endothelial dysfunction, such as hypertension or diabetes, which are themselves associated with decreased endothelial nitric oxide synthase activity and bioavailability of nitric oxide — a major vasodilator and antithrombotic factor74.
The immune and inflammatory response
Several alterations in both innate and adaptive immune responses have been reported following SARS-CoV-2 infection. Immunosenescence — a term used to define the innate and adaptive immunological alterations that occur with ageing — is characterized by inflammageing, a low-grade inflammatory state that may have a key role in determining organ dysfunction, as well as by ineffective T cell responses and antibody production. These features have been reported in COVID-19 and may in part explain the higher mortality among elderly individuals and those with comorbidities as a consequence of inefficient viral clearance, the enhanced release of cytokines and chemokines, endothelial damage and activation of the coagulation and complement cascades75.
Inflammation
The enhanced release of inflammatory mediators by immune and resident kidney cells is likely to be a key mechanism of tissue damage in patients with COVID-19. Inflammatory mediators, such as TNF and FAS, can bind to their specific receptors expressed by renal endothelial and tubule epithelial cells causing a direct injury76,77. Such interactions have been observed in experimental models of sepsis and are supported by measurements of plasma cytokine levels in patients with sepsis-associated AKI78, although their role in COVID-19 AKI is yet to be clearly demonstrated.
Interferon
Other studies have demonstrated a key role for type I interferon responses in suppressing viral replication and regulating the immune response in the context of COVID-19. Available evidence suggests that SARS-CoV-2 infection can lead to suppression of interferon release; moreover, patients treated with interferon demonstrated improved viral clearance with a concomitant reduction in levels of IL-6 and C-reactive protein79. One study demonstrated that patients with inborn errors of type I interferon immunity and extremely low serum levels of IFNα (<1 pg/ml) are at a greater risk of severe COVID-19 than those with higher IFNα levels (1–60 pg/ml))80. The same group of researchers also identified individuals with severe COVID-19 with autoantibodies directed against type I interferon, suggesting a possible autoimmune basis to the inefficient blockade of SARS-CoV-2 infection as a result of low interferon plasma levels81. These findings justify the ongoing clinical trials of therapeutic interferon administration for patients with COVID-19 (refs82,83). However, a note of caution is warranted given that interferons are well-known mediators of glomerular injury. Indeed, IFNα and IFNβ exert differential effects on parietal epithelial cells and podocytes, acting to enhance podocyte loss and promote glomerulosclerosis, respectively84. Moreover, proteinuria occurring in the context of inflammation has been ascribed to podocyte injury following cytokine release and the activation of type I interferon signalling85. Finally, APOL1 risk alleles may promote glomerular damage via a process that involves interferon86.
Complement
The innate immune response to viral infections involves activation of the complement cascade; however, its persistent and uncontrolled activation can promote inflammatory processes that induce tissue injury. As mentioned earlier, plasma levels of soluble C5b–9 and C5a are higher in patients with COVID-19 than in healthy controls, particularly in those with severe disease62. Complement components might act in concert with other factors to trigger inflammation, coagulation and endothelial damage. A number of studies have demonstrated activation of the complement cascade in different organs, including the kidney, in patients with COVID-19. One study detected C3c and C3d in renal arteries and glomerular capillaries, C3d in the tubular compartment, and the membrane attack complex C5b–9 in peritubular capillaries, renal arterioles and the tubular basement membrane60. Another study published in preprint form identified complement deposits in tubular epithelial cells and vessels, with only mild C5b–9 staining in glomeruli61.These findings suggest activation of the lectin and classical pathways in peritubular capillaries and renal arteries, whereas the alternative pathway may have a more prominent role in mediating tubular damage60.
Complement activation seems to have a predominant role in COVID-19-associated endothelial dysfunction: C5a can directly bind to its receptor C5aR on endothelial cells, inducing the upregulation of tissue factor (TF) and the loss of thrombomodulin. These processes induce coagulation, the exocytosis of P-selectin and the formation of ultra-large von Willebrand factor multimers, leading to increased platelet adhesion and aggregation. C5b–9 also contributes to endothelial dysfunction, increased vascular permeability, and triggers inflammation and coagulation87,88. Moreover, the binding of C5a to C5aR on tubule epithelial cells promotes DNA methylation of genes involved in cellular senescence, thus potentially promoting AKI persistence and progression towards CKD owing to the activation of pro-fibrotic processes89. Together, these findings suggest that COVID-19 can be considered a thrombo-inflammatory disease and that blockade of the complement cascade could be a potential therapeutic option to limit COVID-associated AKI, multiple organ failure and disease severity90. In line with this suggestion, a study of a small cohort of haemodialysis patients with COVID-19 identified elevated levels of plasma C3a and C5a prior to the development of severe disease, suggesting that complement activation preceded severe symptoms91.
Adaptive immunity
Several studies indicate that inadequate adaptive immunity can also contribute to poor outcomes in COVID-19, with CD4+ and CD8+ T lymphopenia representing typical features of the most severe forms of COVID-19 (ref.92). Depletion of plasmacytoid dendritic cells (a major source of IFNα), eosinophils and natural killer cells has also been reported93. In addition, nuclear factor erythroid 2-related factor 2 (NRF2) and its downstream signalling components are also suppressed in lung biopsy samples from patients with COVID-19 (ref.94). NRF2 is a transcription factor that regulates cellular antioxidant responses. It is normally maintained in an inactive state in the cytosol by association with its inhibitor protein Kelch-like ECH-associated protein 1 (KEAP1) but in response to oxidative stress, such as that observed in viral infections, KEAP1 is inactivated and NRF2 is released, inducing NRF2-responsive genes to attenuate stress-induced cell death. These functions suggest that NRF2 may act as master regulator of tissue damage during infection — a theory supported by the finding that NRF2 agonist drugs can induce antiviral activity through interferon-independent mechanisms94, and suggesting that a similar approach may have value in the treatment of COVID-19. The suggestion that activation of NRF2 may have a protective role in COVID-19 AKI is at present speculative; however, data from experimental AKI in other settings support this hypothesis. In a mouse model of ischaemia–reperfusion injury, for example, augmentation of T cell-specific expression of NRF2 provided renal functional and histological protection, associated with lower levels of TNF, IFNγ and IL-17 (ref.95). Conversely, NRF2 deficiency enhances susceptibility to ischaemic and nephrotoxic tissue injury, supporting a role for this transcription factor as a potential therapeutic target96.
Humoral immunity
In terms of humoral immunity, it has been noted that patients with COVID-19 can exhibit different phenotypic responses characterized by decreased numbers of circulating memory B cells or an increase in circulating plasmablasts, as also shown for other viral infections such as Ebola97. Induction of a specific antibody response with an adequate IgG component is in general essential to control viral infection; however, in the context of COVID-19, immunosenescence might lead to T cell exhaustion and to the aberrant production of tissue-specific autoantibodies. As reported for anti-interferon autoantibodies, this immunosenescence may underlie an autoimmune response directed against a soluble form of ACE2 (sACE2). sACE2, which is present in the blood and in extracellular fluids, is thought to act as a dummy receptor and an inactivator molecule for SARS-CoV-2, as has been described for other soluble receptors for other pathogenic viruses98. However, the high affinity of the SARS-CoV-2 spike protein for ACE2 may lead to the formation of SARS-CoV-2–sACE2 complexes and to the development of anti-ACE2 autoantibodies that might target tissue ACE2 — the receptor that allows viral entry into cells — creating vasculitis-like lesions after the early infective phase of the virus99,100. Thus, although targeting ACE2 may be useful in the initial stages of disease to prevent viral uptake by cells (discussed later), the presence of ACE2-targeted autoantibodies after the infective phase may be harmful and result in organ damage.
Reports of an IgM autoantibody response against ACE2 provide further support for the notion that COVID-19 can be associated with a robust autoimmune response. Purified anti-ACE2 IgM can activate complement components in endothelial cells — a finding supported by histological analysis of post-mortem lung tissue, and highlighting the angiocentric pathology of severe disease101. Of note, anti-ACE2 IgM production has been associated with a robust anti-SARS-CoV-2 spike protein IgG response, suggesting the presence of an anti-idiotype IgM response cross-reacting with ACE2 (ref.100). Given the wide expression of ACE2 in different organs including the kidney, a role for anti-ACE2 autoantibodies in COVID-19 pathogenesis cannot be excluded, in which autoantibody production may lead to an imbalance in the ratio of ACE to ACE2 (because ACE2 is a negative regulator of ACE), resulting in worsening of tissue oedema, inflammation and damage102. However, this theory is speculative at present and remains to be validated. Furthermore, homology between receptors might lead to the cross-reactivity between ACE2 and ACE receptors103. Finally, evidence of a role of autoimmunity in the pathogenesis of COVID-19 is provided by a study that used a high-throughput autoantibody discovery technique called rapid extracellular antigen profiling, which showed the production of autoantibodies directed against various extracellular and secreted immune-related or tissue-specific proteins104,105,106.
The role of cytokine storm syndrome
Cytokine storm syndrome (CSS) is viewed as a life-threatening condition characterized by organ failure and the rapid proliferation and hyperactivity of all immune system components, including T cells, macrophages, natural killer cells and the increased production and release of numerous chemical mediators and inflammatory cytokines107. The inflammatory response of COVID-19 bears similarities to other conditions that are associated with CSS, including primary haemophagocytic lymphohistiocytosis (HLH)108. Indeed, CSS has been proposed to contribute to the ‘hyperinflammatory state’ of severe COVID-19, contributing to marked elevations in acute phase reactants, lymphopenia and coagulation defects. Elevated levels of cytokines, including IL-6, have been documented in some patients with COVID-19, suggesting hyperactivation of the humoral immune response. Of note, IL-6 is a critical mediator of multiorgan dysfunction, including AKI109,110. A meta-analysis reported that IL-6 levels are elevated and significantly associated with adverse clinical outcomes, including ICU admission, acute respiratory distress syndrome (ARDS) and death, in patients with COVID-19 (ref.111). Serum IL-6 levels were nearly threefold higher in patients with severe disease than in those with non-complicated disease, although variations in the timing of IL-6 measurement, the type of assay, as well as differences in adjuvant immunomodulatory medications, such as corticosteroids, may have affected both IL-6 response and patient outcomes. However, the levels of IL-6 observed in these severe COVID-19 cases (7.9–283 pg/ml) are much lower than those observed in patients with sepsis (frequently >20,000 pg/ml) and non-COVID ARDS (approaching 10,000 pg/ml in cytokine release syndrome)112. These observations are supported by a meta-analysis, which revealed that IL-6 levels in severe COVID-19 were lower than in patients with sepsis, septic shock or hyperinflammatory ARDS112. Similarly, a study from the Netherlands that compared levels of pro-inflammatory cytokines (IL-6, IL-8 and TNF) in critically ill patients with COVID-19 with those in other critically ill individuals113 demonstrated that concentrations of circulating cytokines were lower in patients with COVID-19 than in patients with bacterial sepsis and similar to those of other critically ill patients. However, patients with COVID-19-related ARDS had lower APACHE2 scores than patients with other conditions, suggesting a lower severity of critical illness. These findings suggest that COVID-19 might not be characterized by CSS and its role in the development of COVID-19 AKI is therefore questionable. As discussed later, this proposal has important implications for the use of extracorporeal blood purification techniques. Importantly, the exclusion of a pathogenic role for CSS does not exclude a role for regional inflammation in the pathogenicity of COVID-19, which is supported by evidence of high levels of acute phase reactant inflammatory biomarkers, such as C-reactive protein, in patients with COVID-19 (ref.112).
ACE2 and the renin–angiotensin system
Although ACE2 is considered to be the classic receptor by which SARS-CoV-2 gains entry into cells, a study published in preprint form identified kidney injury molecule 1 (KIM1; also known as T cell immunoglobulin mucin domain 1) as an alternative receptor for SARS-CoV-2 in tubule epithelial cells114. Kidney cells also express transmembrane protease serine 2 (TMPRSS2) — an enzyme that proteolytically cleaves ACE2 and is essential for the viral entry43,115. TMPRSS2 colocalizes to different compartments of the kidney, although its expression is greatest in the distal tubules, whereas ACE2 is predominantly expressed in the proximal tubules116,117,118.
In addition to mediating SARS-CoV-2 entry into cells, ACE2 acts as an enzyme within the renin–angiotensin system, metabolizing angiotensin II by cleaving a terminal peptide to form angiotensin(1–7) (Ang1–7)119,120. Ang(1–7) generally opposes the actions of angiotensin II, which include activation of endothelium and platelets, vasoconstriction and the release of pro-inflammatory cytokines. Following binding of SARS-CoV-2 to human ACE2, ACE2 is thought to be downregulated121, leading to increased levels of angiotensin II and decreased Ang(1–7)122,123,124. This proposal is in line with the observed decrease in plasma levels of angiotensin II observed in a patient with COVID-19 after administration of recombinant human sACE2, which, like endogenous sACE2, can act as a dummy receptor to bind and sequester SARS-CoV-2 (ref.125). Recombinant human sACE2 also led to a marked reduction in IL-6 and IL-8 (ref.125). Lower plasma levels of Ang1 and Ang(1–7) were also reported in patients with COVID-19 than in healthy controls and non-ICU patients111.
Importantly, although in the kidney, formation of Ang(1–7) from angiotensin II is predominantly mediated by ACE2, formation of Ang(1–7) in the plasma and lungs are reportedly largely independent of ACE2 (ref.126). Of note, circulating levels of sACE2 are very low110, which, in theory, makes the kidney more sensitive to ACE2 activity with respect to the angiotensin II and Ang(1–7) balance. Whether an imbalance between angiotensin II and Ang(1–7) has a direct role in endothelial activation and COVID-19 AKI remains speculative at present59.
Polymorphisms in ACE2 have been described but no information exists with regard to their relationship to COVID-19 AKI127. Although some of these polymorphisms might enhance SARS-COV-2 entry into the tubule epithelial cells, future study should explore whether these genetic differences are associated with specific injury patterns.
Together, the available data suggest that the relationship between ACE2 and angiotensin II contributes to kidney injury in COVID-19. This interaction may, however, depend on the severity of the disease and the extent to which it represents an adaptive response to shock, as low levels of angiotensin II may be associated with poor outcomes in critically ill patients128,129. One small, single-centre study of critically ill patients with COVID-19 demonstrated an association between AKI and an increase in plasma renin levels, indicative of low angiotensin II activity130. This association is also observed in other critical care settings, such as distributive shock or cardiac surgery as a consequence of a relative deficit of angiotensin II, which induces renin release via a positive-feedback loop129,130. The existence of a similar mechanism in COVID-19 is suggested by the presence of lower angiotensin II levels in patients with COVID-19 and ARDS than in patients with milder disease131.
The impact of withholding renin–angiotensin system blockers, such as angiotensin-converting enzyme inhibitors and angiotensin-receptor blockers, in patients with COVID-19 has been intensely debated but does not seem to affect outcomes132,133. Studies in mice demonstrate that administration of captopril or telmisartan leads to a decrease in ACE2 expression in isolated kidney membranes, with no effect on ACE2 activity in isolated lung membranes, suggesting differential effects on the kidney and lung134. Furthermore, in a randomized controlled trial of patients admitted to hospital with COVID-19, discontinuation of renin–angiotensin system inhibitors had no impact on disease severity or kidney function135.
Non-specific factors
In addition to virus-specific responses, the pathogenesis of AKI in the context of COVID-19 most likely also involves factors that are not specific to the virus but are part of a general response to critical illness or its treatment, including haemodynamic factors, drug toxicity and the impact of organ support systems.
Organ crosstalk and lung–kidney interactions
Crosstalk between lung and kidney has been identified in critical illnesses; these interactions are complex and comprise several putative mechanisms136 that are also likely to exist in patients with severe COVID-19 (Fig. 1). For example, acute hypoxaemia might alter kidney function and increase renal vascular resistance74,137, which might contribute to renal hypoperfusion138 and acute tubular injury139.
Moreover, following the development of AKI, increases in levels of inflammatory cytokines, such as IL-6, as a consequence of their reduced renal clearance and increased production has been reported, and may contribute to respiratory failure via kidney–lung crosstalk128.
In patients with severe disease, mechanical ventilation can contribute to the development of AKI through immune-mediated processes and haemodynamic effects140. Mechanical ventilation has been associated with an increased risk of AKI among patients with COVID-19. In a cohort of veteran patients with COVID-19 in the USA, AKI was associated with more frequent mechanical ventilation use (OR 6.46; 95% CI 5.52–7.57)23. Whether this association reflects the greater severity of the disease and systemic inflammation or is a direct effect of the impact of mechanical ventilation is uncertain, but it is likely a combination of both.
Haemodynamic factors
Crosstalk between the cardiovascular system and kidneys is also likely to contribute to COVID-19 AKI. Rare cases of acute myocarditis141,142 and myocardial injury143 have been described in patients with COVID-19, which potentially result in impairment of cardiac function and thereby potentially compromise kidney perfusion through a decrease in cardiac output or through renal vein congestion144,145. As in other forms of ARDS, use of high positive end-expiratory pressure and/or tidal volumes increases intrathoracic pressure, right atrial pressure and right ventricular afterload, and can decrease cardiac output140. Right-sided heart dysfunction and increased venous pressures can result in increased interstitial and tubular hydrostatic pressure within the encapsulated kidney, which decreases net GFR and oxygen delivery to the kidney146. The observed association between mechanical ventilation or use of vasopressors with the risk of AKI further suggests that haemodynamic factors contribute to COVID-19 AKI5,147,148.
Nephrotoxins
As with all patients at risk of AKI, drug stewardship with regard to potential nephrotoxic drugs should be paramount. COVID-19 AKI is in this regard no different from AKI from other causes. In particular, administration of antibiotics such as vancomycin and aminoglycosides, especially in the context of critical illness, can have an important role in its aetiology149,150. Administration of nephrotoxins (for example, vancomycin, colistin and aminoglycosides) has also been associated with an increased risk of AKI in patients with COVID-19 (ref.151).
Several uncertainties exist with regard to the safety of antivirals used to treat COVID-19 in patients with AKI. Remdesivir is a nucleotide analogue that inhibits viral RNA-dependent RNA polymerase and is predominantly excreted via the kidneys. Although evidence of its efficacy have been reported by some, but not all studies, remdesivir may exert nephrotoxic effects through the induction of mitochondrial injury in renal tubule epithelial cells. This renal toxicity is most likely to occur after prolonged exposure or at high doses. A randomized controlled trial of 1,062 patients reported a shorter recovery time from COVID-19 symptoms with use of remdesivir — a benefit mainly observed among patients treated early after the onset of symptoms and not in critically ill patients152. A decline in eGFR was observed in 14% of patients in the placebo group and 10% in the treated group; however, patients with an eGFR <30 ml/min/1.73 m2 were excluded from the trial, thereby largely excluding those with AKI. Of note, another randomized controlled trial failed to show a benefit of remdesivir for patient outcome153. In that study, baseline eGFR was 99 ml/min and 110 ml/min in the short (5 days) and extended duration (10 days) treatment groups, respectively, and the trial again excluded patients with evidence of impaired kidney function. A decline in creatinine clearance was seen in 30% of patients in the control group, 15% of patients in the short duration treatment group and 26% of patients in the extended duration treatment group. Thus, available evidence from clinical trials is not suggestive of notable nephrotoxic activity in patients without severely impaired kidney function at baseline. Beyond clinical trials, however, some evidence of renal toxicity has been identified. An analysis of the international pharmacovigilance post-marketing databases of the World Health Organization revealed a statistically significant nephrotoxicity signal, demonstrating a 20-fold higher risk of AKI with remdesivir use than that associated with other drugs frequently used in COVD-19 (hydroxychloroquine, tocilizumab and lopinavir/ritonavir)154. Cases of AKI associated with lopinavir and low-dose ritonavir therapy in the course of COVID-19 management were also reported155. Finally, rhabdomyolysis represents a potential non-pharmacological mechanism of nephrotoxicity in COVID-19 AKI through the precipitation of myoglobin and the release of free radicals, as has been described for other forms of AKI associated with viral infections156,157.
Extracorporeal membrane oxygenation
In two European multicentre cohorts of patients with COVID-19, it was found that 22% and 46% of patients on extracorporeal membrane oxygenation (ECMO) required KRT158,159. Potential mechanisms by which ECMO may contribute to AKI include venous congestion, a higher risk of secondary infections, haemolysis, major bleeding and inflammation. In one cohort of patients with COVID-19 requiring ECMO, major bleeding occurred in 42% of patients, haemolysis in 13%, cannula infection in 23% and ventilator-associated pneumonia in 87%159.
Similarities to non-COVID-19 AKI
Of interest is the extent to which sepsis-associated AKI and COVID-19 AKI share similarities. Sepsis-associated AKI is characterized by a drop in GFR, whereas renal blood flow can be lower or higher than normal rates160. Factors that contribute to sepsis-associated AKI include regional inflammation, microvascular alterations and haemodynamic alterations (including glomerular shunting, activation of tubuloglomerular feedback, and increased interstitial and thus intratubular pressure)161,162. Filtered damage-associated molecular patterns and pathogen-associated molecular patterns are considered to be triggers of interstitial inflammation through activation of TLR2 and TLR4 on the brush border of proximal tubule epithelial cells163,164. In addition, glomerular infiltration of leukocytes and intraglomerular thrombus formation are indicative of endothelial damage, and in animal models, leads to increased filtration barrier permeability and albuminuria165,166. Inflammatory cytokines also promote the release of ultra-large von Willebrand factor multimers from endothelial cells and inhibit the cleavage and clearance of these pro-thrombotic agents by the metalloproteinase ADAMTS13 (ref.166). This mechanism, combined with endothelial injury and shedding of the glycocalyx by inflammatory mediators may increase susceptibility of glomerular and peritubular capillaries to microthrombi formation and occlusion, and prolong the exposure of tubule epithelial cells to inflammation and hypoxia. Of note, histology of post-mortem kidney samples from patients with sepsis-associated AKI shows overall rather modest tubular and glomerular damage despite the profound impairment of renal function165,167,168. The relative dissociation between tissue damage and extensively altered kidney function are consistent with findings in COVID-19 AKI. Thus, the major difference between COVID-19 AKI and other types of sepsis, including viral sepsis169, seems to be the inconsistent finding of virus particles in epithelial cells combined with more prominent vascular alterations in COVID-19 AKI. However, the potential contribution of viral infection and vascular alterations to kidney dysfunction is not yet fully understood.
ARDS is a complication of severe COVID-19. Endothelial injury and the systemic release of pro-inflammatory mediators, such as plasminogen activator inhibitor-1, IL-6 and soluble TNF receptors, have been associated with the development of AKI in patients with non-COVID-19 ARDS170,171, and similar processes are likely to be associated with the development of AKI in patients with COVID-19. In addition and as mentioned earlier, other factors that are associated with ARDS, including hypoxaemia, which can increase renal vascular resistance172, and elevated central venous pressure145, resulting from right-sided heart failure, high intrathoracic pressures or pulmonary vascular thrombi, can lead to increased interstitial and tubular hydrostatic pressure within the encapsulated kidney, compromising renal perfusion and GFR.
Implications for research and therapy
A disease model that centres around regional inflammation, immunothrombosis, vascular pathology and potential direct viral renal toxicity has important implications for the ongoing search for therapeutics173 (Box 2).
Non-kidney-specific strategies
Several non-kidney-specific measures are expected to affect kidney outcomes, particularly in the context of organ crosstalk. Although early liberal intubation and mechanical ventilation had been advocated in the early stages of the pandemic, a more restrictive approach is now typically applied. Limiting indications for invasive ventilation and therefore limiting ventilator-induced lung injury and the consequences of high levels of positive end-expiratory pressure may have contributed to the decreased rate of AKI over the course of the pandemic23,174. Translating insights from non-COVID ARDS to COVID-related ARDS, including strategies to avoid excessive fluid depletion and fluid overload, are also likely to provide kidney protection in COVID-19.
As discussed earlier, regional inflammation may have an important role in the pathogenesis of COVID-19. In line with this proposal, a prospective meta-analysis performed by a WHO working group identified an association between glucocorticoid use and lower 28-day mortality in critically ill patients with COVID-19 (ref.175). The subsequently published RECOVERY trial demonstrated that use of dexamethasone resulted in lower 28-day mortality in patients with COVID-19 requiring ventilation or oxygen176. Among patients who did not require KRT at randomization, those who received dexamethasone were less likely than those in the control group to receive KRT (4.4% versus 7.5%, RR 0.61; 95% CI 0.48–0.76) suggesting a protective effect of dexamethasone on the kidney. Tocilizumab is a recombinant humanized anti-IL-6 receptor monoclonal antibody that inhibits the binding of IL-6 and its receptors and thereby blocks IL-6 signalling and associated inflammation177. Preliminary results from the RECOVERY trial suggest that administration of tocilizumab to hospitalized patients with COVID-19, hypoxia and evidence of inflammation improved survival and chances of hospital discharge at 28 days. Furthermore, the preliminary report demonstrates a significant reduction in the requirement for KRT, suggesting the beneficial effects of tocilizumab on preventing AKI and/or promoting renal recovery178.
Interferon therapy is one of the most promising approaches to improving viral clearance in the early stages of COVID-19. However, although small interventional trials have reported encouraging results with interferon therapy, more robust trials are needed179,180. Of note, and as mentioned earlier, caution is mandated in populations at risk of collapsing glomerulopathy given the role of interferon in podocyte injury32. The use of convalescent plasma therapy so far is not supported by available evidence. Potential safety concerns have also emerged owing to the presence of circulating autoantibodies against type I interferons that can be present in plasma and associated with worse outcomes181. The high incidence of microthrombotic and macrothrombotic events and the demonstration of microvascular thrombi in some patients with COVID-19 calls for a better understanding of anticoagulation strategies in this setting; however, the potential impact of these strategies on COVID AKI is unknown.
As mentioned earlier, CSS is not observed in most patients with COVID-19 and histology findings indicate a complex inflammatory response, making extracorporeal cytokine removal unlikely to be superior to anti-inflammatory drugs in terms of improving renal outcomes in the vast majority of patients. The rate of AKI requiring KRT among patients with severe COVID-19 varies between 5% and 21% — an incidence similar to that observed in other critical illnesses16. Although no trial has specifically investigated the impact of KRT timing in COVID-19, trials in the setting of non-COVID critical illness have demonstrated that liberal use of KRT does not improve survival but is associated with an increased risk of adverse effects and use of resources182,183. Conservative use of KRT is also highly relevant in the setting of a pandemic in which critical resources such as dialysis facilities can be limited.
Specific strategies for COVID-19 AKI
Specific strategies for the treatment or prevention of COVID AKI are currently lacking. As for other critical illnesses, understanding of the pathophysiology of COVID AKI is limited by difficulties in accessing kidney tissue and assessing kidney haemodynamics in humans. Attempts to model COVID-19 in animals has been challenging, mostly because of interspecies characteristics. For example, SARS-CoV-2 is unable to effectively use mouse or rat ACE2 for viral entry into the cell. Several strategies have been developed to overcome this issue, including modification of the virus spike protein to enable binding to mouse ACE2 or the generation of genetically modified mice that express human ACE2 (refs184,185). However, differences in tissue expression levels of human ACE2 might limit the use of these genetic models to explore viral infection of kidney tissue. In addition, these models often fail to induce severe disease, including manifestations of extrapulmonary organ damage, including the kidney. Similarly, neither kidney damage nor viral infection of kidney cells was detected in a hamster model of COVID-19 (ref.184). Other species have also been used to model COVID-19, including non-human primates, but to our knowledge kidney damage has not yet been explored in these.
Finally, the specific interaction between SARS-CoV-2 and ACE2 deserves specific investigation. If kidney damage is caused by direct viral entry into kidney cells, blocking of ACE2 might limit tissue infection and subsequent damage. In line with this proposal, administration of recombinant human sACE2 inhibited SARS-CoV-2 infection of engineered human blood vessel organoids and human kidney organoids186. Its use is currently under investigation in the clinical setting125,187,188.
Conclusions
Acute tubular injury seems to be a common occurrence in patients with COVID-19 AKI, but is often mild, despite severely altered kidney function. Endothelial injury, microvascular thrombi, local inflammation and immune cell infiltration have been repeatedly observed in patients with COVID-19 AKI; however, differences and similarities in the pathophysiology of COVID-19 AKI and non-COVID sepsis-associated AKI remain to be established. A high incidence of thrombi and intravascular coagulation might be one striking difference. Given the interactions between lung and kidney, treatment and strategies that prevent progression of the disease and need for mechanical ventilation are very likely to protect the kidney. Regional inflammation contributes to COVID-19-associated organ injury; in line with this mechanism of organ injury, available data suggest that steroids and IL-6 receptor antagonists may be promising in preventing severe AKI, although further work is required to confirm these findings and assess their impact on renal recovery. Direct viral infection of kidney cells has been observed in several cohorts, including in analyses of tissue samples taken several weeks after disease onset. However, the role of direct viral infection in the development of AKI remains controversial. Of note, an impaired type I interferon response in severely ill patients with COVID-19 has been reported and might contribute to the ineffective clearance of virus from kidney cells in a subset of patients. However, collapsing nephropathy in patients with COVID-19 seems to be associated with the high-risk APOL1 genotype and may involve pathogenic pathways linked to interferon-mediated podocyte injury. Despite advancing insights into the processes underlying kidney injury in COVID-19, however, therapeutic strategies that specifically target the kidney are lacking. Human recombinant sACE2 has been shown to prevent viral infection of kidney cells in vitro, and might represent a promising specific treatment for COVID-19 AKI in the future.
References
Guan, W.-J. et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 382, 1708–1720 (2020).
Batlle, D. et al. Acute kidney injury in COVID-19: emerging evidence of a distinct pathophysiology. J. Am. Soc. Nephrol. 31, 1380–1383 (2020).
Cheng, Y. et al. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 97, 829–838 (2020).
Cheng, Y. et al. The incidence, risk factors, and prognosis of acute kidney injury in adult patients with Coronavirus Disease 2019. Clin. J. Am. Soc. Nephrol. 15, 1394–1402 (2020).
Hirsch, J. S. et al. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. 98, 209–218 (2020).
Mohamed, M. M. B. et al. Acute kidney injury associated with Coronavirus Disease 2019 in urban New Orleans. Kidney360 1, 614–622 (2020).
Cummings, M. J. et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: a prospective cohort study. Lancet 395, 1763–1770 (2020).
Ng, J. H. et al. Outcomes among patients hospitalized with COVID-19 and acute kidney injury. Am. J. Kidney Dis. 77, 204–215.e1 (2021).
Gupta, S. et al. AKI treated with renal replacement therapy in critically ill patients with COVID-19. J. Am. Soc. Nephrol. 32, 161–176 (2021).
Carlson, N. et al. Increased vulnerability to COVID-19 in chronic kidney disease. J. Intern. Med. https://doi.org/10.1111/joim.13239 (2021).
Williamson, E. J. et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature. 584, 430–436 (2020).
Cantaluppi, V. et al. A call to action to evaluate renal functional reserve in patients with COVID-19. Am. J. Physiol. Renal Physiol. 319, F792–F795 (2020).
Peng, S. et al. Early versus late acute kidney injury among patients with COVID-19 — a multicenter study from Wuhan, China. Nephrol. Dial. Transpl. 35, 2095–2102 (2020).
Portolés, J. et al. Chronic kidney disease and acute kidney injury in the COVID-19 Spanish outbreak. Nephrol. Dial. Transpl. 35, 1353–1361 (2020).
Russo, E. et al. Kidney disease and all-cause mortality in patients with COVID-19 hospitalized in Genoa, Northern Italy. J. Nephrol. 34, 173–183 (2021).
Fu, E. L. et al. Acute kidney injury and kidney replacement therapy in COVID-19: a systematic review and meta-analysis. Clin. Kidney J. 13, 550–563 (2020).
Argenziano, M. G. et al. Characterization and clinical course of 1000 patients with COVID-19 in New York: retrospective case series. BMJ 369, m1996 (2020).
Pei, G. et al. Renal involvement and early prognosis in patients with COVID-19 pneumonia. J. Am. Soc. Nephrol. 31, 1157–1165 (2020).
Chan, L. et al. AKI in hospitalized patients with COVID-19. J. Am. Soc. Nephrol. 32, 151–160 (2021).
Heung, M. et al. Acute kidney injury recovery pattern and subsequent risk of CKD: an analysis of Veterans Health Administration Data. Am. J. Kidney Dis. 67, 742–752 (2016).
Xu, Z. et al. Systematic review and subgroup analysis of the incidence of acute kidney injury (AKI) in patients with COVID-19. BMC Nephrol. 22, 52 (2021).
Lin, L. et al. Risk factors and prognosis for COVID-19-induced acute kidney injury: a meta-analysis. BMJ Open 10, e042573 (2020).
Bowe, B. et al. Acute kidney injury in a national cohort of hospitalized US veterans with COVID-19. Clin. J. Am. Soc. Nephrol. 16, 14–25 (2020).
Charytan, D. M. et al. Decreasing incidence of acute kidney injury in patients with COVID-19 critical illness in New York City. Kidney Int. Rep. 6, 916–927 (2021).
Kormann, R. et al. Coronavirus disease 2019: acute Fanconi syndrome precedes acute kidney injury. Clin. Kidney J. 13, 362–370 (2020).
Ostermann, M. et al. Recommendations on acute kidney injury biomarkers from the Acute Disease Quality Initiative consensus conference: a consensus statement. JAMA Netw. Open 3, e2019209 (2020).
Werion, A. et al. SARS-CoV-2 causes a specific dysfunction of the kidney proximal tubule. Kidney Int. 98, 1296–1307 (2020).
Gansevoort, R. T. et al. Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. Lancet 382, 339–352 (2013).
Santoriello, D. et al. Postmortem kidney pathology findings in patients with COVID-19. J. Am. Soc. Nephrol. 31, 2158–2167 (2020).
Kudose, S. et al. Kidney biopsy findings in patients with COVID-19. J. Am. Soc. Nephrol. 31, 1959–1968 (2020).
Sharma, P. et al. COVID-19-associated kidney injury: a case series of kidney biopsy findings. J. Am. Soc. Nephrol. 31, 1948–1958 (2020).
Wu, H. et al. AKI and collapsing glomerulopathy associated with COVID-19 and APOL1 high-risk genotype. J. Am. Soc. Nephrol. 31, 1688–1695 (2020).
Su, H. et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 98, 219–227 (2020).
Ferlicot, S. et al. The spectrum of kidney biopsies in hospitalized patients with COVID-19, acute kidney injury, and/or proteinuria. Nephrol. Dial. Transplant. https://doi.org/10.1093/ndt/gfab042 (2021).
Golmai, P. et al. Histopathologic and ultrastructural findings in postmortem kidney biopsy material in 12 patients with AKI and COVID-19. J. Am. Soc. Nephrol. 31, 1944–1947 (2020).
Miller, S. E. & Brealey, J. K. Visualization of putative coronavirus in kidney. Kidney Int. 98, 231–232 (2020).
Remmelink, M. et al. Unspecific post-mortem findings despite multiorgan viral spread in COVID-19 patients. Crit. Care 24, 495 (2020).
Schurink, B. et al. Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe 1, e290–e299 (2020).
Gomez, H. et al. A unified theory of sepsis-induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock 41, 3–11 (2014).
Hanley, B. et al. Histopathological findings and viral tropism in UK patients with severe fatal COVID-19: a post-mortem study. Lancet Microbe 1, e245–e253 (2020).
Akilesh, S. et al. Multicenter clinicopathologic correlation of kidney biopsies performed in COVID-19 patients presenting with acute kidney injury or proteinuria. Am. J. Kidney Dis. 77, 82–93.e1 (2020).
Bradley, B. T. et al. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: a case series. Lancet 396, 320–332 (2020).
Puelles, V. G. et al. Multiorgan and renal tropism of SARS-CoV-2. N. Engl. J. Med. 83, 590–592 (2020).
Cassol, C. A., Gokden, N., Larsen, C. P. & Bourne, T. D. Appearances can be deceiving — viral-like inclusions in COVID-19 negative renal biopsies by electron microscopy. Kidney360 1, 824–828 (2020).
Braun, F. et al. SARS-CoV-2 renal tropism associates with acute kidney injury. Lancet 396, 597–598 (2020).
Wölfel, R. et al. Virological assessment of hospitalized patients with COVID-2019. Nature 581, 465–469 (2020).
Sun, J. et al. Isolation of infectious SARS-CoV-2 from urine of a COVID-19 patient. Emerg. Microbes Infect. 9, 991–993 (2020).
Yao, H. et al. Molecular architecture of the SARS-CoV-2 virus. Cell 183, 730–738.e13 (2020).
Ackermann, M. et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N. Engl. J. Med. 383, 120–128 (2020).
Rapkiewicz, A. V. et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: a case series. EClinicalMedicine 24, 100434 (2020).
Fox, S. E., Lameira, F. S. & Rinker, E. B. & Vander Heide, R. S. Cardiac endotheliitis and multisystem inflammatory syndrome after COVID-19. Ann. Intern. Med. 173, 1025–1027 (2020).
Patel, B. V. et al. Pulmonary angiopathy in severe COVID-19: physiologic, imaging, and hematologic observations. Am. J. Respir. Crit. Care Med. 202, 690–699 (2020).
Varga, Z. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 95, 1417–1418 (2020).
Goshua, G. et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 7, e575–e582 (2020).
Spadaro, S. et al. Markers of endothelial and epithelial pulmonary injury in mechanically ventilated COVID-19 ICU patients. Crit. Care 25, 74 (2021).
Vassiliou, A. G. et al. ICU admission levels of endothelial biomarkers as predictors of mortality in critically ill COVID-19 patients. Cells 10, 186 (2021).
Escher, R., Breakey, N. & Lämmle, B. Severe COVID-19 infection associated with endothelial activation. Thromb. Res. 190, 62 (2020).
Connors, J. M. & Levy, J. H. COVID-19 and its implications for thrombosis and anticoagulation. Blood 135, 2033–2040 (2020).
Leisman, D. E., Deutschman, C. S. & Legrand, M. Facing COVID-19 in the ICU: vascular dysfunction, thrombosis, and dysregulated inflammation. Intensive Care Med. 46, 1105–1108 (2020).
Pfister, F. et al. Complement activation in kidneys of patients with COVID-19. Front. Immunol. 11, 594849 (2020).
Macor, P. et al. Multi-organ complement deposition in COVID-19 patients. Preprint at medRxiv https://doi.org/10.1101/2021.01.07.21249116 (2021).
Cugno, M. et al. Complement activation in patients with COVID-19: a novel therapeutic target. J. Allergy Clin. Immunol. 146, 215–217 (2020).
Ince, C. The central role of renal microcirculatory dysfunction in the pathogenesis of acute kidney injury. Nephron Clin. Pract. 127, 124–128 (2014).
Zhang, S. et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 13, 120 (2020).
Manne, B. K. et al. Platelet gene expression and function in patients with COVID-19. Blood 136, 1317–1329 (2020).
Hottz, E. D. et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 136, 1330–1341 (2020).
Taha, M. et al. Platelets and renal failure in the SARS-CoV-2 syndrome. Platelets 32, 130–137 (2021).
Nicolai, L. et al. Immunothrombotic dysregulation in COVID-19 pneumonia is associated with respiratory failure and coagulopathy. Circulation 142, 1176–1189 (2020).
Zuo, Y. et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl. Med. 12, eabd3876 (2020).
Shi, H. et al. Endothelial cell-activating antibodies in COVID-19. Preprint at medRxiv https://doi.org/10.1101/2021.01.18.21250041 (2021).
Middleton, E. A. et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 136, 1169–1179 (2020).
Philipponnet, C., Aniort, J., Chabrot, P., Souweine, B. & Heng, A.-E. Renal artery thrombosis induced by COVID-19. Clin. Kidney J. 13, 713 (2020).
El Shamy, O. et al. Bilateral renal artery thrombosis in a patient with COVID-19. Kidney Med. 3, 116–119 (2021).
Legrand, M., Mik, E. G., Johannes, T., Payen, D. & Ince, C. Renal hypoxia and dysoxia after reperfusion of the ischemic kidney. Mol. Med. 14, 502–516 (2008).
Cunha, L. L., Perazzio, S. F., Azzi, J., Cravedi, P. & Riella, L. V. Remodeling of the immune response with aging: immunosenescence and its potential impact on COVID-19 immune response. Front. Immunol. 11, 1748 (2020).
Cantaluppi, V. et al. Interaction between systemic inflammation and renal tubular epithelial cells. Nephrol. Dial. Transpl. 29, 2004–2011 (2014).
Cantaluppi, V. et al. Polymyxin-B hemoperfusion inactivates circulating proapoptotic factors. Intensive Care Med. 34, 1638–1645 (2008).
Zarbock, A. et al. Sepsis-induced acute kidney injury revisited: pathophysiology, prevention and future therapies. Curr. Opin. Crit. Care 20, 588–595 (2014).
Buszko, M. et al. The dynamic changes in cytokine responses in COVID-19: a snapshot of the current state of knowledge. Nat. Immunol. 21, 1146–1151 (2020).
Zhang, Q. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370, eabd4570 (2020).
Bastard, P. et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370, eabd4585 (2020).
U.S. National Library of Medicine Clinicaltrials.gov https://clinicaltrials.gov/ct2/show/NCT04449380 (2021).
U.S. National Library of Medicine Clinicaltrials.gov https://clinicaltrials.gov/ct2/show/NCT04492475 (2021).
Migliorini, A. et al. The antiviral cytokines IFN-α and IFN-β modulate parietal epithelial cells and promote podocyte loss: implications for IFN toxicity, viral glomerulonephritis, and glomerular regeneration. Am. J. Pathol. 183, 431–440 (2013).
Gurkan, S. et al. Inhibition of type I interferon signalling prevents TLR ligand-mediated proteinuria. J. Pathol. 231, 248–256 (2013).
Beckerman, P. et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat. Med. 23, 429–438 (2017).
Perico, L. et al. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat. Rev. Nephrol. 17, 46–64 (2020).
Vinayagam, S. & Sattu, K. SARS-CoV-2 and coagulation disorders in different organs. Life Sci. 260, 118431 (2020).
Castellano, G. et al. Complement component C5a induces aberrant epigenetic modifications in renal tubular epithelial cells accelerating senescence by Wnt4/βcatenin signaling after ischemia/reperfusion injury. Aging 11, 4382–4406 (2019).
Guglielmetti, G. et al. ‘War to the knife’ against thromboinflammation to protect endothelial function of COVID-19 patients. Crit. Care 24, 365 (2020).
Prendecki, M. et al. Temporal changes in complement activation in haemodialysis patients with COVID-19 as a predictor of disease progression. Clin. Kidney J. 13, 889–896 (2020).
Rydyznski Moderbacher, C. et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell 183, 996–1012.e19 (2020).
Laing, A. G. et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 26, 1623–1635 (2020).
Olagnier, D. et al. SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 11, 4938 (2020).
Noel, S. et al. T lymphocyte-specific activation of Nrf2 protects from AKI. J. Am. Soc. Nephrol. 26, 2989–3000 (2015).
Liu, M. et al. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 76, 277–285 (2009).
Mathew, D. et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 369, eabc8511 (2020).
Halpert, G. & Shoenfeld, Y. SARS-CoV-2, the autoimmune virus. Autoimmun. Rev. 19, 102695 (2020).
Arvin, A. M. et al. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature 584, 353–363 (2020).
McMillan, P. & Uhal, B. D. COVID-19 — a theory of autoimmunity to ACE-2. MOJ Immunol. 7, 17–19 (2020).
Casciola-Rosen, L. et al. IgM autoantibodies recognizing ACE2 are associated with severe COVID-19. Preprint at medRxiv https://doi.org/10.1101/2020.10.13.20211664 (2020).
Gemmati, D. et al. COVID-19 and individual genetic susceptibility/receptivity: role of ACE1/ACE2 genes, immunity, inflammation and coagulation. might the double X-chromosome in females be protective against SARS-CoV-2 compared to the single X-chromosome in males? Int. J. Mol. Sci. 21, 3474 (2020).
Kugaevskaya, E. V. et al. Epitope mapping of the domains of human angiotensin converting enzyme. Biochim. Biophys. Acta 1760, 959–965 (2006).
Wang, E. Y. et al. Diverse functional autoantibodies in patients with COVID-19. Nature https://doi.org/10.1038/s41586-021-03631-y (2021).
Khamsi, R. Rogue antibodies could be driving severe COVID-19. Nature 590, 29–31 (2021).
Nalbandian, A. et al. Post-acute COVID-19 syndrome. Nat. Med. 27, 601–615 (2021).
Fajgenbaum, D. C. & June, C. H. Cytokine storm. N. Engl. J. Med. 383, 2255–2273 (2020).
Spittler, A. et al. Relationship between Interleukin-6 plasma concentration in patients with sepsis, monocyte phenotype, monocyte phagocytic properties, and cytokine production. Clin. Infect. Dis. 31, 1338–1342 (2000).
Xia, P. et al. Clinicopathological features and outcomes of acute kidney injury in critically ill COVID-19 with prolonged disease course: a retrospective cohort. J. Am. Soc. Nephrol. 31, 2205–2221 (2020).
Villa, G. et al. Blood purification therapy with a hemodiafilter featuring enhanced adsorptive properties for cytokine removal in patients presenting COVID-19: a pilot study. Crit. Care 24, 605 (2020).
Coomes, E. A. & Haghbayan, H. Interleukin-6 in Covid-19: a systematic review and meta-analysis. Rev. Med. Virol. 30, 1–9 (2020).
Leisman, D. E. et al. Cytokine elevation in severe and critical COVID-19: a rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 8, 1233–1244 (2020).
Kox, M., Waalders, N. J. B., Kooistra, E. J., Gerretsen, J. & Pickkers, P. Cytokine levels in critically ill patients with COVID-19 and other conditions. JAMA 324, 1565–1567 (2020).
Ichimura, T. et al. KIM-1/TIM-1 is a receptor for SARS-CoV-2 in lung and kidney. Preprint at medRxiv https://doi.org/10.1101/2020.09.16.20190694 (2020).
Dargelos, M. et al. Severe acute respiratory syndrome coronavirus 2 indirectly damages kidney structures. Clin. Kidney J. 13, 1101–1104 (2020).
Pan, X. et al. Identification of a potential mechanism of acute kidney injury during the COVID-19 outbreak: a study based on single-cell transcriptome analysis. Intensive Care Med. 46, 1114–1116 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020).
Hikmet, F. et al. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 16, e9610 (2020).
South, A. M., Tomlinson, L., Edmonston, D., Hiremath, S. & Sparks, M. A. Controversies of renin–angiotensin system inhibition during the COVID-19 pandemic. Nat. Rev. Nephrol. 16, 305–307 (2020).
Hamming, I. et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 203, 631–637 (2004).
Jiang, F. et al. Angiotensin-converting enzyme 2 and angiotensin 1–7: novel therapeutic targets. Nat. Rev. Cardiol. 11, 413–426 (2014).
Groß, S., Jahn, C., Cushman, S., Bär, C. & Thum, T. SARS-CoV-2 receptor ACE2-dependent implications on the cardiovascular system: From basic science to clinical implications. J. Mol. Cell Cardiol. 144, 47–53 (2020).
Davidson, A. M., Wysocki, J. & Batlle, D. Interaction of SARS-CoV-2 and other coronavirus with ACE (angiotensin-converting enzyme)-2 as their main receptor: therapeutic implications. Hypertension 76, 1339–1349 (2020).
Henry, B. M. et al. Coronavirus disease 2019 is associated with low circulating plasma levels of angiotensin 1 and angiotensin 1,7. J. Med. Virol. 93, 678–680 (2021).
Zoufaly, A. et al. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir. Med. 8, 1154–1158 (2020).
Serfozo, P. et al. Ang II (Angiotensin II) conversion to angiotensin-(1–7) in the circulation is POP (prolyloligopeptidase)-dependent and ACE2 (angiotensin-converting enzyme 2)-independent. Hypertension 75, 173–182 (2020).
Lippi, G., Lavie, C. J., Henry, B. M. & Sanchis-Gomar, F. Do genetic polymorphisms in angiotensin converting enzyme 2 (ACE2) gene play a role in coronavirus disease 2019 (COVID-19)? Clin. Chem. Lab. Med. 58, 1415–1422 (2020).
Hall, A., Busse, L. W. & Ostermann, M. Angiotensin in critical care. Crit. Care 22, 69 (2018).
Legrand, M. & Bokoch, M. P. The Yin and Yang of the renin-angiotensin-aldosterone system in AKI. Am. J. Respir. Crit. Care Med. 203, 1053–1055 (2020).
Dudoignon, E. et al. Activation of the renin-angiotensin-aldosterone system is associated with acute kidney injury in COVID-19. Anaesth. Crit. Care Pain. Med. 39, 453–455 (2020).
Ozkan, S. et al. Efficacy of serum angiotensin II levels in prognosis of patients with Coronavirus Disease 2019. Crit. Care Med. 49, e613–e623 (2021).
Liu, X. et al. Association of angiotensin converting enzyme inhibitors and angiotensin II receptor blockers with risk of COVID-19, inflammation level, severity, and death in patients with COVID-19: a rapid systematic review and meta-analysis. Clin. Cardiol. https://doi.org/10.1002/clc.23421 (2020).
Lopes, R. D. et al. Continuing versus suspending angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: impact on adverse outcomes in hospitalized patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) — The BRACE CORONA Trial. Am. Heart J. 226, 49–59 (2020).
Wysocki, J., Lores, E., Ye, M., Soler, M. J. & Batlle, D. Kidney and lung ACE2 expression after an ACE inhibitor or an Ang II receptor blocker: implications for COVID-19. J. Am. Soc. Nephrol. 31, 1941–1943 (2020).
Cohen, J. B. et al. Continuation versus discontinuation of renin-angiotensin system inhibitors in patients admitted to hospital with COVID-19: a prospective, randomised, open-label trial. Lancet Respir. Med. 9, 275–284 (2021).
Joannidis, M. et al. Lung–kidney interactions in critically ill patients: consensus report of the acute disease quality initiative (ADQI) 21 Workgroup. Intensive Care Med. 46, 654–672 (2020).
Darmon, M. et al. Diagnostic accuracy of Doppler renal resistive index for reversibility of acute kidney injury in critically ill patients. Intensive Care Med. 37, 68–76 (2011).
Fogagnolo, A. et al. Focus on renal blood flow in mechanically ventilated patients with SARS-CoV-2: a prospective pilot study. J. Clin. Monit. Comput. https://doi.org/10.1007/s10877-020-00633-5 (2021).
Vasquez-Bonilla, W. O. et al. A review of the main histopathological findings in coronavirus disease 2019. Hum. Pathol. 105, 74–83 (2020).
Barthélémy, R. et al. Haemodynamic impact of positive end-expiratory pressure in SARS-CoV-2 acute respiratory distress syndrome: oxygenation versus oxygen delivery. Br. J. Anaesth. 126, e70–e72 (2021).
Kawakami, R. et al. Pathological evidence for SARS-CoV-2 as a cause of myocarditis: JACC review topic of the week. J. Am. Coll. Cardiol. 77, 314–325 (2021).
Bajaj, R. et al. Delayed-onset myocarditis following COVID-19. Lancet Respir. Med. 9, e32–e34 (2021).
Kotecha, T. et al. Patterns of myocardial injury in recovered troponin-positive COVID-19 patients assessed by cardiovascular magnetic resonance. Eur. Heart J. 42, 1866–1878 (2021).
Legrand, M., Mebazaa, A., Ronco, C. & Januzzi, J. L. When cardiac failure, kidney dysfunction, and kidney injury intersect in acute conditions: the case of cardiorenal syndrome. Crit. Care Med. 42, 2109–2117 (2014).
Legrand, M. et al. Association between systemic hemodynamics and septic acute kidney injury in critically ill patients: a retrospective observational study. Crit. Care 17, R278 (2013).
Cruces, P. et al. Renal decapsulation prevents intrinsic renal compartment syndrome in ischemia-reperfusion-induced acute kidney injury: a physiologic approach. Crit. Care Med. 46, 216–222 (2018).
Chaibi, K. et al. Severe acute kidney injury in patients with COVID-19 and acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 202, 1299–1301 (2020).
Hansrivijit, P., Gadhiya, K. P., Gangireddy, M. & Goldman, J. D. Risk factors, clinical characteristics, and prognosis of acute kidney injury in hospitalized COVID-19 patients: a retrospective cohort study. Medicines 8, 4 (2021).
Bell, S. et al. Risk of AKI with gentamicin as surgical prophylaxis. J. Am. Soc. Nephrol. 25, 2625–2632 (2014).
Sinha Ray, A., Haikal, A., Hammoud, K. A. & Yu, A. S. L. Vancomycin and the risk of AKI: a systematic review and meta-analysis. Clin. J. Am. Soc. Nephrol. 11, 2132–2140 (2016).
Sang, L. et al. The incidence, risk factors and prognosis of acute kidney injury in severe and critically ill patients with COVID-19 in mainland China: a retrospective study. BMC Pulm. Med. 20, 290 (2020).
Beigel, J. H. et al. Remdesivir for the treatment of Covid-19 — final report. N. Engl. J. Med. 383, 1813–1826 (2020).
Spinner, C. D. et al. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19: a randomized clinical trial. JAMA 324, 1048–1057 (2020).
Gérard, A. O. et al. Remdesivir and acute renal failure: a potential safety signal from disproportionality analysis of the WHO safety database. Clin. Pharmacol. Ther. 109, 1021–1024 (2020).
Binois, Y. et al. Acute kidney injury associated with lopinavir/ritonavir combined therapy in patients with COVID-19. Kidney Int. Rep. 5, 1787–1790 (2020).
Geng, Y. et al. Rhabdomyolysis is associated with in-hospital mortality in patients with COVID-19. Shock https://doi.org/10.1097/SHK.0000000000001725 (2021).
Joannidis, M. & Forni, L. G. Severe viral infection and the kidney: lessons learned from the H1N1 pandemic. Intensive Care Med. 37, 729–731 (2011).
Shaefi, S. et al. Extracorporeal membrane oxygenation in patients with severe respiratory failure from COVID-19. Intensive Care Med. 47, 208–221 (2021).
Schmidt, M. et al. Extracorporeal membrane oxygenation for severe acute respiratory distress syndrome associated with COVID-19: a retrospective cohort study. Lancet Respir. Med. 8, 1121–1131 (2020).
Langenberg, C., Wan, L., Egi, M., May, C. N. & Bellomo, R. Renal blood flow and function during recovery from experimental septic acute kidney injury. Intensive Care Med. 33, 1614–1618 (2007).
Joannidis, M. et al. Neutrophil transmigration in renal proximal tubular LLC-PK1 cells. Cell Physiol. Biochem. 14, 101–112 (2004).
Bellomo, R. et al. Acute kidney injury in sepsis. Intensive Care Med. 43, 816–828 (2017).
Xu, C. et al. TNF-mediated damage to glomerular endothelium is an important determinant of acute kidney injury in sepsis. Kidney Int. 85, 72–81 (2014).
Netti, G. S. et al. LPS removal reduces CD80-mediated albuminuria in critically ill patients with Gram-negative sepsis. Am. J. Physiol. Renal Physiol 316, F723–F731 (2019).
Lerolle, N. et al. Histopathology of septic shock induced acute kidney injury: apoptosis and leukocytic infiltration. Intensive Care Med. 36, 471–478 (2010).
Bernardo, A., Ball, C., Nolasco, L., Moake, J. F. & Dong, J. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood 104, 100–106 (2004).
Peerapornratana, S., Manrique-Caballero, C. L., Gómez, H. & Kellum, J. A. Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 96, 1083–1099 (2019).
Langenberg, C., Bagshaw, S. M., May, C. N. & Bellomo, R. The histopathology of septic acute kidney injury: a systematic review. Crit. Care 12, R38 (2008).
Nin, N. et al. Kidney histopathological findings in fatal pandemic 2009 influenza A (H1N1). Intensive Care Med. 37, 880–881 (2011).
Liu, K. D. et al. Acute kidney injury in patients with acute lung injury: impact of fluid accumulation on classification of acute kidney injury and associated outcomes. Crit. Care Med. 39, 2665–2671 (2011).
Liu, K. D. et al. Predictive and pathogenetic value of plasma biomarkers for acute kidney injury in patients with acute lung injury. Crit. Care Med. 35, 2755–2761 (2007).
Darmon, M. et al. Impact of mild hypoxemia on renal function and renal resistive index during mechanical ventilation. Intensive Care Med. 35, 1031–1038 (2009).
Nadim, M. K. et al. COVID-19-associated acute kidney injury: consensus report of the 25th acute disease quality initiative (ADQI) Workgroup. Nat. Rev. Nephrol. 16, 747–764 (2020).
Chiumello, D. et al. Physiological and quantitative CT-scan characterization of COVID-19 and typical ARDS: a matched cohort study. Intensive Care Med. 46, 2187–2196 (2020).
WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group. Association between administration of systemic corticosteroids and mortality among critically ill patients with COVID-19: a meta-analysis. JAMA 324, 1330–1341 (2020).
RECOVERY Collaborative Group. et al. Dexamethasone in hospitalized patients with Covid-19. N. Engl. J. Med. 384, 693–704 (2021).
REMAP-CAP Investigators et al. Interleukin-6 receptor antagonists in critically ill patients with Covid-19. N. Engl. J. Med. 384, 1491–1502 (2021).
RECOVERY Collaborative Group. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet 397, 1637–1645 (2021).
Rahmani, H. et al. Interferon β-1b in treatment of severe COVID-19: a randomized clinical trial. Int. Immunopharmacol. 88, 106903 (2020).
Davoudi-Monfared, E. et al. A randomized clinical trial of the efficacy and safety of interferon β-1a in treatment of severe COVID-19. Antimicrob. Agents Chemother. 64, e01061–20 (2020).
Hadjadj, J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724 (2020).
STARRT-AKI Investigators. et al. Timing of initiation of renal-replacement therapy in acute kidney injury. N. Engl. J. Med. 383, 240–251 (2020).
Gaudry, S. et al. Initiation strategies for renal-replacement therapy in the intensive care unit. N. Engl. J. Med. 375, 122–133 (2016).
Sia, S. F. et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 583, 834–838 (2020).
Bao, L. et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 583, 830–833 (2020).
Monteil, V. et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 181, 905–913.e7 (2020).
Jiang, S., Zhang, X., Yang, Y., Hotez, P. J. & Du, L. Neutralizing antibodies for the treatment of COVID-19. Nat. Biomed. Eng. 4, 1134–1139 (2020).
U.S. National Library of Medicine. Clinicaltrials.gov https://clinicaltrials.gov/ct2/show/NCT04335136 (2021).
Acknowledgements
This manuscript was written by members and on behalf of the Acute Disease Quality Initiative (ADQI) group.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Nephrology thanks D. Batlle, A. Salama and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Glossary
- Autolysis
-
The destruction of cells through the action of self-produced enzymes after death.
- Damage-associated molecular patterns
-
Endogenous danger molecules that are released from damaged or dying cells and activate the innate immune system by interacting with pattern recognition receptors.
- Anti-idiotype
-
Antibody that binds to the antigen-combining site of another antibody, either suppressing or enhancing the immune response.
- Positive end-expiratory pressure
-
Pressure maintained in the airway at the end of expiration during mechanical ventilation to prevent the lung from collapse
- Tidal volumes
-
Volume of air insufflated during a respiratory cycle (here in mechanical ventilation).
- Glomerular shunting
-
The flow of blood from preglomerular arterial vessels to post-glomerular venous vessels that does not contribute to the perfusion of peritubular capillaries.
- Pathogen-associated molecular patterns
-
Small molecular motifs that are associated with pathogens; they are recognized by pattern-recognition receptors and initiate an innate immune response.
Rights and permissions
About this article

Cite this article
Legrand, M., Bell, S., Forni, L. et al. Pathophysiology of COVID-19-associated acute kidney injury. Nat Rev Nephrol 17, 751–764 (2021). https://doi.org/10.1038/s41581-021-00452-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41581-021-00452-0
