
Abstract
Pseudopodoces humilis is a small passerine bird predominantly found in the mid-latitude regions of the Tibetan Plateau in Asia. A chromosome-level reference genome assembly for P. humilis was generated using PacBio CLR with Hi-C. The final genome assembly spans approximately 1.096 Gb, consisting of 1,968 contigs with a Contig N50 of 32.246 Mb, and was evaluated to be 95.60% complete using BUSCO. Hi-C chromosome mapping resulted in 33 chromosome sequences, which enabled the ordering and orientation of 329 contigs, with chromosome lengths ranging from 2.08 Mb to 152.13 Mb, covering 95.85% of the total genome sequence. Repetitive sequences comprised 144.91 Mb of the genome. A total of 381 tRNA, 507 non-coding RNA (ncRNA), and 205 rRNA were identified. In addition, we identified 17,108 protein-coding genes and 29,473 proteins, comprising a total of 17,236,726 amino acids. This high-quality genome assembly provides a strong genomic foundation for exploring critical questions in evolutionary genetics, phylogenomics, and the molecular mechanisms of adaptation - key areas for understanding biodiversity and species resilience amidst changing environments.
Similar content being viewed by others
Background & Summary
The ground tit (Pseudopodoces humilis) is a small passerine bird endemic to the Tibetan plateau. The birds are sexually monomorphic in plumage coloration but slightly dimorphic in body size, with adult males (38.7 g) being 3.75% heavier than adult females (37.3 g)1. It is the only species within the genus Pseudopodoces and belongs to the family Paridae2. This bird was misclassified under the genus Podoces within the Corvidae family. However, later research recognized it as a distinct genus, Pseudopodoces, although it initially remained classified within Corvidae2. Comparative morphological studies, combined with molecular analyses—including nuclear and mitochondrial DNA sequencing—and skeletal morphology, demonstrated its closer evolutionary relationship to species in the Paridae family, prompting its reclassification from Corvidae to Paridae3.
Unlike other members of the Paridae family, the ground tit exhibits several unique morphological features. It has a long, curved beak, specialized for digging and foraging in the harsh environment of the Tibetan Plateau4, and elongated tarsi that aid in terrestrial mobility5. These distinctive traits are directly linked to its ecological niche and have been crucial for its survival in high-altitude environments. Beyond these physical adaptations, the ground tit also exhibits complex social behaviors, making it an emerging model species for research in avian behavioral ecology and evolution6,7,8,9,10,11,12. Molecular studies have revealed that the ground tit’s genome contains numerous adaptations for high-altitude survival, such as enhanced oxygen utilization and resistance to hypoxia - critical traits for thriving on the Tibetan Plateau13. Although passerines are one of the most species-diverse avian groups14, high-quality, chromosome-level genome assemblies are still rare within this clade. Having a high-resolution genome assembly for the ground tit is not only vital for understanding its unique adaptations but also serves as a valuable resource for investigating broader evolutionary questions. Furthermore, this genome assembly provides an essential foundation for comparative genomics, enabling scientists to study how evolutionary pressures have shaped the genomes of species within the Paridae family and beyond.
The high-quality chromosome-level assembly presented in this study significantly advances our understanding of avian genome evolution and paves the way for future research into the evolutionary biology of high-altitude species. This assembly provides a robust genomic foundation for investigating key questions in evolutionary genetics, phylogenomics, and the molecular mechanisms underlying adaptation - crucial areas for comprehending biodiversity and species resilience in the face of changing environments.
Methods
Ethics statement
All animal handling and experimental procedures were approved by the IACUC of Wuhan University Center for Animal Experiment with the code of WP20240520.
Sample collection and sequencing
Muscle tissue from P. humilis was collected for chromosome-level whole genome sequencing. Total RNA was isolated from the muscle using TRIzol reagent (Invitrogen, CA, USA). The purity and integrity of the isolated RNA were assessed with spectrophotometer (NanoDrop Technologies, DE, USA) and Bioanalyzer 2100 system (Agilent Technologies, CA, USA). To confirm the absence of RNA contamination, samples were also analyzed on a 1.5% agarose gel. The SMRTbell library was prepared using the SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, CA, USA). Briefly, 5 µg of genomic DNA was first subjected to enzymatic treatment to remove single-stranded overhangs, followed by DNA backbone repair to address any existing damage, then polished and given an A-overhang. Ligation with T-overhang SMRTbell adapters was carried out at 20 °C for 60 min. The library’s size distribution and concentration were assessed with the FEMTO Pulse automated pulsed-field capillary electrophoresis system (Agilent Technologies, CA, USA) and a Qubit 3.0 Fluorometer (Life Technologies, CA, USA). 3 µg of the library underwent size selection using the BluePippin system (Sage Science, MA, USA) and removing SMRTbells ≤ 25 kb in size. The library was purified once again with 1X AMPure PB beads, and its final size and concentration were confirmed using the FEMTO Pulse and Qubit dsDNA HS Reagents Assay Kit. Prior to sequencing, a primer and Sequel II DNA polymerase were annealed and bound to the final SMRTbell library. The library was then loaded at a concentration of 35 pM via diffusion loading onto a single 8 M SMRT Cell. Sequencing was performed on the Sequel II System using the Sequel II Sequencing Kit, with 900-minute runtime movies conducted by Frasergen Bioinformatics Co., Ltd. (Wuhan, China). The sequencing generated 147.20 Gb of polymerase reads (Table 1) and 147.07 Gb of subreads (Table 2). The distributions of polymerase read lengths and subread lengths are depicted in Fig. 1A,B, respectively.

The reads number and length distribution of P. humilis genome. (A) The distributions of polymerase read lengths (bp) and reads number. (B) The distributions of subread lengths (bp) and reads number.
Genome assembly and quality evaluation
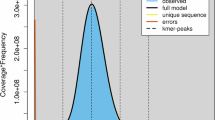
The draft genome was initially constructed using MECAT2 (version 20.190.226). Following this, error correction was performed using the GCPP tool with further corrections applied to any residual errors using short-read data in conjunction with Pilon (v1.22)15. The final assembly of P. humilis genome spans approximately 1.096 Gb and consists of 1,968 contigs, with a Contig N50 of 32.246 Mb (Table 3). CLR subreads were realigned to the final genome using minimap2 (v2.5) to access the accuracy of the assemblied genome. The completeness of the assembled genome was evaluated using BUSCO16 with the vertebrata_odb10 database, resulting 95.60% of the BUSCO evaluation, indicating a highly complete genome assembly (Fig. 2). A total of 1.096 Gb of clean read were generated from the Hi-C library and aligned to P. humilis genome using BWA (v0.7.17). Hicup software was used to filtered out Invalid reads - including those resulting from self-ligation, non-ligation, Start Near Rsite errors, PCR artifacts, random breaks, as well as excessively large or small fragments. The 3D-DNA tool was then employed to order and orient these contigs17,18, 1,591 contigs were grouped into 33 clusters, as illustrated in Fig. 3. This approach successfully ordered and oriented 329 contigs, covering a total of 1,051,101,716 bp. As a result, we achieved the first high-quality chromosome-level genome assembly, with chromosome lengths ranging from 2.08 Mb to 152.13 Mb, accounting for 95.85% of the total genome sequence.

The BUSCO assessment results for P. humilis genome. It showed 95.60% of the BUSCO genes were complete, indicating a highly complete genome assembly. The indicators used in the assessment are as follows: C represents complete BUSCOs, S indicates single-copy BUSCOs, D refers to complete and duplicated BUSCOs, F denotes fragmented BUSCOs, and M indicates missing BUSCOs. The total number of BUSCO groups analyzed is denoted by n.

P. humilis genome contig contact matrix was constructed using Hi-C data. The intensity of the red coloration indicates the contact density, where darker shades correspond to higher interaction frequencies.
Analysis of repetitive sequences
For the homology-based analysis, known transposable elements (TEs) were identified using RepeatMasker (version open-4.0.9) with the Repbase TE library19. Additionally, RepeatProteinMask was employed to search using the TE protein database as a query. For de novo prediction, a custom repeat library for P. humilis genome was constructed using RepeatModeler (http://www.repeatmasker.org/RepeatModeler/), which integrates RECON (v1.08)20 and RepeatScout (v1.0.5)21. In addition, we performed de novo searches for long terminal repeat (LTR) retrotransposons using LTR_FINDER (v1.0.7)22. Tandem repeats were identified using the Tandem Repeat Finder (TRF) package23. Those non-interspersed repeat sequences, such as low-complexity repeats, satellites, and simple repeats, were detected with RepeatMasker. Overall, 144.91 Mb of repeat sequences were identified in P. humilis genome. A summary of these repeat sequences is provided in Table 4.
Annotation and analysis of non-coding RNA
The tRNA genes were identified using the tRNAscan-SE algorithm (v1.3.1)24 with default settings. For rRNA identification, rRNA sequences from closely related species were first retrieved from the Ensembl database and then aligned to our genome using blastn25,26, applying an E-value cutoff of <1e−5, identity ≥85.00%, and match length ≥50 bp. The miRNAs and snRNAs were detected using Infernal (v1.1.2)27 by searching against the Rfam (v14.1) database28 with default parameters. Our analysis led to the identification of 381 tRNA sequences, with a total length of 28,060 bp and an average sequence length of 73 bp. We also identified 507 non-coding RNA (ncRNA) sequences and 205 rRNA sequences. Among the rRNA genes, the 5S rRNA was the most prevalent, with 98 sequences totaling 11,584 bp and an average length of 118.2 bp. Within the ncRNA category, 197 miRNAs were identified, with a combined length of 16,343 bp and an average length of 82.96 bp. Detailed statistics for the identified rRNA and ncRNA in P. humilis genome are summarized in Tables 5, 6, respectively.
Annotation and analysis of protein coding genes
Protein-coding genes in P. humilis genome were predicted using three complementary approaches: ab initio prediction, homology-based prediction, and RNA-Seq-assisted prediction. For ab initio prediction, Augustus (v3.3.1)29,30,31 and Genescan32 were used, utilizing gene models trained on high-quality proteins derived from the RNA-Seq dataset. Homology-based prediction was carried out with Exonerate (v2.2.0)33 by aligning known protein sequences to the genome and predicting coding genes using default settings. Finally, Maker (v3.00)34 was used to integrate the results from all three approaches, producing consistent, non-overlapping gene models to define the final gene structures. In total, 17,108 protein-coding genes were identified in the assembled P. humilis genome, with an average gene length of 28,058.77 bp. Additionally, 29,473 proteins were identified, comprising a total of 17,236,726 aa, with an average protein length of 584.83 aa, and a coding sequence GC content (CDS_GC) ratio of 0.54. The summary of the predicted protein-coding genes is provided in Table 7. The completeness of the predicted protein-coding genes was assessed using BUSCO with the vertebrata_odb10 database, resulting in a completeness score of 95.40% (Fig. 4).

The protein-coding gene BUSCO assessment for P. humilis genome using vertebrata_odb10. It showed a completeness score of 95.40%. Indicator: C, indicates complete BUSCOs; S, single-copy BUSCOs; D, complete and duplicated BUSCOs; F, fragmented BUSCOs; and M, missing BUSCOs; n, total number of BUSCO groups analyzed.
Functional annotation and analysis of protein coding genes
Gene functions were inferred based on alignments with the best matches from multiple databases, including the NCBI Non-Redundant (NR) protein database, SwissProt35, TrEMBL35, and InterPro36, using BLASTP (NCBI BLAST v2.6.0+)25,26 with an E-value threshold of 1E−5. In addition, gene function annotations were conducted using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database37. Gene Ontology (GO) IDs were assigned through Blast2GO38. Protein domain identification was performed using PfamScan36 and InterProScan (v5.35–74.00)39 by referencing the InterPro protein database. Motifs and domains in the gene models were identified using the Pfam database28. In total, approximately 27,368 protein-coding genes (around 92.00%) of P. humilis genome were successfully annotated with known functions, conserved domains, and Gene Ontology terms. The detailed functional annotation of protein-coding genes is summarized in Table 8.
For KOG annotation, the top five categories with the highest number of annotated genes were: signal transduction mechanisms (3,428), posttranslational modification, protein turnover, chaperones (1,481), transcription (1,488), intracellular trafficking, secretion, and vesicular transport (1,072), and cytoskeleton (775). In the GO functional annotation, the molecular function category was dominated by the terms binding (57.91%) and catalytic activity (28.46%). In the cellular component category, membrane part and cell represented 39.74% and 22.07% of annotated genes, respectively. In the biological process category, cellular process (20.51%) and metabolic process’ (31.52%) were the most highly represented terms. Figure 5 illustrates the top five GO terms with the highest percentage of annotated genes across the molecular function, cellular component, and biological process. For KEGG annotation, 1,165 genes were categorized under cellular community – eukaryotes. The signal transduction category included 6,137 annotated genes, while 767 genes were linked to folding, sorting, and degradation. In the metabolism classification, 903 genes were related to lipid metabolism, and 818 were involved in carbohydrate metabolism. Moreover, 2,522 genes were annotated under the endocrine system and 2,015 genes under the immune system. Figure 6 depicts the top KEGG classifications with the highest number of annotated genes across these five categories.

The top five GO terms with the highest number of annotated genes across the molecular function, cellular component, and biological process categories in P. humilis genome.

The top KEGG classifications with the higher number of annotated genes across these five categories in P. humilis genome.
Data Records
The chromosome assembly data has been uploaded and deposited in NCBI GeneBank under accession number GCA_049639895.140. The genome assembly and annotation files have been submitted to figshare database41. The BioProject and BioSample accession number for P. humilis was PRJNA118206942 and SAMN4458034543, respectively. Furthermore, the RNA-sequencing data was deposited with accession number of SRR3148548144. The whole genome shotgun data was deposied in NCBI with accession number of JBJMBD000000000.145. In addition, PacBio and Hi-C data have been uploaded and deposited in the NCBI SRA under accession numbers of SRR3148548246 and SRR3148548347, respectively.
Technical Validation
To ensure the accuracy of the sequencing data, a series of quality control checks were performed on the samples prior to library construction. The genomic DNA concentration was precisely measured and the Qubit readings were compared with NanoDrop measurements to further verify sample purity. Only samples that met all quality standards proceeded to library preparation. The completeness of the assembled genome was evaluated using BUSCO (v3.1) with the vertebrata_odb10 database.
Code availability
No custom scripts or codes were used in the management and verification of the data sets in this study. All software and pipelines used for data processing were executed according to the manuals and protocols of the bioinformatics software cited above. The specific parameters were described if the default parameters were not applied for data analysis.
References
Fan, H., Guo, W. & Lu, X. How do helpers slow senescence in a Tibetan cooperatively breeding bird? Ecosphere 13, e4314 (2022).
Cai, Q. et al. Genome sequence of ground tit Pseudopodoces humilis and its adaptation to high altitude. Genome Biol. 14, 1–2 (2013).
James, H. F. et al. Pseudopodoces humilis, a misclassified terrestrial tit (Paridae) of the Tibetan Plateau: evolutionary consequences of shifting adaptive zones. Ibis 145(2), 185–202 (2003).
Cheng, Y. et al. Evolution of beak morphology in the Ground Tit revealed by comparative transcriptomics. Front. Zool. 14, 1–13 (2017).
Huang, M., Liu, Y. & Lu, X. Genomic basis of adaptive divergence in leg length between ground-and tree-dwelling species within a bird family. Genome Biol. Evol. 15(9), evad166 (2023).
Du, B. & Lu, X. Bi-parental vs. cooperative breeding in a passerine: fitness-maximizing strategies of males in response to risk of extra-pair paternity? Mol. Ecol. 18, 3929–3939 (2009).
Ke, D. & Lu, X. Burrow use by Tibetan Ground Tits Pseudopodoces humilis: coping with life at high altitudes. Ibis 151, 321–331 (2009).
Li, Z., Chen, S., Wei, S., Komdeur, J. & Lu, X. Should sons breed independently or help? Local relatedness matters. J. Anim. Ecol. 92, 2189–2200 (2003).
Lu, X., Huo, R., Li, Y., Liao, W. & Wang, C. Breeding ecology of ground tits in northeastern Tibetan plateau, with special reference to cooperative breeding system. Curr. Zool. 57, 751–757 (2011).
Tang, S. et al. Social organization, demography and genetic mating system of a Tibetan cooperative breeder. Ibis 159, 687–692 (2017).
Wang, C. & Lu, X. Female ground tits prefer relatives as extra-pair partners: driven by kin-selection? Mol. Ecol. 20, 2851–2863 (2011).
Wang, C. & Lu, X. Hamilton’s inclusive fitness maintains heritable altruism polymorphism through rb = c. Proc. Natl. Acad. Sci. USA. 115, 1860–1864 (2018).
Qu, Y. et al. Ground tit genome reveals avian adaptation to living at high altitudes in the Tibetan plateau. Nat. Commun. 4(1), 2071 (2013).
Barker, F. K., Cibois, A., Schikler, P., Feinstein, J. & Cracraft, J. Phylogeny and diversification of the largest avian radiation. Proc. Natl. Acad. Sci. U.S.A. 101(30), 11040–5 (2004).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One. 9(11), e112963 (2014).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31(19), 3210–3212 (2015).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 1–11 (2015).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Jurka, J. Repbase Update: a database and an electronic journal of repetitive elements. Trends Genet. 16(9), 418–20 (2000).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12(8), 1269–76 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics. 21(suppl_1), i351–358 (2005).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 25, 955–964 (1997).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25(17), 3389–3402 (1997).
Camacho, C. BLAST plus: architecture and applications. BMC Bioinform. 10(421), 1 (2009).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics. 25(10), 1335–1337 (2009).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42(D1), D222–230 (2014).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–312 (2004).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–W467 (2005).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 6, 31 (2005).
Cantarel, B. L. et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31(1), 365–370 (2003).
Mitchell, A. et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43(D1), D213–21 (2015).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40(D1), D109–114 (2012).
Conesa, A. & Götz, S. Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics. 1, 619832 (2008).
Mistry, J., Bateman, A. & Finn, R. D. Predicting active site residue annotations in the Pfam database. BMC Bioinform. 8, 1–4 (2007).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 30(9), 1236–1240 (2014).
NCBI Genome. https://identifiers.org/ncbi/insdc.gca:GCA_049639895.1 (2025).
Da, X. W., Liu, Y. R., Jin, X. & Lu, X. The genome assembly and annotation of Pseudopodoces humilis. Figshare dataset. https://figshare.com/s/7df335612015d9d188e6 (2025).
NCBI BioProject. https://www.ncbi.nlm.nih.gov/bioproject/1182069 (2025).
NCBI BioSample. https://www.ncbi.nlm.nih.gov/biosample/44580345 (2025).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR31485481 (2025).
NCBI Sequence Read Archive. https://www.ncbi.nlm.nih.gov/nuccore/JBJMBD000000000 (2025).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR31485482 (2025).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR31485483 (2025).
Acknowledgements
We thank Supercomputing Center of Wuhan University for assistance with data analyses. This research was supported by the National Natural Science Foundation of China (31830085) and the Second Tibetan Plateau Scientific Expedition and Research program (2019QZKK0501).
Author information
Authors and Affiliations
Contributions
X.L. designed the research; X.D., Y.L. and X.J. collected the samples and performed the data analysis; X.L., X.D., Y.L. and X.J. wrote the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Da, X., Liu, Y., Jin, X. et al. Chromosome-level assembly of Pseudopodoces humilis genome: A resource for avian evolutionary studies. Sci Data 12, 799 (2025). https://doi.org/10.1038/s41597-025-05171-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05171-w
