
Abstract
The pearlspot, Etroplus suratensis is a climate resilient cichlid fish that exhibits unusual adaptation to salinity. The fish is able to complete full life cycle in diverse salinity habitats ranging from fresh water to marine environments. High-quality primary and phased genome assemblies were generated for pearlspot fish using PacBio HiFi and Arima HiC sequencing technologies, for the first time. The primary assembly is highly contiguous with contig N50 length of 36 Mb. The final assembly is of 1.247 Gb with N50 length of 51.57 Mb and 98% of the genome length anchored to 24 chromosomes. The genome was assessed to be 99.9% complete based on BUSCO evaluation and was predicted to contain 52.96% repeat elements. We have predicted 27,192 protein encoding genes, of which 21,580 were functionally annotated. The genome offers an invaluable resource to understand adaptation of pearlspot fish to diverse salinity habitats.
Similar content being viewed by others


Background and Summary
Cichlid fishes characterized by rapid adaptive radiation and sympatric speciation1 serve as excellent model species to understand divergent evolution. Existence of vast number of closely related cichlid species within the confines of a single geographical environment2 displaying wide phenotypic variation makes them an ideal model system to understand the genetic basis of vertebrate speciation3. While the genomic resources of African cichlid fishes of the subfamily Pseudocrenilabrinae are being extensively used to understand vertebrate speciation, the resources for Asian cichlid fish of subfamily Etroplinae are scanty.
The Etroplus suratensis (Bloch, 1790) commonly known as pearlspot or green chromide is an edible fish of the subfamily Etroplinae. This substrate spawning cichlid fish (Fig. 1) is characterized by elaborate courtship and multiple parental care4. Though brackishwater is principal habitat for this herbivorous fish, it displays great adaptations to salinity by surviving and breeding in freshwater habitats. Only two species of the subfamily Etroplinae, Etroplus canarensis and Paratilapia polleni have draft genome assemblies available in public repository5. Both of these assemblies were generated with short DNA reads and have thousands of scaffolds with N50 lengths around 20 Kb. Therefore, at present, we do not have a chromosome-scale reference genome for the subfamily Etroplinae.

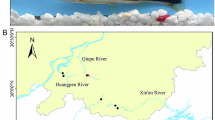
A specimen of Etroplus suratensis.
In this study, we have used PacBio HiFi technology to generate a highly contiguous genome assembly contigs for pearlspot fish with assembly length of 1.276 Gb and N50 length of 36.16 Mb. Then, the Arima HiC technology was used to order and orient contigs to 24 chromosome-scale scaffolds (Fig. 2). The combination of these two sequencing technologies resulted in the generation of a chromosome-scale genome assembly which is of 1.247 Gb length in 117 scaffolds with N50 length of 51.57 Mb (Table 1). The assembly length is closer to the estimate obtained with flow cytometry method (1.195 Gb) than the estimate obtained with k-mer based analysis (1.103 Gb) using short DNA reads (Fig. 3). A k-mer based analyses estimated the consensus quality value (QV) of 60 for the genome assembly and 98% of assembly length was anchored to 24 chromosomes with telomere ends. Additionally, the haplotype-resolved assemblies generated using a combination of HiFi and HiC reads are of 1.242 Gb and 1.225 Gb with N50 statistic of 51.89 Mb and 51.40 Mb, respectively.

(a) Hi-C map representing the 24 Chromosomes of Etroplus suratensis genome assembly (b) Circos plot representation of the 24 chromosomes of Etroplus suratensis. From the outermost: Track1: The 24 chromosomes of the pearlspot genome. Track2: Contigs corresponding to the chromosomes represented as tiles. Track3: Representation of telomeric repeats in log scale at chromosome ends. Track4: Quality values (QV) across the chromosomes shown as line plot with red, yellow and green backgrounds representing ranges 0 - 20, 20 – 30 and 30 -100 respectively. Track5 - Track8: Major repeat elements, SINEs, LINEs, LTRs and DNA Transposons, respectively, shown as scatter plot with a 2 kb sliding window. Track9: Predicted protein-coding genes in chromosomes depicted as highlights with incremental gene lengths of 20 Kb [viz. <20 kb (Very dark orange), 20 kb - 40 kb (dark orange), 40 kb - 60 kb (orange), 60 kb - 80 kb (Light orange) and >80 kb (Green)]. Track10: GC content of pearlspot genome shown as line diagram plotted with 50 kb sliding window. The GC values below 35 and above 45 are shown in dark red color, and remaining in orange color.

Genome size estimation profiles of Etroplus suratensis. (a) Flow cytometry principle. Histogram depicting the count of events for Etroplus suratensis blood cells and chicken erythrocytes. (b) Genome length assessment of Etroplus suratensis by k-mer frequency generated using Jellyfish and Genomescope.
A custom repeat library consisting of 2,112 repeat families obtained through de novo modelling of repeat elements in the assembly was used to identify and classify the repeat elements in the pearlspot genome. The repeat elements accounted for 52.96% (Table 2) of the genome predominated by LINEs (20.2%), DNA transposons (16.71%) and LTR elements (3.85%). A strategy that combines evidence generated using Illumina RNAseq reads, PacBio Iso-Seq reads, ab initio methods and predicted proteins in related-species genomes resulted in the prediction of 27,192 protein-encoding genes (PEGs) in pearlspot genome. (Table 3). Further, 18,089 non-coding RNAs were detected, with abundant presence of tRNA, ribosomal RNA, spliceosomal RNA, microRNA and Small nucleolar RNA (Supplementary Table 1). The high-quality genome resource would help in specific understanding of salinity tolerance and parental care of pearlspot fish and also the evolution of cichlid fish in general.
Methods
Specimen for generating sequence data
A single specimen of male pearlspot fish was used to generate the sequence data required for building the genome assembly. The lineage of the specimen was confirmed based on the analysis of the barcoding gene, Cytochrome C Oxidase I (CO I). Briefly, sequence of partial CO I gene of the specimen was generated (MG923355) following amplification with universal primers6. The CO I sequence of other accessions under subfamily Etroplinae were sourced from BOLD system v4 database7 along with Oreochromis niloticus accession as outgroup (Supplementary Table 2). The sequences aligned with MUSCLE module of MEGA X8,9 were used to build a Maximum Likelihood tree with HKY + G + I model and 1000 bootstrap iterations in MEGA X9. (Supplementary Figure S1).
DNA sequence reads
In this study, three types of DNA sequence data, short reads, long high-fidelity (HiFi) reads and chromatin linked reads (HiC) were generated. The short reads were used to assess the genome properties, the HiFi reads were used for generating genome assembly contigs and HiC reads were used for building assembly scaffolds. Briefly, high molecular weight genomic DNA was isolated from muscle tissue of a single male fish using QIAGEN Genomic-tip 100/G Midi kit (Qiagen, Hilden, Germany). DNA quantity was measured with Qubit 3.0 fluorometer (Thermofisher Scientific, Massachusetts, USA) using DNA HS assay kit (Thermofisher Scientific, Massachusetts, USA) and DNA purity was checked with NanoDrop 2000 (Thermofisher Scientific, Massachusetts, USA). DNA integrity was evaluated on 1% agarose gel and on Femto pulse system (Agilent Technologies, California, USA). DNA shearing was performed on Megaruptor 3 system (Diagenode, Belgium). Three separate sequencing libraries were constructed using the SMRTbell Express template Preparation Kit 2.0 (Pacific Biosciences, California, USA). The libraries were purified using AMPure PB beads (Pacific Biosciences, California, USA) and the purified libraries were treated with SMRTbell Enzyme cleanup kit 2.0 to remove any unbound adapters and damaged DNA. The libraries were size selected using BluePippin (Sage Science, USA) with 0.75% DF Marker S1 High pass Cassette. The size selected libraries were subjected to primer annealing and polymerase binding using Sequel II binding kit 2.2. About 75 to 80 pM of each library was loaded onto individual 8 M SMRT cells (n = 3) and sequenced on PacBio Sequel II system in CCS/HiFi mode to generate polymerase read sequences. Later, the raw polymerase reads were processed with ccs algorithm v6.4.0 (–min-passes = 3;–min-snr = 2.5;–min-rq = 0.99) to generate HiFi reads. The HiFi read recovery from polymerase read bases was 50.7% and 4.87% for HiFi read number and HiFi read bases, respectively (Table 4).
The same DNA was used to construct a sequencing library with KAPA HyperPlus kit (Basel, Switzerland) as per manufacturers’ protocol. The quality of the library was assessed using Agilent 2100 bioanalyzer (Agilent Technologies, California, USA). The libraries with average insert size of 571 bp were sequenced on Illumina Novaseq6000. These short DNA reads were only used to understand the properties of the genome (Table 5).
The HiC library was constructed using Proximo HiC Kit, animal (Phase genomics, USA) as per the manufacturer’s instructions. About 10 nM of library was sequenced using S4 flow cell on Illumina Novaseq6000 in paired-end mode to generate 150 bp linked reads. The restrictions enzymes used to prepare HiC library from fish sample were DpnII, DdeI, HinfI, and MseI (Table 6).
In total, 9.975 million HiFi reads (98.18 Gb, 78.7 X), 992.5 million short reads (149.87 Gb, 120.2 X, 92.3% Q30 bases) and 4.398 billion HiC reads (659.83 Gb, 529.1 X, 89% Q30 bases) of DNA sequence data has been generated.
RNA sequence reads
The RNA sequence reads were generated using specimens of various development stages (1-, 3-, and 15-day old larvae) and tissues (muscle, skin, kidney, liver, stomach, intestine, gill, brain, spleen, testis, and heart) collected from the same adult male Pearlspot fish. Briefly, the total RNA was isolated by using Trizol (DSS Takara, CA, USA) and purified with Nucleospin RNA cleanup kit (Macherey-Nagel, Germany). RNA quantification was performed with Qubit3.0 fluorometer using RNA HS assay kit (ThermoFisher Scientific, Massachusetts, USA) and on Nanodrop 2000. The quality and integrity of RNA was checked on Agilent 2100 bioanalyzer. The cDNA library was prepared with KAPA HyperPrep kit (Roche, Basel, Switzerland) and sequenced on Illumina Novaseq6000 to generate 2 × 150 bp paired-end reads. The raw reads were trimmed with Trimmomatic v0.3910 to obtain clean reads with Q30 bases above 90% (Table 7).
Genome size assessment
An assessment of genome size was made on flow cytometry principle with the blood sample following propidium iodide staining in BD AccuriTM C6 flow cytometer11,12. The Chicken erythrocytes from BDTM DNA QC Particles kit (BD Biosciences, California, USA) was used as control. The histogram data analyzed with BD AccuriTM C6 Plus software v1.0.23.1 indicated the estimated genome size as 1.22 pg (1.195 Gb) for pearlspot fish (Fig. 3a). The assessment of genome size was also made with DNA sequence reads on k-mer principle. The DNA short reads were subjected to quality trimming with Trimmomatic v0.3910 to obtain 78.6 Gb (79X) of clean reads with 96.2% Q30 bases. An assessment of genome properties was made with these clean reads using jellyfish v2.3.013 and GenomeScope v2.014 based on k-mer count and coverage principle. The 21-mer based histogram indicated that the estimated genome length, repeat content and heterozygosity of Pearlspot genome is 1.103 Gb, 28.3% and 0.228%, respectively (Fig. 3b).
Genome assembly
The HiFi reads were initially screened with NCBI foreign contamination screen15 to discard contaminants originating from adaptor/vector and foreign organisms. About 78 X coverage of HiFi reads were used to generate assembly contigs with Hifiasm v0.16.1 tool16. There were 375 contigs with a total length of 1.276 Gb and N50 length of 36.16 Mb. Then the haplotigs and the overlaps in the primary assembly were removed with purge-dups17. Thereafter, about 3.2 billion HiC reads (473.99 Gb, 380 X, 94% Q30 bases) obtained after quality trimming of raw reads with fastp v0.12.418 were used for ordering and orienting the assembly contigs to final scaffolds with YaHS v1.119. The final assembly consisted of 117 scaffolds with a total length of 1.247 Gb and N50 length of 51.57 Mb. The assembly was assessed for its completeness by benchmarking with single-copy orthologs of actinopterygii_odb10 (2021-02-19) using BUSCO v5.7.020. Of the 3,640 BUSCO orthologs, 3,584 were complete and single-copy genes, 29 were complete and duplicated genes, 21 were fragmented genes and 6 were missing genes which indicated that the genome assembly has 99.9% completeness with 0.1% missing genomic regions. The phased assemblies obtained by following the similar methodology were assessed to be 99.2% and 98.7% complete based on BUSCO scores.
Repeat prediction
The RepeatModeler v 2.0.5 (http://repeatmasker.org/RepeatModeler/) enabling LTR structural analysis was used with rmblast v2.14.1 search engine to model and find de novo repeat elements in the pearlspot genome. The analysis identified 1,924 RepeatScout/RECON families and 281 LTRPipeline families. After removing redundant LTR families, a custom repeat library with 2,112 repeat families was established. Then, RepeatMasker v 4.1.521 with rmblast v2.14.1 search engine was used with custom repeat library to identify and classify the repeat elements in pearlspot genome assembly. The repeat elements accounted for 52.96% (Table 2) of the genome predominated by LINEs (20.2%), DNA transposons (16.71%) and LTR elements (3.85%).
Genome annotation
A strategy described earlier22,23 that combines evidence generated using Illumina RNAseq reads (generated in this study), PacBio Iso-Seq reads (GenBank accessions, SRR28827909-916), ab initio methods and predicted proteins from related-species genomes (Supplementary Table 3) has been used to predict protein-encoding genes (PEGs). Overall, five different evidences were used which were, (1) ab initio predictions obtained with AUGUSTUS v3.4.024; (2) predictions with AUGUSTUS v3.4.024 based on hints generated with Iso-Seq reads using GMAP v2017.11.15-425; (3) predictions obtained based on predicted proteins from genomes of related species using BRAKER v2.0.426 and GenomeThreader 1.7.327; (4) Iso-Seq reads derived transcript evidence obtained with GMAP v2017.11.15-425; and (5) RNAseq reads derived transcript evidence obtained with Hisat v2.2.1-428, Stringtie v2.2.129 and TransDecoder v5.7.0. All the five evidences were combined using EVidenceModeler v2.0.030 to arrive at the consensus prediction of PEGs. The Pearlspot genome assembly was predicted to contain 27,192 PEGs with mean exon number of 9 per gene (Table 3). Annotation and pathway analysis31,32 of PEGs (Table 8 and Supplementary Figures S2 to S5) were performed by combining results from blastx tool against Actinopterygii (txid7898) dataset of non-redundant database from NCBI, and InterProScan33 and EggNOG34 mapper module of OmicsBox Tool v3.0.2535.
The identification of noncoding genes in the Pearlspot genome involved aligning the repeat-masked assembly with the Rfam database [http://rfam.xfam.org/], using cmscan from infernal v1.1.236. A total of 18,089 non-coding RNAs were detected, with abundant presence of tRNA, ribosomal RNA, spliceosomal RNA, microRNA and Small nucleolar RNA (Supplementary Table 1).
Data Records
The raw datasets were deposited under Sequence Read Archive (SRA) at NCBI with the accession numbers, SRR27970333, SRR27999027-029, SRR28233220, SRR28003587-595, SRR28003597-599, SRR28003601-602. The genome was submitted under Genome category at NCBI with the Genome assembly accession number, GCA_041004005.137. All the raw datasets were linked to the Bioproject, PRJNA107666238 and SRA study SRP48980339. The genome annotations were submitted to the Figshare repository with the https://doi.org/10.6084/m9.figshare.26303968.v332.
Technical Validation
The full-length mitochondrial DNA genome (Fig. 4) of 16,467 bp was obtained as a single scaffold in the final assembly suggesting the sufficiency of 78 X coverage of HiFi reads and 380 X coverage of HiC reads. The assembly generated for pearlspot fish is highly contiguous as indicated by contig N50 of 36.16 Mb. The assembly was assessed to be containing 99.3% of complete and 0.6% of the fragmented genes when benchmarked with actinopterygii_odb10 (2021-02-19) lineage using BUSCO v5.7.020 (Fig. 5). About 98.03% of the assembly length is represented in the longest 24 scaffolds indicating chromosome-scale nature of the assembly. The consensus quality value and error rate of the genome assembly were assessed to be 60.0762 and 9.82596e-07, respectively when validated with k-mer (31-mer) based procedure executed in Merqury v1.340 indicating high base accuracy of the assembly. The good alignment statistics (Table 9) obtained by aligning RNAseq reads and DNA short reads on to the genome further validated the accuracy of the assembly. The chromosome-scale scaffolds were searched for the presence of telemore repeat sequences using tidk v0.2.041 with Cypriniformes clade (AACCCT). All the scaffolds were observed to be having telomere ends (Fig. 2b,Track 3). The genome assembly has shown good synteny (Fig. 6) with other closely related cichlid fish genomes. About 21,580 (79.36%) of the protein-encoding genes could be annotated functionally (Table 8).

Etroplus suratensis mitochondrial genome map depicting the protein coding genes, D-loop, rRNA genes, tRNA genes, GC content and GC Skew.

Completeness analysis using BUSCO for Etroplus suratensis genome against Actinopterygii_odb10 orthologous database.

(a) Synteny map between Pearlspot and Spotted tilapia (b) Synteny map between Pearlspot and Nile tilapia.
Code availability
All data processing programs were executed with default parameters unless otherwise specified in the Methods section. There were no custom scripts or code utilized in this study.
References
Schliewen, U. K., Tautz, D. & Pääbo, S. Sympatric speciation suggested by monophyly of crater lake cichlids. Nature 368, 629–632 (1994).
Ronco, F. et al. Drivers and dynamics of a massive adaptive radiation in cichlid fishes. Nature 589, 76–81 (2021).
Kocher, T. D. Adaptive evolution and explosive speciation: the cichlid fish model. Nat Rev Genet 5, 288–298 (2004).
Ward, J. A. & Wyman, R. L. Ethology and ecology of cichlid fishes of the genus Etroplus in Sri Lanka: preliminary findings. Environ Biol Fishes 2, 137–145 (1977).
Matschiner, M., Böhne, A., Ronco, F. & Salzburger, W. The genomic timeline of cichlid fish diversification across continents. Nat Commun 11 (2020).
Ward, R. D., Zemlak, T. S., Innes, B. H., Last, P. R. & Hebert, P. D. N. DNA barcoding Australia’s fish species. Philosophical Transactions of the Royal Society B: Biological Sciences 360, 1847–1857 (2005).
Ratnasingham, S. & Hebert, P. D. N. BOLD: The Barcode of Life Data System: Barcoding. Mol Ecol Notes 7, 355–364 (2007).
Edgar, R. C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797 (2004).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35, 1547–1549 (2018).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Swathi, A., Shekhar, M. S., Katneni, V. K. & Vijayan, K. K. Genome size estimation of brackishwater fishes and penaeid shrimps by flow cytometry. Mol Biol Rep 45, 951–960 (2018).
Raymond, J. A. J. et al. Comparative genome size estimation of different life stages of grey mullet, Mugil cephalus Linnaeus, 1758 by flow cytometry. Aquac Res 53, 1151–1158 (2022).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun 11 (2020).
Astashyn, A. et al. Rapid and sensitive detection of genome contamination at scale with FCS-GX. Genome Biol 25 (2024).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39 (2023).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol 38, 4647–4654 (2021).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. 2013–2016.
Katneni, V. K. et al. A Superior Contiguous Whole Genome Assembly for Shrimp (Penaeus indicus). Front Mar Sci 8 (2022).
Shekhar, M. S. et al. First Report of Chromosome-Level Genome Assembly for Flathead Grey Mullet, Mugil cephalus (Linnaeus, 1758). Front Genet 13, 911446 (2022).
Hoff, K. J. & Stanke, M. Predicting Genes in Single Genomes with AUGUSTUS. Curr Protoc Bioinformatics 65, 1–54 (2019).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom Bioinform 3, 1–11 (2021).
Gremme, G. Computational gene structure prediction. Staats-und Universitätsbibliothek Hamburg Carl von Ossietzky, (2012).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295 (2015).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, 1–22 (2008).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45, D353–D361 (2017).
Vinaya Kumar Katneni et al. Etroplus suratensis genome and annotation. Figshare at https://doi.org/10.6084/m9.figshare.26303968.v3 (2024).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol Biol Evol 34, 2115–2122 (2017).
Omicsbox. OmicsBox-Bioinformatics made easy (Version 3.0.25) (2019).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
NCBI Genome Assembly Database http://identifiers.org/assembly:GCA_041004005.1 (2024).
NCBI BioProject http://identifiers.org/bioproject:PRJNA1076662 (2024).
NCBI Sequence Read Archive http://identifiers.org/insdc.sra:SRP489803 (2024).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: Reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21 (2020).
Brown, M., la Rosa, P. M. & Mark, B. A Telomere Identification Toolkit. Zenodo https://doi.org/10.5281/zenodo.10091385 (2023).
Acknowledgements
This work was carried out in the project entitled ‘Unravelling signatures of growth and salinity adaptation in Etroplus suratensis through omics approaches’ funded by Department of Biotechnology, Government of India (BT/PR34518/AAQ/3/965/2019). The authors are thankful to Director, ICAR-CIBA for providing necessary support in executing this research work. We acknowledge Nucleome Informatics Private Limited for help in generating the sequence data.
Author information
Authors and Affiliations
Contributions
V.K.K. and M.S.S. conceived the study. V.K.K., K.K., S.K.P., R.J., A.K.J., N.K. and K.J. performed analysis. N.Q., K.V. and J.R.J.A. prepared the material. V.K.K., A.K.J., K.K. and S.K.P. drafted the manuscript. All authors contributed to final manuscript editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Katneni, V.K., Krishnan, K., Prabhudas, S.K. et al. Genome assembly at chromosome scale with telomere ends for Pearlspot, Etroplus suratensis. Sci Data 11, 1226 (2024). https://doi.org/10.1038/s41597-024-04096-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-04096-0
