
Summary
Crofton weed (Ageratina adenophora), a significant invasive species, extensively disrupts ecosystem stability, leading to considerable economic losses. However, genetic insights into its invasive mechanisms have been limited by a lack of genomic data. In this study, we present the successful de novo assembly of the triploid genome of A. adenophora, leveraging long-read PacBio Sequel, optical mapping, and Hi-C sequencing. Our assembly resolved into a haplotype-resolved genome comprising 51 chromosomes, with a total size of ~3.82 Gb and a scaffold N50 of 70.8 Mb. BUSCO analysis confirmed the completeness of 97.71% of genes. Genome annotation revealed 3.16 Gb (76.44%) of repetitive sequences and predicted 123,134 protein-coding genes, with 99.03% functionally annotated. The high-quality reference genome will provide valuable genomic resources for future studies on the evolutionary dynamics and invasive adaptations of A. adenophora.
Similar content being viewed by others


Background & Summary
Biological invasions represent a widespread global issue, posing challenges for conserving biodiversity, ecological stability, and environmental resources, as well as causing significant social challenges and economic losses1,2. The proliferation of alien plants, notably driven by the rapid expansion of international trade, tourism, and transportation, constitutes a pivotal element in the broader scope of biological invasion, holding significance in social economics, ecology, and evolutionary studies.
Ageratina adenophora (Spreng.) R.King & H.Rob., commonly known as crofton weed, is a perennial, evergreen, semi-shrub in the Asteraceae family3, indigenous to Central America. It was firstly introduced to Europe as an ornamental plant in the 19th century and later spread to Australia and Asia4,5. Nowadays, it emerged as highly destructive alien invasive weed globally, widely problematic in the United States, Australia, Indian Ocean islands, and Pacific Ocean islands as well as Southern Asia and Eastern Asia. In China, it ranks among the top 10 most destructive invasive species6,7. Crofton weed exhibits various biological characteristics contributing to its success as an invasive species. Firstly, A. adenophora contains many active substances exerting strong allelopathic effects on other plant species and poisoning animals, enabling it to dominate ecological niche. For example, flavonoids, serve as a protective mechanism against biotic and abiotic stressors, including herbivores, pathogens, ultraviolet radiation, and high temperatures. Furthermore, A. adenophora has a high reproduction coefficient, producing ~10,000 small, widely dispersed seeds per plant. Its exceptional adaptation to harsh conditions and efficient soil nutrient absorption, coupled with a robust rhizome system, facilitate rapid invasion, colonization, and expansion8. Therefore, a comprehensive understanding of the genetic basis underlying these biological characteristics is promising for not only efficiently controlling A. adenophora, but also providing valuable genes for crop improvement through genetic engineering.
Although previous studies have explored the biological characteristics of crofton weed, its genetic resource and genomic data are limited, hindering in-depth investigations into its invasive mechanism. Here, we employed a diverse array of sequencing technologies and assembly strategies (Fig. 1) to successfully construct a chromosome-level reference genome of A. adenophora with genome size ~3.82 Gb and a scaffold N50 of 70.8 Mb. In summary, our study provides the valuable genomic resources for further exploring the invasion mechanisms and control strategies of Crofton weed.

Flowchart of genome assembly and quality control for the A. adenophora. Various sequencing technologies, including Illumina, BAC, PacBio, BioNano, and Hi-C, was utilized to achieve accurate genome assembly. Multiple quality control methods were employed for evaluation.
Methods
Plant materials and sequencing
The crofton weed samples in this study were obtained from Tengchong County (N 25°529″ 204″, E 98°45″220″) in Yunnan Province, China. Emphasis was on young leaves, providing high-quality genomic DNA for subsequent genome sequencing and Hi-C analysis. Genomic DNA from A. adenophora was extracted using the modified CTAB method9. DNA concentration and quality were assessed using the NanoDrop 2000 (Thermo Fisher, USA) and Qubit fluorometer (Thermo Fisher, USA). Subsequently, libraries with 270 bp insert fragments were constructed following standard Illumina procedures, and sequenced on the Illumina platform (Illumina, USA) with a PE150 strategy, generating a total of 92.83 Gb (~34×) clean Illumina reads. PacBio DNA sequencing libraries with 30 kb insert size were constructed following PacBio’s recommended protocol (Pacific Biosciences, CA, USA) and sequenced on the PacBio Sequel platform, resulting in 206 Gb (~54×) of raw data with an average length of 13,666 bp. Simultaneously, 28.81 Gb of PacBio HiFi reads (average length 10,505 bp) were generated for genome polishing analysis. The BAC library was constructed and sequenced by Nanjing Hong-Yuan Biotechnology Company Limited (Nanjing, China). Hi-C libraries were constructed following previous protocol10, and sequenced with a 2 × 150 bp read length on the Illumina platform, resulting in a total of 398.39 Gb (~104×) of high-quality Hi-C clean data. The BioNano Genomics Irys system (BioNano Genomics, USA) was employed to generate optical maps and about 797.39 Gb (~209×) of high-quality optical molecular data (length >100 kb, label signal-to-noise ratio of 3.0, average molecule intensity <0.6) were obtained.
The method by Yang et al.11 was utilized for total RNA extraction and cDNA synthesis from A. Adenophora roots, leaves, and flower. mRNA sequencing libraries, constructed on the Illumina NovaSeq platform with 150 bp paired-end sequencing technology, underwent three biological replicate experiments per sample. For full-length transcriptome sequencing, root, leave, and flower samples were prepared following the PacBio Iso-seq experimental workflow and sequenced on the PacBio Sequel platform.
De novo assembly of a phased triploid genome
Fluorescence in situ hybridization (FISH) confirmed 51 chromosomes in A. adenophora (Fig. 2a). Smudgeplot12 analysis indicated an “AAB” genome structure for A. adenophora (Fig. 2b). The genome size estimation was conducted using flow cytometry, with rice serving as the standard reference genome. The measured fluorescence intensity ratio between A. adenophora and rice was approximately 5.1, which was used to estimate the genome size of A. adenophora at 3.96 gigabases (Gb) (Fig. 2c). We delineated the intricacies of the A. adenophora genome by employing multiple sequencing technologies and a meticulous assembly strategy (Fig. 1). Initially, PacBio Sequel long reads were used for contig-level assembly by Canu v1.913 (the parameter set as genomeSize = 3.96 g), and polished by Pilon14 (v1.2229)(parameters:–min depth 10–changes–fix bases) and Arrow v7.01 (Pacific BioScience). The assembled contig size is 3.46 Gb with an N50 of 1.52 Mb. Hybrid genome assembly was performed by utilizing BioNano Solve v3.0.13 (https://bionanogenomics.com/support/software-downloads/) with the parameters “-B 2 -N 2”, and over 93.35% of contig sequences were assembled into super scaffolds, achieving scaffold N50 of 27.33 Mb. Subsequently, we use LACHESIS15 (version 20171221) software to make chromosomal assembly with optimized parameters CLUSTER_MIN_RE_SITES = 100, CLUSTER NONINFORMATIVE RATIO = 1.5, CLUSTER_MAX_LINK_DENSITY = 2, ORDER MIN N RES IN SHREDS = 60, ORDER MIN N RES IN TRUNK = 60. Analysis of the anchored chromosome interaction heatmap revealed a distinct pattern, with one haplotype exhibiting a stronger interaction signal than the other two. Accordingly, we isolated the haplotype with the stronger interaction signals from the three chimeric haplotypes by Juicebox Assembly Tools (JBAT v1.1)16 software, resulting in a non-redundant haplotype-resolved assembly comprising 17 chromosomes, designated as haplotype B. Utilizing haplotype B as the reference, we applied the ALLHIC17 method to distinguish the remaining haplotypes, A1 and A2. Consequently, we obtained a haplotype-resolved, chromosome-level genome assembly for A. adenophora, with a total size of ~3.82 Gb and a scaffold N50 of 70.8 Mb (Fig. 2d and Table 1). Notably, this assembly represents 96.46% of the genome size estimated through flow cytometry measurement (Fig. 2c). A significant portion of scaffolds, totaling 3.55 Gb (92.98%), successfully anchored to 51 pseudo-chromosomes. These pseudochromosomes form 17 homologous groups, each comprising three allelic chromosomes (A1: 1.16 G, A2: 1.17 G, and B: 1.22 G) (Table 2).

Overview of A. adenophora genome. (a) The A. adenophora karyotype. DAPI-stained metaphase chromosomes are shown in the left two parts and demonstrated 51 chromosomes in A. adenophora. Three homologous chromosomes signal was detected by the hybridization sites of 18S (red) and the 5S (green) rDNA probes and shown in the right two parts. Bar 10 μm. (b) Smudgeplots analysis based on density of k-mer for A. adenophora. (c) Genome size estimation by utilizing flow cytometry. The rice with genome size 385.7 Mb was used as the standard sample. The fluorescence intensity fold between A. adenophora and rice was ~5.1, and the genome size of A. adenophora was estimated to be 3.96 Gb. (d) Chromosomal features of A. adenophora genomes: a chromosome ideograms. b Transposable element in each chromosome. c Gene density content in each chromosome. d 5 mC DNA methylation levels obtained from the Pacbio HiFi data. e SNP density. f InDel density. g gene expression levels. (e) BUSCO scores of the three haplotypes of A. adenophora.
Benchmarking Universal Single-Copy Orthologs (BUSCO)18 analysis revealed that ~97.71% of BUSCO genes were completed in our assemblies (Fig. 2e). The annotation of LTRs revealed an LTR Assembly Index19 (LAI) score of 18.53 for the A. adenophora genome (Table 1). The package Merqury20 (v1.3) was used to assess the quality and completeness of the genome using short sequencing reads of A. adenophora and the result indicated a base accuracy of the genome was over 99.97% (QV > 35.43), and k-mer completeness estimated at 97.30% (Table 1). Collectively, these results indicate a high quality of the A. adenophora genome assembly.
Structural variations among allelic chromosomes
Structural variation analysis between haplotypes was conducted using MUMMER v4.021 software. The “nucmer” command facilitated genome alignment, with parameters set as “–maxmatch -c 500 -b 500 -l 100”. Subsequently, the delta-filter command was applied to refine alignment results, using parameters “-i 90 -l 1000 -m” to ensure a minimum 1 Kb matching and at least 90% similarity. The SyRI (v1.6)22 software was then employed to identify structural variations among three haplotypes, with parameters set as “–allow-offset 100 –unic 2000”. To ensure result accuracy, only DUP types with a minimum 50% overlap with SYN were retained. All variations were required to have a minimum length of 30 Kb. Finally, a total of 1,352 structural variations >30 kb in length between haplotypes were detected. Notably, 46 extra-large inversions >1 Mb were detected, comprised ~57.80% of the cumulative structural variation length. The most of largest inversion between haplotype was localized on chromosome 14, spanning ~40 Mb and constituting 57.88% of its length (Fig. 3a). Hi-C and optical mapping method were confirmed the accuracy of the inversion (Fig. 3b and c).

Characterization of genomic structural variations between the A. adenophora haplotypes. (a) Overview of syntenic blocks across the three haplotypes. Gray lines represent the syntenic regions, while orange lines represent the inversions. (b) Identification of large inversions in Chr14. The upper two heatmaps show a chromatin 300-kb interaction matrix, including mapping Hi-C data of A1_14 against the haplotype B genome (A1_map_B), mapping Hi-C data of B_14 against the haplotype A1 genome (B_map_A1). The middle panel shows the syntenic blocks across A1, B and A2 in chromosome 14. The largest inversion regions are shown by orange lines. The lower two heatmaps for A2_14 show a chromatin interaction heatmap with a similar mapping strategy as the upper track. (c) Consistent alignment of the BioNano contigs with the PacBio assembly demonstrates the inversion accuracy in the PacBio assembly. The red arrows indicate inversion breakpoints, while the black lines shown collinearity between the PacBio assembly and BioNano maps.
Transposable elements annotation
To build a comprehensive repeat sequence library for A. adenophora, RepeatModeler23 (v2.02), LTR-FINDER24 (v1.05), MITE-hunter(20100819)25, and PILER-DF26 (v1.0) were used with default parameters. This sequence library was merged with the Repbase27 database, and sequences were classified into different categories using PASTEClassifier.py28. Finally, RepeatMasker v.4.1.129 was used to mask the genome with the finalized repeat library.). Finally, a total of 3.16 Gb (76.44%) repetitive sequences were identified in the A. adenophora genome (Table 3). Among these, 881.04 Mb (76.00%), 893.83 Mb (76.34%), and 982.94 Mb (80.55%) of TEs were in the haplotype A1, A2, and B, respectively. We conducted a comparative analysis among several closely related Asteraceae family species, revealing a notably elevated LTR ratio in A. adenophora (Fig. 4a). To elucidate LTR expansion, we identified 55,232 full-length LTR-RTs (A1:17,721, A2:17,646 and B:19,865) in A. adenophora using LTRretriever30 (v2.9.8), with 61.71% in the Gypsy subfamily and 16.89% in the Copia subfamily.

LTR retrotransposon accumulation and insertion analysis. (a) Comparison of repetitive sequences contents in Asterids. (b) The insertion time distribution of intact LTRs in different Asteraceae species. Mya indicates million years ago.
Furthermore, we calculated the LTR insertion time. Flanking sequences on both sides of LTRs were aligned using MAFFT31 (v7.205) (parameters:–local pair–max iterate 1000). Subsequently, the Kimura model in EMBOSS32 (v6.6.0) was employed to calculate the distance (K). The formula for LTR insertion time is T = K / (2 × r), with the molecular clock rate (r) set at 7 × 109. The results showed a recent burst expansion [<0.5 million years ago (Mya)] shared among all three haplotypes, consistent with H. annuus and M. micrantha, but later than L. sativa (~1.9 Mya) and C. cardunculus (~2.4 Mya) (Fig. 4b).
Gene annotation
We integrated homology-based, de novo, and transcriptome prediction methods gor gene annotation. GenScan33 (v1.0), Augustus34 (v2.4), GlimmerHMM35 (v3.0.4), GeneID36 (v1.4), and SNAP37 were utilized for ab initio prediction. Homology-based prediction employed GeMoMa38 (v1.4.2) with genome information from Oryza sativa L. ssp. japonica, Arabidopsis thaliana, M. micrantha, and H. annuus. Additionally, the clean RNA-seq reads from different tissues were mapped to the genome using HISAT39 (v2.1.0), and the alignments were then input to Trinity40 (v. 2.2.0) by running genome-guided mode to make de novo transcriptome assembly into unigenes. The unigenes plus with the PacBio full-length cDNA were then aligned to the genome using BLAT41 (v35), and then followed by PASA v2.0.242 for transcriptome prediction. EVidenceModeler v1.1.143 integrated homologous-based, transcriptome, and ab initio predictions, producing a unified gene model updated by PASA. The maximum intron length was set to 20 kilobases (kb) for all software tools mentioned above. Finally, we identified 123,134 protein-coding genes in the A. adenophora genome (Table 1). For gene function annotation, DIAMOND v0.9.2844 was used to align the predicted protein sequences against NCBI non-redundant protein (NR), eggNOG45, Swissprot and TrEMBL46 databases with a cutoff value of 1e-5. HMMER47 (v3.1b2) was used for search Pfam48 database for protein domain annotation, and Gene Ontology49 (GO) term annotation was obtained from InterProScan50 (v4.3). Additionally, Kyoto Encyclopedia of Genes and Genomes51 (KEGG) pathway annotation used the KEGG Automatic Annotation Server52 (KAAS). Approximately 99.03% (121,934) of gene models were functionally annotated in Swissprot, TrEMBL, NR, KEGG, GO, eggNOG, or Pfam databases (Table 4).
Data Records
All sequencing raw data used in this study53 and the Whole Genome Shotgun (WGS) assembly54 have been submitted to the National Center for Biotechnology Information (NCBI) via BioProject ID PRJNA1096832. The genomic Illumina sequencing data were deposited in the Sequence Read Archive at NCBI SRR28607150 and SRR28607151. PacBio CLR DNA sequencing data is available under the NCBI Sequence Read Archive accession number SRR28607117 and PacBio HiFi DNA sequencing data are under SRR28607109 and SRR28607110. PacBio Iso-Seq data for all tissues (flowers, root and leaf) are available under the NCBI Sequence Read Archive accession numbers SRR28607137, SRR28607138 and SRR28607140-SRR28607143. The RNA-seq data (flowers, root and leaf) are available under the NCBI Sequence Read Archive accession number SRR28607106-SRR28607108 and SRR28607144-SRR28607149. The short read sequences for Hi-C sequencing have been deposited in the SRA accessions SRR28607128 and SRR28607139. The final genome assembly, structural variations, transposable elements, gene structure and function annotation were deposited in the Figshare55 database.
Technical Validation
We evaluated the continuity of the genome, and the results indicated that the contig N50 value reached 1.22 Mb (Table 1). This is a significant improvement compared to the previously reported triploid banana genome, which had a contig N50 of 1.08 Mb56. Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis revealed that ~97.71% of BUSCO genes were completed in our assemblies (Table 1and Fig. 2e), comparable to percentage reported in cultivated hexaploidy of C. morifolium57. Long Terminal Repeats (LTRs), crucial for assessing assembly quality in repetitive sequences and intergenic regions. The annotation of LTRs revealed an LTR Assembly Index (LAI) score of 18.53 for the A. adenophora genome (Table 1), meeting standard expected for a reference genome58. Comparison with whole genome sequencing short reads of A. adenophora indicated a base accuracy of the A. adenophora genome was over 99.97% (QV > 35.43), and k-mer completeness estimated at 97.30% (Table 1).
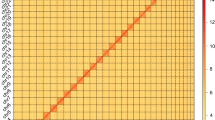
The chromosome interaction heatmap highlighted the grouping of 51 pseudochromosomes into 17 homoeologous clusters, each cluster comprising three allelic chromosomes (Fig. 5). To further affirm the phased genome’s accuracy, we utilized 971 Bacterial Artificial Chromosome (BAC) sequences (N50 of 114.26 kb), calculating a switch error of 4.20% between haplotypes (Fig. 6). The most of largest inversion between haplotype B and A was localized on chromosome 14, spanning ~40 Mb and constituting 57.88% of its length (Fig. 3a). Discrete chromatin interaction signals around breakpoints were observed through inter-haplotypes mapping of Hi-C data (Fig. 3b), and the inversions were also confirmed by BioNano optical maps (Fig. 3c).

Hi-C interaction heatmap of 17 homoeologous chromosome groups in A. adenophora. The Hi-C data was aligned to the A. adenophora genome. The heatmaps for haplotype A1, A2, and B of each group are shown at a resolution of 300 Kb. The dark red dots indicate a high probability of interaction, while the light-yellow dots indicate a low probability of interaction.

Consistency plot of BAC sequences with each chromosome of the A. adenophora genome. The BAC sequences ID were drawn in Y axis.
Code availability
No specific codes or scripts were used in our work. All operations of the data processing were performed according to the manuals and protocols of the corresponding bioinformatics software, some of the parameters are described in the Methods section, and those not mentioned are set as default parameters.
References
Young, A. M. & Larson, B. M. Clarifying debates in invasion biology: a survey of invasion biologists. Environ Res 111, 893–898 (2011).
Mack, M. C. & D’Antonio, C. M. Impacts of biological invasions on disturbance regimes. Trends Ecol Evol 13, 195–198 (1998).
Yang, G., Gui, F., Liu, W. & Wan, F. in Biological Invasions and Its Management in China: Volume 2. (eds. F. Wan, M. Jiang & A. Zhan) 111-129 (Springer Singapore, Singapore; 2017).
Xiao-yu, S., Zhao-hua, L. & Wei-guo, S. Review on studies ofEupatorium adenophoruman important invasive species in China. Journal of Forestry Research 15, 319–322 (2004).
Nie, X. et al. Complete Chloroplast Genome Sequence of a Major Invasive Species, Crofton Weed (Ageratina adenophora). PLOS ONE 7, e36869 (2012).
Wang, R. Invasion dynamics and potential spread of the invasive alien plant species Ageratina adenophora (Asteraceae) in China. Diversity and Distributions, (2006).
Hao, Q. & Ma, J. S. Invasive alien plants in China: An update. Plant Divers 45, 117–121 (2023).
Wang, R. et al. Multiple mechanisms underlie rapid expansion of an invasive alien plant. New Phytol 191, 828–839 (2011).
Hu, W. et al. Genetic and evolution analysis of extrafloral nectary in cotton. Plant biotechnology journal 18, 2081–2095 (2020).
Koren, S. et al. De novo assembly of haplotype-resolved genomes with trio binning. Nat Biotechnol, (2018).
Yang, Z. et al. Extensive intraspecific gene order and gene structural variations in upland cotton cultivars. Nat Commun 10, 2989 (2019).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun 11, 1432 (2020).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), i351–358 (2005).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 31, 1119–1125 (2013).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell systems 3, 99–101 (2016).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat Plants 5, 833–845 (2019).
Waterhouse, R. M. et al. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol Biol Evol 35, 543–548 (2018).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic acids research 46, e126–e126 (2018).
Rhie, A. et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, 245 (2020).
Marçais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput Biol 14, e1005944 (2018).
Goel, M., Sun, H., Jiao, W. B. & Schneeberger, K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol 20, 277 (2019).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117.17, 9451–9457 (2020).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35, W265–268 (2007).
Han, Y. & Wessler, S. R. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res 38, e199 (2010).
Edgar, R. C. & Myers, E. W. PILER: identification and classification of genomic repeats. Bioinformatics 21(Suppl 1), i152–158 (2005).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6, 11 (2015).
Hoede, C. et al. PASTEC: an automatic transposable element classification tool. PLoS One 9, e91929 (2014).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, 4.10.11–14.10.14 (2009).
Ou, S. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol 176, 1410–1422 (2018).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772–780 (2013).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16, 276–277 (2000).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J Mol Biol 268, 78–94 (1997).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–439 (2006).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Guigo, R. Assembling genes from predicted exons in linear time with dynamic programming. J. Comput. Biol. 5, 681–702 (1998).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res 44, e89 (2016).
Kim, D. et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915 (2019).
Haas, B. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8, 1494–1512 (2013).
Kent, W. J. BLAT–the BLAST-like alignment tool. Genome Res 12, 656–664 (2002).
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res 43, e78 (2015).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology 9, R7 (2008).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60 (2015).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic acids research 47, D309–d314 (2019).
Bairoch, A. & Apweiler, R. Te SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic acids research 28, 45–48 (2000).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, 29–37 (2011).
J, Mistry, et al. Pfam: The protein families database in 2021. Nucleic Acids Research (2020).
Ashburner, M. et al. Gene Ontology: tool for the unifcation of biology. Nat genetics 25, 25–29 (2000).
Jones, P. et al. InterProScan 5: genome-scale protein function classifcation. Bioinformatics 30, 1236–1240 (2014).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research 28, 27–30 (2000).
Moriya, Y. et al. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Research 35, W182–5 (2007).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP500679 (2024).
Liu, F. et al. Ageratina adenophora isolate FL-2024a, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JBCNYI000000000 (2024).
Liu, F. et al. Chromosome-level genome assembly of the crofton weed (Ageratina adenophora). figshare https://doi.org/10.6084/m9.figshare.24872130 (2025).
Li, X. et al. Origin and evolution of the triploid cultivated banana genome. Nat Genetics 56.1, 136–142 (2024).
Song, A. et al. Analyses of a chromosome-scale genome assembly reveal the origin and evolution of cultivated chrysanthemum. Nat Commun 14, 2021 (2023).
Mokhtar, M. M., Abd-Elhalim, H. M. & El Allali, A. A large-scale assessment of the quality of plant genome assemblies using the LTR assembly index. AoB Plants 15, plad015 (2023).
Acknowledgements
This work was funded by the National Key Research and Development Program of China (grant numbers 2022YFC2601003, 2021YFC2600100, 2021YFC2600101, and 2021YFD1400200) and the Shenzhen Science and Technology Program (grant numbers JCYJ20190813144407666 and KQTD20180411143628272) as well as partially supported by the Programmer of Introduction Talents of Innovative Discipline to Universities (Project 111) from the State Administration of Foreign Experts Affairs, China (Grant No. #B18042). We thank Prof. Furong Gui and Zujun Yang for advice in plant material collecting and are grateful to the High-Performance Computing Center of Northwest A&F University for providing computational resources in this work.
Author information
Authors and Affiliations
Contributions
F.W. and W.S. conceived and supervised the project. G.Z., and B.L. collected samples for DNA and RNA sequencing. L.D., T.L. and F.L. performed genome sequencing, assembly, and annotation. T.L. and J.G. conducted BAC and Bionano analysis. R.L. performed TE analysis. H.W., X.L., Y.Z. and B.L. carried out RNA-seq analysis and updated all images. L.D., Y.P. and F.L. prepared the results and methods sections. L.D. and F.L. wrote the initial draft of the manuscript with input from all other authors. F.W., L.D., F.L. and W.S. reviewed and revised the manuscript together.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, F., Du, L., Li, T. et al. Chromosome-level genome assembly of the crofton weed (Ageratina adenophora). Sci Data 12, 560 (2025). https://doi.org/10.1038/s41597-025-04637-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04637-1
