
Abstract
Vigna radiata (L.) R. Wilczek var. radiata (mungbean) is a pulse crop important for both the global protein security and sustainable crop production. Here, to facilitate genomics-assisted breeding programs in mungbean, we present a high-quality reference genome originating from the crop’s centre of origin, India. In this study, we present a significantly continuous genome assembly of V. radiata Indian cultivar, achieved through a combination of long-read PacBio HiFi sequencing and Hi-C sequencing. The total assembled genome size is ~596 Mb equating to ~98% of the predicted genome size complemented by a contig N50 value of 10.35 Mb and a BUSCO score of 98.5%. Around 502 Mb of the assembled genome is anchored on 11 pseudochromosomes conforming to the chromosome count in the crop with distinctly identified telomeres and centromeres. We predicted a total of 43,147 gene models of which 39,144 protein coding genes were functionally annotated. The present assembly was able to resolve several gaps in the genome and provides a high-quality genomic resource for accelerating mungbean breeding programs.
Similar content being viewed by others


Background & Summary
Vigna radiata (L.) R. Wilczek var. radiata (NCBI: txid3916), commonly known as greengram or mungbean is a member of subfamily Papilionoideae of Fabaceae, holding a taxonomical position in genera Vigna, subgenus Ceratotropis and sectionA Ceratotropis. The legume crop is diploid in nature with a chromosome number of 2n = 2x = 22 and an estimated genome size varying in the range of 494 Mb1 to 579 Mb2. Mungbean is believed to have undergone domestication from its wild progenitor, V. radiata var. sublobata (Roxb.) Vercourt approximately 4,000 to 4,500 years ago in India3. At present, the crop is majorly cultivated in South, East to Southeast Asian regions due to its adaptability to diverse ecological conditions with a growth habitat favouring semiarid and subtropical regions. It is a crucial legume crop functioning centrally as one of the major sources of dietary proteins in a cereal-based society4. Moreover, mungbean plays a vital role in enhancing soil nitrogen owing to its nitrogen fixing capabilities in symbiosis with members of the Rhizobium sp. The soil enrichment capabilities complemented by its short life span encourages its regular deployment in the crop rotation programmes5. The conventional mungbean breeding methods have faced challenges in achieving significant yield advancements. Despite emphasis on traits like short duration, photo and thermo-insensitivity, synchronous maturity, and disease resistance, the genetic limitations of the single cross pedigree method have become apparent6,7. The loss of variability in early generations and the highly homozygous nature hinder further productivity gains. To overcome these challenges, strategic parental selection, evaluation based on genetic diversity and combining ability, and intermittent selections in later generations are suggested to harness a broader spectrum of desirable genes. Additionally, this lack of progress is also attributed to the influence of abiotic and biotic factors4,7. Emphasizing the need for resource development and innovative strategies to harness the full potential of mungbean breeding, a shift towards high-quality genome sequencing becomes imperative for comprehensive understanding and targeted improvement of this vital crop.
Over the years, legume genomics has experienced a significant advancement marked by establishment of new draft genome assemblies for various members of genus Vigna including cowpea8, adzuki bean9, asparagus bean10, lima bean11, blackgram12, rice bean13, as well as the allopolyploid, Vigna reflex pilosa and its progenitors, Vigna hirtella and Vigna trinervia14. The first draft genome of Vigna radiata var. radiata VC1973A (Origin: Thailand) was reported in 2014 from AVRDC-The World Vegetable Centre Taiwan wherein short reads from Illumina and long read GL Flex + were combined with a linkage map to anchor the scaffolds into 11 chromosomes15. This was followed by an improved version of the assembly from a recombinant inbred line originating from V. radiata cultivar VC1973A and a Korean landrace V2984 with greater contiguity rectifying few mis-assembled regions of the previous assembly16. Recently, a major advancement in the mungbean genomics has been achieved by introduction of more complete genome Jilv 7 (Vrad JL7 Origin: China) and a pangenome which encapsulates a broad spectrum of diversity existing in the species17. While these reference genomes are valuable resources, a notable gap is the absence of a reference genome assembly derived from India, the centre of origin of mungbean. A reference genome is indispensable not only for comprehending the evolutionary history, domestication and selection processes that shaped the current genetic composition but also for mitigating reference biases while utilizing the data for improvement of local varieties and cultivars. Moreover, given that India is the largest producer and consumer of mungbean (Directorate of Pulses Development (DPD) Annual Report 2021–2022 https://dpd.gov.in/AbtRep.html), a reference genome derived from a local line would be a more advantageous choice for implementation in breeding programs.
In the present study, we established a chromosomal-level genome assembly of Indian mungbean cultivar, IPM02-03 using HiFi reads from Pacific Biosciences of California Inc. (PacBio) Sequel II system combined with in vivo chromosome fixation Hi-C sequencing. The cultivar IPM02-03 is prized for exhibiting high resistance against mungbean yellow mosaic virus (MYMV) and tolerance to major pests. The present genome assembly displays high quality metrics across all aspects of the genomic features including contiguity and annotation. The sequenced genome (hereafter referred to as “VR_IPM02-03”) is anchored to 11 pseudochromosomes encompassing ~502 Mb (84.22%) of the total genome size with a cumulative length of ~596 Mb and a scaffold N50 of 46.42 Mb. Our final assembly incorporated an additional ~121 Mb to the total genome with an augmentation of ~33 Mb onto the chromosomes compared to the earlier published mungbean genome Vrad_JL717, which is currently regarded as the most contiguous assembly available for V. radiata. Transposable elements (TEs) constitute 56.38% (~336.36 Mb) of the total genome with long terminal repeats (LTRs) of RNA transposons emerging as the most predominant TE type in mungbean. Based on ab initio and evidence-based exploration, we predicted a total of 43,147 gene models in mungbean, of which 39,144 protein coding genes were functionally annotated. We identified 3,475 unique protein coding genes from the Indian cultivar, of which 1,980 were not captured even in the reported pangenome of Vigna radiata17. In addition, the present mungbean assembly VR_IPM02-03 uncovered end to end telomere signatures and centromeric repeat signals in almost all the pseudo-chromosomes. We were able to identify telomeric and centromeric repeats in all the pseudo-chromosomes of VR_IPM02-03 except chromosome number 9 (VR_pChr9). We identified two main CEN repeats of sizes 177 bp (CEN177) and 174 bp (CEN174) wherein, the CEN174 repeats were observed to delineate the starting junction for centromeric regions in mungbean. Further, our findings were able identify and resolve several junctions of mis-assembled regions in Vrad_JL717. Thus, our assembly reveals the challenging structural characteristics of the intricate mungbean genome.
Methods
Plant material and sample collection
A kharif cultivar of Indian greengram, IPM02-03 exhibiting high resistance against the mungbean yellow mosaic virus (MYMV) and significant tolerance against major pests was selected for genome sequencing (Mungbean varieties.pdf; Fig. 1). Young expanding tri-foliate leaves were harvested from an individual plant grown under controlled environment (30 −35 °C, 14-10 hrs day-night cycle, 55% relative humidity) in the growth chamber. The harvested tissue was immediately frozen in liquid nitrogen and subsequently stored at −80 °C until further use. For transcriptome sequencing, various developmental tissues including roots, shoots, leaves, and flowers covering most of the vegetative and reproductive parts of mungbean were harvested, immediately frozen in liquid nitrogen and preserved at −80 °C for future use.

Mungbean Growth Habitat and Plant Parts. (a) Under greenhouse conditions (b) Open field conditions (c) Flower (d) Fruit Pod (e) Seeds.
Genomic DNA isolation, PacBio library construction and sequencing
For long-read PacBio sequencing, high-molecular weight (HMW) genomic DNA was extracted using Monarch HMW DNA Extraction Kit following manufacturer’s instructions (New England Biolabs). Genomic DNA for HiC sequencing was extracted using the CTAB method18,19.
For genome sequencing, HMW genomic DNA was utilized for generating a ~20 kb insert size library for PacBio Sequel II system using SMRTbell express template prep kit 2.0 following the manufacturer recommendations. The library was sequenced on single SMRT cell in the PacBio Sequel II system (PacBio Pacific, CA, USA). The long-read sequencing executed on the PacBio Sequel II system yielded 30,322,508 subreads with an average length of 11,136 bp. From the subreads, a total of 1,581,400 HiFi reads were generated using the circular consensus sequencing (CCS) workflow embedded in the SMRT package. In total, we generated ~18 Gb (30x coverage) of PacBio HiFi long reads with a mean length of 11.4 kb (Supplementary Table 1).
Hi-C library preparation and sequencing
Hi-C library construction was performed using Arima-HiC kit following the manufacturer’s recommendations. Briefly, crosslinked DNA was first digested with Arima restriction enzyme cocktail which included DdeI and DpnII. The digested products with 5′ overhangs were further mixed with biotinylated nucleotides followed by ligation of the blunt ends. The DNA was purified and pulled down using streptavidin beads. Sequencing libraries were constructed using Next Ultra enzymes Kit (NEB) and compatible Illumina adapters. The prepared libraries were sequenced in a paired-end mode on Illumina NovaSeq 6000 platform (Illumina, USA). We generated a total of 28 Gb (45.2x coverage) Hi-C data. Concisely, the raw Hi-C paired-end reads were processed using HiC-Pro with default parameters20. HiC-Pro allowed removal of low-quality pairs, multiple pairs alignment, pairs with singletons and unmapped reads with default parameters. The analysis yielded 4,979,118 pairs of valid interactions, out of which 28% were subsequently identified as duplicates and removed. Within the final set of valid interactions, we identified trans contacts (30%), cis long-range contacts (22%), and cis short-range contacts (19%) (Supplementary Table 2).
Assessment of genome size
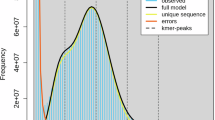
Two methods, flow cytometry and k-mer frequency analysis were used to determine the total nuclear DNA content of V. radiata L. For flow cytometry, young fresh leaves of VR_IPM02-03 and Solanum lycopersicum L. (‘Stupickeť polnıť raneť’; internal reference control with an established genome size of 1.96 pg/2 C DNA)21 were harvested. The samples were processed using CyStain PI Absolute P kit (Sysmex, Germany) following the manufacturer’s guidelines. The stained nuclei samples were evaluated in CyFlow Cube 8 flow cytometer (Sysmex, Germany) in two replicates with a minimum of 3,000 nuclei counts per replicate (Fig. 2). The HiFi PacBio reads were utilized for frequency-based k-mer analysis using Kmerfreq22 with a k-mer size of 17. Next, Genomic Character Estimator was employed to estimate the genome size23.

Genome Size Estimation of VR_IPM02-03; Flow cytometery based fluorescence intensity plot of (a) Solanum lycopersicum L. ‘Stupicke´polnı´ rane (reference) and (b) Vigna radiata IPM02-03 (sample). (c) k-mer based genome size estimation at 17-mer depth distribution using PacBio HiFi reads.
Flowcytometry based on linear relationships of 2 C peaks revealed an approximate haploid genome size of ~605 Mb for mungbean which is slightly higher than the previously reported size of 579 Mb2. A k-mer based approach using k = 17 from long HiFi reads unveiled a genome size of 574 Mb with k-mer peak at 30. Based on the present investigation, we suggest an approximate genome size range of ~579 to 605 Mb for mungbean (Table 1 and Fig. 2).
De novo genome assembly and chromosome anchoring
The HiFi reads were applied to generate the primary de novo assembly using Hifiasm v0.16.124 with the following parameters:–primary (generate a primary assembly) and -l 2 (purge the haplotigs as the species is highly homozygous in nature). All the other parameters were set at defaults. Notably, de novo assembly displayed a contig N50 value of 10.35 Mb and the largest contig measured ~36.5 Mb. A total of ~596 Mb of the genome was assembled covering 98% of the overall estimated genome size of 605 Mb (based on flow cytometry analysis performed in the present study). Next, the filtered Hi-C data was utilized for scaffolding the primary contig-level assembly using a sequential analysis with HiC-Pro20 and EndHiC25. Valid paired reads were extracted to generate intra- and inter- chromosomal contact maps stored in a multiple matrix file at varying resolution (20 Kb to 1 Mb). The matrix file at 40 Kb resolution was utilized as an input for scaffolding with EndHiC in an iterative mode (3 runs) under default parameters25. The resulting assembly was subjected to manual curation based on signal intensity of chromosome interactions using the Juicebox Assembly Tools v1.11.0826. The final chromosome-level assembly was reviewed and obtained with 3D-DNA pipeline (https://github.com/aidenlab/3d-dna), gaps were filled via incorporating 100Ns using parameter: -g 100. A total of ~502 Mb (84%) was successfully anchored to 11 pseudomolecules of the initial primary assembly (Fig. 3a). The longest pseudochromosome obtained in the final genome assembled at a size of 75 Mb while the shortest pseudochromosome comprised of 27 Mb. The assembly exhibited a scaffold N50 of 46.42 Mb. The final assembly (“VR_IPM02-03”) exhibited merely 100 gaps, and 2,237 contigs (~94 Mb) could not be aligned on the pseudomolecules and remained as scaffolds (Fig. 3b and Table 1). The final pseudomolecules were ordered based on size and named accordingly (Table 2).

Overview of VR_IPM02-03 genome assembly (a) Vigna radiata IPM02-03 genome features visualized in a circular plot66. Concentric rings, from outside to inside, depict: A: chromosome physical map, B: Density of Simple Sequence Repeats (SSRs), C: Gene density, D: Abundance of transposable elements, and E: GC content. The y-axis for (B) and (C) represents min-max normalized data, while (D) and (E) are density plots, with darker colors indicating regions of higher density. (b) HiC-interaction map of mungbean of Vigna radiata IPM02-03 genome assembly after manual curation. The x- and y-axes represent genomic coordinates of chromosomes and unplaced scaffolds.
Delineation of telomeric and centromeric regions
Telomeric regions within the genome assembly were delineated through identification of telomeric signature repeats using tidk (Telomere identification toolkit; https://github.com/tolkit/telomeric-identifier)27. We utilized the option -c Fabales limiting the identification to repeats of 5 bp and 7 bp which is a characteristic feature of Fabaceae members. Telomeric regions in the mungbean genome assembly revealed the presence of telomeric repeats at both ends across all chromosomes except VR_pChr9 where it was observed only at one end (Fig. 4). Centromeric repeats were identified using TRASH v1.2 (Tandem Repeat Annotation and Structural Hierarchy28) with default parameters. TRASH algorithm is based on a k-mer counting method for identifying repetitive regions which occur in a tandem manner within a specific genomic window. Each window is evaluated by their ratio of recurrent k-mers and genomic windows are considered as harbouring repeats if they surpass a default threshold set by the tool. Centromeric repeats were examined highlighting strong signals on all chromosomes except VR_pChr9, which notably lacked the signal. This observation again suggests that Chromosome 9 may benefit from further refinement and improvement as unplaced scaffolds showed presence of centromeric repeat signals which might be the unassembled pieces of this chromosome. Further, exploration of centromeric repeats revealed two distinct types of monomeric repeats, namely CEN 174 bp and CEN 177 bp representing the centromeric repeats in VR_IPM02-03 (Fig. 5 and Supplementary Table 3). A detailed examination of the organizational pattern of these repeat sequences revealed a notable structural arrangement wherein the CEN 174 bp repeat functions as one of the pillars or starting point for the subsequent occurrence of the CEN 177 bp repeats. This unique structural organization, involving the tandem occurrence of different repeat lengths is poised to contribute to future investigations focused on the intricate organization and dynamics of centromeric regions and potentially influencing broader perspectives on centromere evolution and function.

Telomeric repeat signals identified and compared between VR_IPM02-03 and Vrad_JL7 (a) Vigna radiata IPM02-03 (b) Vigna radiata JL7.

Centromeric repeat count of both the identified repeats i.e., CEN174 and CEN177 in the Vigna radiata IPM02-03 genome.
Identification and annotation of repetitive regions
The de novo prediction of repeat elements was performed using RepeatModeler2 v2.04 with an enabled LTRstruct pipeline29. The de novo prediction is performed using three individual programs (RECON30, RepeatScout31,32, LTR harvester33) packaged within the RepeatModeller pipeline. All the identified repeat sequences are then classified using RepeatClassifier embedded in RepeatModeller. Additionally, RepeatMasker was employed to perform a homology-based search using search-engine rmblast v2.13.0 to annotate repeats identified from RepeatModeller using Repbase library (Edition:20181026). Overall, comprehensive repeatome analysis of the mungbean draft genome revealed a rich repertoire with approximately 56% (~337 Mb) of the genome constituted with repetitive elements, providing insights into the intricate genomic landscape. The RNA transposons contributed a substantial portion of repeat elements, representing 30.57% of the genome. Long Terminal Repeat (LTR) elements, a prominent subclass of retroelements, comprised 30.10% of the genome, featuring contributions from LTR/Copia (13.35%) and LTR/Gypsy (14.79%) elements. DNA transposons accounted for 7.35% of the genome. Other transposable elements classes are summarised in the Supplementary Table 4. A separate search was performed for identification of Miniature inverted repeat transposable elements (MITEs) in the mungbean genome using MiteFinderII v1.0.00634. Ultimately, it led to identification of ~30,306 MITES loci in the mungbean genome. Further, the full-length transposon sequences were investigated by deploying EDTA v2.1.2 (with parameters: –sensitive 1 –anno 1)35. As a result, a total of ~3,192 intact LTR RT were identified in the mungbean genome (Table 1).
The genome assembly was surveyed for simple sequence repeats (SSRs or microsatellites) using MISA (MIcroSAtellite identification tool)36. The SSR search was performed to identify perfect mononucleotide to hexanucleotide repeats, as well as complex microsatellites with 100 bp gap within SSRs. The customized parameters utilized for microsatellites detection included unit size of 1–6 and minimum repeats for each unit length as 10, 9, 6, 5, 5, and 5, respectively. Overall, our SSR search provided a tally of 2,46,634 SSRs, including 45,455 loci in the compound configuration. The tandem repeats in the mungbean genome assembly were identified using Look4TRs37 which indicated presence of 291,484 tandem repeats contributing further to the enrichment of mungbean repetitive landscape.
RNA isolation, transcriptome sequencing, and assembly
Total RNA was extracted using Trizol method (Molecular Research Center, Inc.). Four independent cDNA libraries, each for respective tissues (described above in Section Plant Material and Sample Collection) were constructed using Illumina TruSeq RNA library preparation kit and sequenced on Illumina HiSeq 2000 sequencer platform (Illumina, Illumina Way, San Diego, CA, USA) in a paired-end mode following manufacturer’s instructions (Illumina, Illumina Way, San Diego, CA, USA).
The quality check on raw transcriptome data was performed using NGSQC toolkit to remove adapter-contaminated and low-quality reads (Q value ≤ 20)38. A total of ~12 Gb of filtered sequencing data was generated from four vegetative and reproductive plant tissues including, roots, shoots, total seedlings, and flowers. The filtered high-quality reads were assembled to generate a de novo transcriptome using Trinity v2.0.639 with a minimum contig length set at 200 base pairs and a minimum count for k-mer to be assembled set at 10 through the Inchworm algorithm. The initial assembly was processed using CD-HIT-EST v4.6.140 to eliminate redundant contigs. The output of the de novo assembly was a comprehensive set of 55,213 transcripts spanning approximately ~43 Mb. The average length of these assembled transcripts measured ~784 bp, with an N50 value of 1,122 bp.The quality of the transcriptome assembly was analyzed by mapping back filtered high-quality RNA-seq reads onto the assembled transcriptome using bowtie241 with the following parameters:–no-unal (to suppress SAM records for reads that fail to align) while the other parameters were used at default values. The alignment of high-quality RNA-seq reads to the assembled transcriptome demonstrated an overall alignment rate of 96%. The transcriptome assembly was subsequently used for genome annotation.
Gene prediction and functional annotation
The structural gene prediction was performed with the BRAKER3 pipeline on the soft-masked mungbean genome42. BRAKER3 utilizes a suite of ab initio and homology-based prediction methods. For RNA evidence, the assembled transcripts from our RNAseq data were utilized. Additionally, the protein sequences from V. mungo12, V. umbellata13, Arabidopsis thaliana Araprot 1143 and Lotus japonicus v3.0 (https://www.kazusa.or.jp/lotus/) were downloaded from their public databases and used as extrinsic evidence to aid structural gene identification. Further, the predicted gene models were filtered to remove transcripts with length shorter than 300 bp and/or protein sequences smaller than 100 amino acids. Finally, we obtained a total of 43,147 genes models in the mungbean genome. The average coding sequence (CDS) length measured 1,128 bp with an N50 value of 1,482 bp.
The functional annotation of the predicted gene models was conducted through homology-based search with blastp against the NCBI-Refseq database using OmixBox v3.0.30 suite (https://www.biobam.com/omicsbox/). The GO, KOG, COG, and Enzyme Code (EC) were assigned to the genes utilizing various functional modules of OmixBox v3.0.30 suite following the developer’s guidelines. KEGG (Kyoto Encyclopedia of Genes and Genomes) annotation was performed utilizing ghost koala module loaded within the online KEGG automatic annotation servers44. Multiple databases including CDD, Panther, Pfam, PRINTS, PROSITE, SUPERFAMILY, Coils, SFLD, FunFam, ProSiteProfiles, PRISR, Gene3D, and SMART were explored to identify conserved domains and motifs in the predicted protein-coding genes using Interproscan v5.62.9445. Out of 43,147 predicted gene models, 48,100 transcripts were functionally annotated. A total of ~39,144 (81%) protein coding genes were annotated with at least one public database (Fig. 6a). Briefly, functional analysis through sequence homology against NCBI RefSeq protein database annotated 39,144 (81%) protein coding genes. The Gene Ontology (GO) annotations provided insights into biological, molecular, and cellular processes for 25,903 (53%) protein coding genes (Fig. 6b), while KEGG annotations categorized 18,118 (37%) protein coding genes, unveiling intricate pathways of mungbean. Conserved functionalities were highlighted in 46% (22,127) of protein coding genes through euKaryotic Orthologous Groups (KOG). Clusters of Orthologous Groups (COG) assigned specific functions to 33% (16,130) of protein coding genes (Fig. 6c). The Enzyme Code (EC) annotations spotlighted enzymatic functions in 22% (10,630) of protein coding genes. InterProScan domain analysis assigned functional annotations to 34,921 protein coding genes (72%) in the mungbean genome (Fig. 6a). These annotations collectively deepen our understanding of the mungbean genome, providing a foundation for further exploration.

Functional annotation of the Vigna radiata IPM02-03 protein-coding genes. (a) Upset plot detailing the unique and overlapping annotations contributed by RefSeq, Interproscan, GO, KOG, KEGG, COG, and EC. (b) Distribution of GO level 2 terms within Biological, Molecular and Cellular Process categories. (c) Annotated COG frequency distribution for each functional class identified in VR_IPM02-03.
We further extended our analysis identifying noncoding RNA genes, particularly ribosomal RNA (rRNA) and transfer RNA (tRNA) genes in the mungbean genome. The rRNA genes were predicted employing barrnap (https://github.com/tseemann/barrnap)46 with predefined parameters for the kingdom eukaryotes (euk) revealing a total of 6,959 rRNA loci. For tRNA gene identification, tRNASCAn-SE v2.047 was used in conjunction with Infernal v1.1.548. The option -E was applied to limit the search to eukaryotic tRNAs while other parameters were kept at defaults. In total, we identified 5,747 tRNAs loci in mungbean genome.
Data Records
The PacBio HiFi Reads, Illumina short reads RNA-seq, and Hi-C sequencing datasets have been submitted to the NCBI Sequence Read Archive (SRA) database under the following accession numbers: SRR2730867249, SRR2730866950, SRR2730866851, SRR2730867052, SRR2730867153 and SRR2730866754. The final genome assembly has been deposited in GenBank with the assembly accession number GCA_043793165.155. Genome annotation files can be accessed on the Figshare database (https://doi.org/10.6084/m9.figshare.25043495)56.
Technical Validation
Nucleic acid quality
Quality control of extracted DNA samples was performed using agarose gel electrophoresis and a Nanodrop-1000 spectrophotometer (Thermo Scientific). Additionally, DNA was quantified using a Qubit fluorometer (Invitrogen). The quality and quantity of the extracted RNA was evaluated using Bioanalyzer 2100 (Agilent Technologies) and NanoDrop-1000 spectrophotometer (Thermo Scientific).
Investigation of genome assembly quality
The quality of VR_IPM02-03 assembly was surveyed through multiple approaches. Four mapping strategies were opted for assessing the integrity and continuity of the generated genome assembly. First, raw HiFi data (long reads) were aligned to the assembled genome through minimap2 v2.26 with the parameters “-ax map-pb”57 which demonstrated an overall alignment rate of ~99.96%. Second, the filtered raw high quality, paired end Illumina reads from the transcriptome data were aligned to the genome using a spliced aligner STARv2.7.10b58 with default parameters and an overall alignment rate of 100% was obtained. Next, we explored mapping of the assembled transcriptome to genome using GMAP v2021-08-2559 with default parameters. This mapping process revealed a comprehensive representation of the assembled transcriptomic data, with an alignment rate of ~99.89%. Lastly, we performed alignment of 1,39,186 nucleotide sequences retrieved from National Center for Biotechnology Information (NCBI) database using the search string “Vigna radiata” (Access on: July 2023) to the draft genome with GMAP v2021-08-2559. We obtained an alignment rate of 99% onto the genome. All the above alignment stats were calculated using the argument -flagstat in samtools v1.960. Further, the genome assembly was examined using Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.4.7 to evaluate the proportion of orthologous gene models of two lineages, Eudicots and Fabales captured in the genome assembly61. BUSCO score revealed a substantial proportion of orthologous gene models from Eudicots (98.5%) and Fabales (98.6%) were represented in the genome (Table 1). Finally, LTR Assembly Index (LAI) was calculated to determine the assembly completeness based on evaluation of intact LTR-RTs and total LTR-RTs present in the assembled genome using LTR_retriever62. Our results indicate a comprehensive assembly completeness with a LAI score of 16.58 (Table 1).
Identifying and correcting multiple mis-assemblies in mungbean genome
We benchmarked our genome assembly against the previously published genome assembly of Vrad_JL7 cultivar of mungbean (Table 1)17. The pseudomolecules of VR_IPM02-03 were sorted and named according to their size aligning with the already established international karyotyping rules63. The size distribution and nomenclature of pseudochromosomes from VR_IPM02-03 and Vrad_JL7 is presented in Table 2.
Briefly, D-GENIES64 tool was implemented for aligning both the genomes in local mode under default parameters using minimap2 v2.2657. The alignment file was extracted as PAF (Pairwise mApping Format) format and genome synteny was plotted using R package syntenyPlotteR65. Our comparative analysis of the present assembly (VR_IPM02-03) with the recently published mungbean genome Vrad_JL7 highlighted four instances of disagreement between the two assemblies (Fig. 7). These were recognised as cases of mis-assembly in Vrad_JL717. The published Vrad_JL7 chromosomes 1, 6, 2 and 3 showed intra-chromosomal inversion events (Fig. 8 upper panel). Most of the breakpoints identified in the assembly were arising at the putative sites of centromere therefore, we implemented the tool TRASH28 on Vrad_JL7 genome17 for identification of repetitive centromeric repeats (Fig. 9). Strikingly, we did not identify any centromeric repeats (CEN174 bp and CEN177 bp) in the Vrad_JL7 chromosomes contrary to delineation of these regions in the VR_IPM02-03 chromosomes (Fig. 9(a,b)). To further resolve these breakpoint events, we mapped the PacBio HiFi reads on both the assemblies and closely inspected the alignment of reads on these breakpoint coordinates. Alignment of long reads, identified events of inversions and deletions on three chromosomes Chromosome 1, 6, and 2) of Vrad_JL7 (Fig. 8 lower panel). We were not able to resolve the breakpoint events on Chromosome 3 of Vrad_JL7 based on mapping results. This complexity arises from inversion events occurring at locations that span both the end and the start of the chromosome, rendering traditional mapping methods less effective in deciphering these intricate miss-assembly. However, we observed absence of the centromeric signals from chromosome 3 Vrad_JL7 (Fig. 9(b)); instead the telomere signals were localized in the center of this chromosome indicating the mis-assembly events at the loci (Fig. 4). On the other hand, all the four chromosomes on VR_IPM02-03 (VR_pChr4, VR_pChr5, VR_pChr7 and VR_pChr11) showed no inversion and deletion events on these breakpoint coordinates and exhibited a constant coverage across these regions indicating an accurately aligned assembly (Fig. 8 lower panel). Additional support for the correct alignment of the pseudo molecules of VR_IPM02-03 is contributed by identification of centromeric repeats and telomeric signals from these chromosomes (Figs. 4, 7, 9(a)). Additionally, in comparison to Vrad_JL7 we could identify 3,475 protein coding genes unique to the sequenced Indian cultivar (VR_IPM02-03), of which 1,980 were also absent from the reported pangenome of V. radiata17. This highlights the importance of sequencing Indian germplasm VR_IPM02-03 which is inadequately represented among the sequenced accessions.

Whole-genome synteny plot depicting the genomic alignment between Vigna radiata IPM02-03 (VR_IPM02-03) and Vigna radiata JL7 (Vrad_JL7). Green arrowheads depicts the telomeric/sub-telomeric signals and red arrowheads indicates the centromeric regions on the VR_IPM02-03 chromosomes.

Comparative analysis highlighting misassemblies in Vigna radiata JL7 (Vrad_JL7) compared to VR_IPM02-03. Four misassembled regions on chromosomes of Vrad_JL7: Chr 1 (corresponding to VR_pChr4), Chr 6 (corresponding to VR_pChr5), Chr 2 (corresponding to VR_pChr7), and Chr 3 (corresponding to VR_pChr11) have been shown. The upper panel shows whole-genome alignment indicating signals of inversions and putative misassemblies, while the lower panel confirms these observations through the mapping of HiFi reads onto the genomes.

Circos plot representing centromeric repeats identified in (a) Vigna radiata IPM02-03: CEN174 bp (brown) and CEN177 bp (red) satellite repeats (b) Vigna radiata JL7: CEN174 bp (green) signal observed only in unplaced scaffolds while CEN177 bp (blue) present only on Chr9.
Code availability
No custom code was used in this study. We processed the data using established pipelines and software, following manuals and protocols. The Methods section provides details on the versions and parameters used. Default parameters, as suggested by developers, were applied in the pipelines and software, unless otherwise specified in this work.
References
Liu, M.-S. et al. Genomic and transcriptomic comparison of nucleotide variations for insights into bruchid resistance of mungbean (Vigna radiata [L.] R. Wilczek). BMC Plant Biol 16, 46 (2016).
Arumuganathan, K. & Earle, E. D. Nuclear DNA content of some important plant species. Plant Mol Biol Report 9, 208–218 (1991).
Fuller, D. Q. & Harvey, E. L. The archaeobotany of Indian pulses: identification, processing and evidence for cultivation. Environmental Archaeology 11, 219–246 (2006).
Mehandi, S. et al. Mungbean (Vigna radiataL. Wilczek): Retrospect and Prospects. in (2019).
Lengwati, D. M., Mathews, C. & Dakora, F. D. Rotation Benefits From N2-Fixing Grain Legumes to Cereals: From Increases in Seed Yield and Quality to Greater Household Cash-Income by a Following Maize Crop. Front Sustain Food Syst 4, (2020).
Mogali, S. C. & Hegde, G. M. Recent Advances in Mungbean Breeding: A Perspective. in Accelerated Plant Breeding, 3 235–282. https://doi.org/10.1007/978-3-030-47306-8_9 (Springer International Publishing, Cham, 2020).
Somta, P., Laosatit, K., Yuan, X. & Chen, X. Thirty Years of Mungbean Genome Research: Where Do We Stand and What Have We Learned? Front Plant Sci 13, (2022).
Lonardi, S. et al. The genome of cowpea (Vigna unguiculata [L.] Walp. The Plant Journal 98, 767–782 (2019).
Kang, Y. J. et al. Draft genome sequence of adzuki bean, Vigna angularis. Sci Rep 5, 8069 (2015).
Xia, Q. et al. The genome assembly of asparagus bean, Vigna unguiculata ssp. sesquipedialis. Sci Data 6, 124 (2019).
Garcia, T. et al. Comprehensive genomic resources related to domestication and crop improvement traits in Lima bean. Nat Commun 12, 702 (2021).
Ambreen, H. et al. Long‐read‐based draft genome sequence of Indian black gram IPU‐94‐1 ‘Uttara’: Insights into disease resistance and seed storage protein genes. Plant Genome 15, (2022).
Guan, J. et al. Genomic analyses of rice bean landraces reveal adaptation and yield related loci to accelerate breeding. Nat Commun 13, 5707 (2022).
Pootakham, W. et al. Genome assemblies of Vigna reflexo-pilosa (créole bean) and its progenitors, Vigna hirtella and Vigna trinervia, revealed homoeolog expression bias and expression-level dominance in the allotetraploid. Gigascience 12, (2022).
Kang, Y. J. et al. Genome sequence of mungbean and insights into evolution within Vigna species. Nat Commun 5, 5443 (2014).
Ha, J. et al. A near‐complete genome sequence of mungbean (Vigna radiata L.) provides key insights into the modern breeding program. Plant Genome 14, (2021).
Liu, C. et al. High-quality genome assembly and pan-genome studies facilitate genetic discovery in mung bean and its improvement. Plant Commun 3, 100352 (2022).
Doyle, J. J. Isolation of plant DNA from fresh tissue. Focus (Madison) 12, 13–15 (1990).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin vol. v.19(1):11–15 Preprint at http://worldveg.tind.io/record/33886/files/d015081.pdf (1987).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16, 259 (2015).
Doležel, J., Greilhuber, J. & Suda, J. Estimation of nuclear DNA content in plants using flow cytometry. Nat Protoc 2, 2233–2244 (2007).
Wang, H. et al. Estimation of genome size using k-mer frequencies from corrected long reads. arXiv preprint arXiv:2003.11817 (2020).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv preprint arXiv:1308.2012 (2013).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Wang, S. et al. EndHiC: assemble large contigs into chromosome-level scaffolds using the Hi-C links from contig ends. BMC Bioinformatics 23, 528 (2022).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98 (2016).
Telomere identification toolkit, tidk https://github.com/tolkit/telomeric-identifier.
Wlodzimierz, P., Hong, M. & Henderson, I. R. TRASH: Tandem Repeat Annotation and Structural Hierarchy. Bioinformatics 39, (2023).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
Bao, Z. & Eddy, S. R. Automated De Novo Identification of Repeat Sequence Families in Sequenced Genomes. Genome Res 12, 1269–1276 (2002).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Wootton, J. C. & Federhen, S. Statistics of local complexity in amino acid sequences and sequence databases. Comput Chem 17, 149–163 (1993).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008).
Hu, J., Zheng, Y. & Shang, X. MiteFinderII: a novel tool to identify miniature inverted-repeat transposable elements hidden in eukaryotic genomes. BMC Med Genomics 11, 101 (2018).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol 20, 275 (2019).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585 (2017).
Velasco, A., James, B. T., Wells, V. D. & Girgis, H. Z. Look4TRs: a de novo tool for detecting simple tandem repeats using self-supervised hidden Markov models. Bioinformatics 36, 380–387 (2020).
Patel, R. K. & Jain, M. NGS QC toolkit: A toolkit for quality control of next generation sequencing data. PLoS One 7, (2012).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–652 (2011).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012).
Gabriel, L. et al. BRAKER3: Fully Automated Genome Annotation Using RNA-Seq and Protein Evidence with GeneMark-ETP, Augustus and Tsebra. https://doi.org/10.1101/2023.06.10.544449.
Cheng, C. et al. Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. The Plant Journal 89, 789–804 (2017).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44, D457–D462 (2016).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Barrnap https://github.com/tseemann/barrnap.
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res 49, 9077–9096 (2021).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX22985932 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX22985935 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX22985936 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX22985934 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX22985933 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRX22985937 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_043793165.1/ (2024).
Oraon, P. K. An improved chromosome-scale genome assembly of Vigna radiata enables delineation of centromeres and telomeres. Figshare https://doi.org/10.6084/m9.figshare.25043495 (2024).
Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 37, 4572–4574 (2021).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10 (2021).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol 38, 4647–4654 (2021).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res https://doi.org/10.1093/nar/gky730 (2018).
Stevens-Kroef, M., Simons, A., Rack, K. & Hastings, R. J. Cytogenetic Nomenclature and Reporting. 303–309, https://doi.org/10.1007/978-1-4939-6703-2_24 (2017).
Cabanettes, F. & Klopp, C. D-GENIES: dot plot large genomes in an interactive, efficient and simple way. PeerJ 6, e4958 (2018).
Quigley, S., Damas, J., Larkin, D. M. & Farré, M. syntenyPlotteR: a user-friendly R package to visualize genome synteny, ideal for both experienced and novice bioinformaticians. Bioinformatics Advances 3, (2023).
Wang, Y. et al. shinyCircos‐V2.0: Leveraging the creation of Circos plot with enhanced usability and advanced features. iMeta 2, (2023).
Acknowledgements
We express gratitude for the grant support from the Department of Biotechnology, Government of India, awarded to the University of Delhi and North-Eastern Hill University under the sanction orders “BT/190/NE/TBP/2011” and “BT/PR24637/NER/95/787/2017” through NER Biotechnology Management Cell (NER-BPMC). Additionally, we acknowledge the grant from the University of Delhi, granted under the “Institute of Eminence” scheme with sanction order IOE/FRP/LS/2020/27 through the Faculty Research Programme Grant – IoE. P.K.O. and P.Y. acknowledges the fellowship provided by the Council of Scientific and Industrial Research, Ministry of Science and Technology, Government of India. We acknowledge Bionivid Pvt. Ltd. for their support in sequencing and data analysis.
Author information
Authors and Affiliations
Contributions
S.G. and R.S. designed the study and arranged the funding and resources required for the study. P.K.O. performed all the experiments, analysed, and visualised the data and wrote the original manuscript. H.A. contributed to conceiving and supervising the study, designing analysis and interpreting the results. P.Y. engaged in extraction of DNA and RNA, and library preparations. All authors reviewed, edited, and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Oraon, P.K., Ambreen, H., Yadav, P. et al. A chromosome-scale reference assembly of Vigna radiata enables delineation of centromeres and telomeres. Sci Data 12, 305 (2025). https://doi.org/10.1038/s41597-025-04436-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04436-8
