
Abstract
Graphite carbon nitride (g-C3N4) is a two-dimensional nano-sheet with electronic properties, which shows unique characteristics with high chemical and thermal stability in its structure. The functionalization of these compounds through covalent bonding is an important step towards significantly improving their properties and capabilities. To achieve this goal, a novel strategy for the covalent functionalization of Fe3O4@g-C3N4 with thiamine hydrochloride (vitamin B1) via cyanuric chloride (TCT), which is a divalent covalent linker, was presented. The efficiency of Fe3O4@gC3N4@Thiamine as a heterogeneous organic catalyst in the synthesis of spirooxindole-pyran derivatives and 2-amino-4H-pyran under solvent-free conditions was evaluated and the yields of high-purity products were presented. In addition, easy recycling and reuse for seven consecutive cycles without significant reduction in catalytic activity are other features of this catalyst. Moreover, the performance of the prepared sorbent in the microextraction technique (herein, magnetic solid phase extraction) was studied. The tebuconazole was selected as the target analyte. The target analyte was extracted and determined by HPLC–UV. Under the optimum condition, the linear range of the method (LDR) was estimated in the range of 0.2–100 μg L−1 (the coefficient of determination of 0.9962 for tebuconazole). The detection limit (LOD) of the method for tebuconazole was calculated to be 0.05 µg L−1. The limit of quantification (LOQ) of the method was also estimated to be 0.16 µg L−1. In order to check the precision of the proposed method, the intra-day and inter-day relative standard deviations (RSD%) were calculated, which were in the range of 1.5- 2.8%. The method was used for the successful extraction and determination of tebuconazole in tomato, cucumber, and carrot samples.
Similar content being viewed by others

Introduction
One of the fundamental goals of researchers today is to develop, produce, and study modern catalysts that not only selectively promote organic reactions, but also produce the target compounds with high efficiency from an economic perspective. The simple separation and the reusability of the catalyst in several stages bring the researchers closer to this goal1,2.
In recent decades, concern about the environmental and health risks associated with catalytic processes has increased, and much attention has been paid to efforts to reduce their impact. One of the areas of research being discussed today is the use of heterogeneous catalysts, which are spreading quickly in chemistry. Recyclable green heterogeneous catalysts are often used for environmentally friendly conditions in multi-component reactions in synthesis and industry3,4,5,6.
Nanoparticles are highly attractive for use as catalysts due to their large surface-to-volume ratio in comparison to bulk materials. One of the pioneering applications of nanoparticles is in catalysts. Nanocatalysis plays a crucial role in both academic and industrial research and development. The superiority of nanocatalyst synthesis technology lies in its ability to create highly efficient catalysts with enhanced surface area and reactivity. Nanoparticles have a high surface area to volume ratio, allowing for more active sites where catalytic reactions can occur. This results in faster reaction rates, lower energy requirements, and increased selectivity in catalytic processes7,8,9. Moreover, they can be intricately designed and customized at the molecular level to enhance their performance for particular reactions. This precise control over the structure and composition of the catalyst results in improved catalytic activity and selectivity. The synthesis technology for nanocatalysts provides a potent tool for creating more sustainable and efficient chemical processes across various industries, such as pharmaceuticals, petrochemicals, and environmental remediation10,11,12.
The g-C3N4 or graphitic carbon nitride is a suitable substrate with chemical stability and low cost, with a quasi-graphite structure as a polymer semiconductor; recently, this topic has garnered the interest of numerous researchers13,14,15. The existence of active positions on the surface of the two-dimensional monolayer created by –NH2 or –NH functional groups in this compound causes easy modification of the g-C3N4 surface through cross-linking or linking with other functional groups13. Due to its remarkable high thermal stability, this compound can be used for high temperatures16. The g-C3N4 is synthesized from various raw materials for instance dicyandiamide13, melamine17, thiourea18, trithiocyanuric acid19, and urea 19, most of which are toxic and explosive in addition to being costly. Amongst them, urea is an inexpensive, safe, and plentiful material 14.
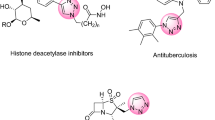
Spirooxindole-pyran derivatives are among the heterocyclic compounds that exist as the core of many natural, biological, or pharmaceutical products due to their unique structures. This group of compounds has diuretic, anti-anaphylactic, anti-cancer, antioxidant, and antimicrobial properties. Therefore, finding facile and efficient synthetic methods for heterocyclic groups in organic synthesis has become extremely important (Fig. 1)20.

Some biological structures containing spirooxindole and 4H-pyran skeletons.
Another group of heterocyclic compounds are substituted derivatives of 2-amino-4H-pyran, which play a vital part in the life of humans21. They are present in the structure of many agricultural chemicals and dyes. They also have various biological activities, such as anticancer antioxidant, antimicrobial, and rheumatic activities. They are used in the synthesis of many useful and important intermediates like pyridones, pyranopyrazoles, lactones, and imidoesters (Fig. 1)22,23.
Pesticides are widely used across the globe to increase the productivity of agricultural practices24,25. It should be noted that pesticides used in farms may not only affect target pest populations but also have consequences for other non-target species. They are classified into organic and inorganic substance groups. Based on their chemical nature and efficiency, pesticides are classified into three groups: fungicides, insecticides, and herbicides. These substances are also classified according to their toxicity, the most toxic of which are insecticides, followed by fungicides and herbicides. As a result of inappropriate use of these poisons in agriculture, these substances enter the human body through ingestion, skin contact or inhalation and cause serious damage to individual health. Therefore, controlling the residues of these chemicals in food and agricultural products such as fruits and vegetables has an important role in maintaining human health. Tebuconazole is among the widely used fungicides in agricultural products26 (Fig. 2). One of the techniques of dispersed solid phase extraction is magnetic solid phase extraction (MSPE). Thus, a magnetic sorbent is introduced into an aqueous sample in order to capture the desired analytes. By applying a magnetic field, the sorbent can be separated27,28,29,30,31,32,33,34.

General structure of tebuconazole.
Herein, the synthesized sorbent was characterized by different methods and after approval, the ability of the sorbent for magnetic solid phase extraction of Tebuconazole was investigated. The extracted analyte was quantified by HPLC–UV and various parameters such as sorbent amount, eluent type, extraction time, pH, desorption volume, and desorption time have been optimized. In the following, the catalytic performance of the sorbent was studied. Also, taking into account the importance of the synthesis of spirooxindole-pyran and 2-amino-4H-pyran compounds in line with our effort in designing all kinds of green catalysts 35,36,37,38,39,40, in light of the singular qualities of g-C3N4 and the effectiveness of thiamine (vitamin B1), we designed a new and efficient catalyst, in which thiamine using We report that the cyanuric chloride linker was immobilized on g-C3N4 magnetic substrate through covalent bonding and used in the preparation of heterocyclic compounds.
Experimental
General
All chemicals were obtained from Sigma Aldrich and Merck. An electrothermal apparatus was used to determine melting points, the IA9100. Shimadzu IR-470 spectrophotometer was used to detect FT-IR spectra. A Bruker Avance DRX-400 spectrometer (Bruker, Germany) was used to acquire 1H and 13C NMR spectra. The SEM images were recorded on MIRA 3-XMU and the TEM images were taken on a Philips EM-208S TEMVSM model MDKB from Danesh Pajohan Kavir Co was used to obtain magnetic analysis curves. Philips PW-1830 was used to record X-ray diffraction (XRD) patterns. To determine the concentration of the target analyte by HPLC, a Waters dual pump (model 1525), a Rheodyne injection valve containing a 20 μL loop, a C18 analytical column (250 mm × 4.6 mm I.D., particle size 5.0 μm) and an ultraviolet detector (Waters) were used. The mobile phase of H2O and MeCN (30:70) with a flow rate of 1 mL min−1 and a wavelength of 220 nm was used for measuring the selected analytes.
Preparation of g-C3N4 nanosheets
g-C3N4 samples were synthesized using the previous reports through thermal polymerization method14. 1.0 g of g-C3N4 in bulk was stirred in a mixture of H2SO4 and HNO3 (1:1 ratio) for 2 h at room temperature. The solution was diluted by adding 1 L of deionized water, and the resulting solid was filtered, rinsed with deionized water multiple times, and then dried at 60 °C. Then, 0.1 g of processed treated bulk g-C3N4 was dispersed in 100 ml of deionized water and subjected to ultrasonic waves for 6 h. Finally, the formed suspension was centrifuged at 5000 rpm to separate the residual unexfoliated g-C3N4 nanoparticles.
Preparation of Fe3O4@g-C3N4
According to the reported method 41. After dispersing g-C3N4 (0.4 g) in deionized water for 4 h under ultrasonic conditions, FeCl2.4H2O (2.3 g, 18.14 mmol) and FeCl3.6H2O (4.68 g, 17.3 mmol) were added to the solution. Then, dropwise additions of 15ml aqueous ammonia solution (NH4OH, 25%) were made to the previous mixture until a pH of 9–10 was reached. The mixture was heated to 80 ℃ and stirred for 2 h in a nitrogen atmosphere. An external magnet separated the black solid, and washed thoroughly in EtOH and H2O; then, Fe3O4@g-C3N4 was dried for overnight at 50 °C.
Preparation of Fe3O4@g-C3N4@Thiamine
Initially, dissolve 1.5 g of Fe3O4@g-C3N4 in 30 mL of dry THF. On an ice bath, the reaction mixture was mixed with 2 mmol of cyanuric chloride (TCT). At 0–5 °C for 3 h, the reaction was stirred. Eventually, the Fe3O4@g-C3N4@TCT was magnetically separated and washed repeatedly 42. Next, the obtained product was dispersed in 50 mL of dry THF for 25 min. Following, 3 mmol of thiamine hydrochloride and 2 mmol of K2CO3 were added to this mixture, placed under nitrogen gas, and refluxed for 48 h. The resulting mixture was cooled to room temperature (Fe3O4@g-C3N4@thiamine) washed with ethyl acetate/H2O/EtOH and dried after magnetic field separation (Fig. 3).

Synthesis of Fe3O4@g-C3N4@Thiamine.
MSPE performance
The synthesized magnetic sorbent was added to the sample solution (5 mL) in order to determine its extraction efficiency. The solution was sonicated at pH = 5 for 8 min at 25 °C. Then, the magnetic adsorbent including the target analyte was collected by a magnetic (1.6 Tesla), and the liquid above it was poured off. 50 μL of MeOH (as a suitable detergent) was added to desorb the extracted analytes, then it was subjected to ultrasound (2 min). At last, an external magnet collected the adsorbent and the desorbed analytes were injected into the HPLC–UV (Fig. 4).

Overview of isolation and identification of tebuconazole with the help of Fe3O4@g-C3N4@Thiamine nanocatalyst.
With respect to the amino groups of the as-synthesized sorbent, the extracting device has the ability of to establish a hydrogen bond with the selected model analyte. Moreover, the elements of the prepared sorbent were analyzed by EDX analysis. The obtained result approved the presence of O, C, N, S, and Cl atoms. Based on the data, it is proposed that hydrogen bonding is used to transport the selected tebuconazole to the sorbent. Additionally, it can be considered that the chosen analyte can form Vander Waals bonds with the analyte due to the dispersion of amine group on the surface of the sorbent. Therefore, the prepared sorbent has the ability to establish hydrogen bonds and π-π bonds. Since the model analyte has benzene rings and OH, N, and Cl groups, it has the ability to establish hydrogen and π-π bonds with the desired adsorbent, and in this way, it is extracted on the prepared magnetic sorbent. According to the extraction mechanism, any species that has π-electrons and groups participating in hydrogen bonding can be selected as a candidate for extraction by the proposed sorbent (Fig. 5). Moreover, it is worth mentioning that the extraction process after the screening step was improved by optimizing the effective factors using the Box-Behnken (BBD) design. The experimental design utilized MINITAB software. To optimize this factor, the one-at-a-time strategy was also used as a result of the discrete nature of the eluent solvent type. Finally, the optimal conditions for optimization were determined as follows: pH = 5, extraction time of 8 min, desorption time of 2 min, sorbent amount of 5 mg, and the MeOH volume of 50 µL.

A possible mechanism for tebuconazole adsorption onto Fe3O4@g-C3N4@Thiamine surface.
Optimization by experimental design
To achieve a high extraction yield, an optimization step is essential. Optimization was performed using a multivariate approach. The peak area was selected as the response. In the first step, various solvents such as isopropanol, MeCN, ethyl acetate, EtOH, and MeOH were tested. The results indicated that MeOH has a better response. The extraction performance of the method can be influenced by multiple parameters containing the pH of the sample solution, type and volume of desorption solvent, time of desorption, extraction time, and amounts of sorbent. In order to determine the important factors, we used the Placket Burman Design (PBD) to screen the six parameters. The results indicated that extraction time, pH, desorption time, and the amount of the sorbent have significant impacts on extraction efficiency. So, these parameters were measured by BBD for further study. The extraction process after the screening step was improved by optimizing the effective factors using the Box-Behnken (BBD) design. The experimental design utilized MINITAB software. To optimize this factor, the one-at-a-time strategy was also used as a result of the discrete nature of the eluent solvent type. Finally, the optimal conditions for optimization were determined as follows: pH = 5, extraction time of 8 min, desorption time of 2 min, sorbent amount of 5 mg, and the MeOH volume of 50 µL.
Method evaluation
To assess the ability of this method to perform quantitative analytical work, figures of merit including the limit of quantification (LOQ), the limit of detection (LOD), linear dynamic range (LDR), relative standard deviations (RSDs), and enrichment factor (EF) were calculated. Testing was conducted using various concentrations of spiked aqueous samples, including 0.2, 0.50, 1.00, 2.00, 5.00, 10.00, 20.00, 50.00, and 100.00 μg L−1 To evaluate the LDR. The determination of LOD and LOQ was made by analyzing the signal-to-noise ratio of 3 and 10, respectively. A %RSD calculation was performed at three concentration levels of 2.00, 10.00, and 100.00 g L−1 to demonstrate the reproducibility of the method. EF was obtained as the analyte concentration in the optimal eluent compared to the initial concentration. The estimated EF of tebuconazole was 72. In ideal conditions, the linear range of the method (LDR) was estimated in the range of 0.2–100 μg L−1 (the coefficient of determination of 0.9962 for tebuconazole). The detection limit (LOD) of the method for tebuconazole was calculated to be 0.05 µg L−1. The limit of quantification (LOQ) of the method was also estimated to be 0.16 µg L−1. To verify the precision of the proposed method, the intra-day and inter-day relative standard deviations (RSD%) were calculated, which were in the range of 1.5–2.8%. The matrix effect was also calculated according to the comment. The Matrix effect is an alteration of the analyte response due to matrix component(s) in the sample. The Matrix effect is evaluated by calculating the matrix factor (MF). The MF is determined by comparing the analyte response in the presence of matrix with that in the absence of matrix. The MF may be normalized by its internal standard. The precision of the MF calculated should not exceed 15%. Matrix effect can also be evaluated by analyzing QC samples, each prepared using matrix from at least 6 different sources. The precision of determined concentrations should not be greater than 15%. So, herein, the MF is determined by comparing the analyte response in the presence of matrix with that in the absence of matrix. In this study the 10 µg L−1 of the target analyte was selected. The analyte response in the presence of matrix was 2153. The analyte response in the absence of matrix (DI-water) was 2175 (n = 3 for both experiments). The T-Test at the confidence limit of 95% was carried out. The obtained results of T-test indicated that there is no difference between the response of the analyte in the real sample and the DI-water. Moreover, the relative recoveries were also measured to study the matrix effect. The relative recoveries were measured using the following equation:
In the equation, Creal, Cadded, and Cfound are the concentrations of the analyte (µg L−1) in the real sample, the spiked concentration into the real sample and the concentration after MSPE, respectively. According to the data, the relative recoveries from the real sample analysis were higher than 95% which approves the acceptable accuracy of the presented method.
Real sample analysis
Thorough monitoring of pesticide residues is crucial for proper assessment of human exposure to pesticides through foods. Therefore, maximum residue levels (MRLs) for pesticides in foodstuffs are established in most countries to guarantee consumer safety and to regulate. MRLs in foodstuffs have been set by Government agencies and European Union Commission to guarantee consumer safety and to regulate international trade. In this context, the existing MRLs from 0.05 mg/kg to 0.5 mg/kg for olives for oil production and table olives; to 6 mg/kg for fresh herbs, to 40 mg/kg for herbal infusions and from 1 to 4 mg/kg for rice has been presented for tebuconazole. In order to protect consumers, Codex Alimentarius Commission (CAC) have established standards/regulations for the MRL of tebuconazole between 0.02–5.0 mg kg−1 1,32,34. Moreover, the MRL of tebuconazole in tomato, carrot and cucumber is 0.7 mg/kg, 0.4 mg/kg and 0.2 mg/kg, respectively.
The selected analyte was measured in some real samples such as tomato, cucumber, and carrot. The standard addition method was utilized for the quantification. The desired samples were prepared from a local market and after washing with H2O, and then fully peeled and crushed. In order to prepare cucumber and tomato, 10 g of the samples were crushed and mixed with MeCN (15 mL) and H2O (5 mL). Subsequently, NaCl (1 g) was added to the solution and the mixture was subjected to ultrasound for 1 min. Eventually, MgSO4 (4 g) was added and centrifuged for 5 min. Afterward, the supernatant (5 mL) was removed and centrifuged for an additional 3 min. The extract of the target analytes was extracted using the obtained solution. To prepare the carrot sample, the following steps were done: 5 g of sample was crushed and mixed with MeCN (10 mL). The resulting mixture was shaken for 1 min and then centrifuged at 6000 rpm for 10 min. Following that, the clear supernatant solution (1 mL) was removed and vortexed for 1 min followed by centrifugation. The supernatant was passed through a 0.22 μm syringe filter and the final solution was used for the extraction process. The spiked samples contained three various concentration levels (2, 5, and 10 gL−1), each of which was done with three replications. Additionally, spiking and relative recoveries were evaluated. Three repeated experiments revealed RSDs between 2.2% and 3.9%, this approach is appropriate for the extraction and identification of target analytes in real samples (Table 1).
General procedure for the synthesis of spirooxindole pyran derivatives
1 mmol of isatin derivatives, 1.2 mmol of malononitrile, 1 mmol of 1,3-dicarbonyl compounds and 15 mg of Fe3O4@g-C3N4@Thiamine were stirred in solvent-free conditions at 25 °C. Once the reaction had been completed (checked by TLC), hot EtOH was added, Fe3O4@g-C3N4@Thiamine was collected by the magnetic field and the product was separated (Fig. 6).

The synthesis of spirooxindole-pyrans.
General procedure for the synthesis of 2-amino-4Hchromene derivatives
1 mmol of aromatic aldehydes, 1 mmol of active methylenes, 1 mmol of OH-acids, and 20 mg of Fe3O4@g-C3N4@Thiamine were mixed under solvent-free conditions at 25 °C. As soon as the reaction was complete, the magnetic field was used to separate Fe3O4@g-C3N4@Thiamine. By recrystallizing in EtOH, the products were obtained (Fig. 7).

The synthesis of 2-amino-4Hchromene.
Selected spectral data
2-amino-7,7-dimethyl-2′,5-dioxo-5,6,7,8-tetrahydrospiro[chromene-4,3′-indoline]-3-carbonitrile (4a)
1H NMR (CDCl3, 400 MHz): δ 1.00 (s, 3H), 1.04 (s, 3H), 2.14 (q, 2H), 2.54 (d, 2H), 6.78 (d, 1H), 6.88 (t, 1H), 6.97 (d, 1H), 7.15 (t, 1H), 7.24 (s, 2H), 10.42 (s, 1H). 13C NMR (CDCl3, 100 MHz): δ 28.4, 29.0, 32.4, 47.2, 50.4, 57.9, 109.6, 111.2, 117.7, 122.1, 123.4, 128.6, 134.8, 142.4, 159.2, 164.5,178.5, 195.3 ppm.
7′-amino-5-chloro-1′,3′-dimethyl-2,2′,4′-trioxo-1′,2′,3′,4′-tetrahydrospiro[indoline-3,5′-pyrano[2,3-d] pyrimidine]-6′-carbonitrile (4d)
1H NMR (DMSO, 400 MHz): δ 3.05 (s, 3H), 3.7 (s, 3H), 6.83 (d, 1H), 7.22 (d, 2H), 7.29 (s, 1H), 7.66 (s, 2H), 10.66 (s, 1H). 13C NMR (CDCl3, 100 MHz): 28.11, 29.81, 48.07, 57.43, 86.96, 111.13, 117.17, 124.52, 126.24, 128.82, 136.16, 141.50, 150.13, 152.70, 158.64, 159.98, 177.85 ppm.
2′-amino-2,5′-dioxo-5′H-spiro[indoline-3,4′-pyrano[3,2-c] chromene]-3′-carbonitrile (4i)
1H NMR (DMSO, 400 MHz): δ 6.87 (d, 1H), 6.95 (t, 1H), 7.22 (d, 2H), 7.51 (t, 1H), 7.55 (t,1H), 7.67 (s, 1H), 7.76 (t, 1H), 7.95 (d, 1H), 10.7 (s, 1H). 13C NMR (CDCl3, 100 MHz): 20.15, 27.22, 36.73, 47.61, 57.25, 111.01, 111.70, 123.92, 126.08, 128.50, 137.03, 141.38, 159.18, 167.053, 178.40, 195.71 ppm.
2-amino-5′-chloro-2′,5-dioxo-5,6,7,8-tetrahydrospiro[chromene-4,3′-indoline]-3-carbonitrile (4m)
1H NMR (DMSO, 400 MHz): δ 1.87–198 (m, 2H), 2.25 (t, 2H), 2.66 (t, 2H), 6.81 (d, 1H), 7.15 (s, 1 H), 7.20 (d, 1H), 7.31 (s, 2H), 10.57 (s, 1H). 13C NMR (CDCl3, 100 MHz): 20.15, 27.22, 36.73, 47.61, 57.25, 111.01, 111.70, 123.92, 126.08, 128.50, 137.03, 141.38, 159.18, 167.053, 178.40, 195.71 ppm.
7′-amino-5-chloro-2,4′-dioxo-2′-thioxo-1′,2′,3′,4′ tetrahydrospiro[indoline-3,5′-pyrano[2,3-d] pyrimidine]-6′-carbonitrile (4r)
1H NMR (DMSO, 400 MHz): δ 2.74 (s,1H), 2.89 (s,1H), 6.81 (d, 1H), 7.22 (d, 1H), 7.39 (s, 1H), 7.49 (s, 2H), 10.67 (s, 1H). 13C NMR (CDCl3, 100 MHz): 47.47, 57.19, 91.42, 111.12, 117.20, 124.84, 126.36, 128.91, 135.51, 141.50, 153.59, 158.72, 159.75, 174.53, 177.56 ppm.
2-Amino-7,7-dimethyl-5-oxo-4-phenyl-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6a)
1H NMR (DMSO‐d6, 400 MHz): δ (s, 3H, CH3), 1.04 (s, 3H, CH3), 2.11—2.26 (, 2H, CH2), 2.49—2.55 (2H, CH2), 4.18 (s, 1H, CH), 7.00 (s, 2H, NH2), 7.16 (d, 2H), 7.06 (d, 2H), 7.18 (t, 2H), 7.29 (t, 2H). 13C NMR (DMSO‐d6, 100 MHz): 27.27, 28.87, 32.27, 36.04, 40.15, 5.44, 58.77, 113.20, 120.19, 127.62, 128.79, 145.21, 158.95, 162.95, 196.10 ppm.
Ethyl 2-amino-7,7-dimethyl-5-oxo-4-phenyl-5,6,7,8-tetrahydro-4H-chromene-3carboxylate (6b)
1H NMR (DMSO‐d6, 400 MHz): δ 1.00 (s, 3H, CH3), 1.12 (s, 3H, CH3), 1.19 (t, 3H, CH3), 2.17–2.25 (2H, CH2), 2.46 (s, 2H, CH2), 4.06 (m, 2H, CH2), 4.73 (s, 1H, CH), 6.18 (s, 2H, NH2), 7.13 (tt, 1H, CH), 7.23 (t, 1H, CH), 7.28 (m, 1H, CH). 13C NMR (DMSO‐d6, 100 MHz): 14.21, 27.42, 29.10, 32.23, 33.83, 40.67, 50.73, 59.69, 80.89, 116.84, 126.03, 127.78, 128.26, 145.78, 158.31, 161.34, 169.15, 196.41 ppm.
3-Amino-1-(p-tolyl)-1H-benzo[f]chromene-2-carbonitrile (6i)
1H NMR (DMSO‐d6, 400 MHz): δ 2.20 (s, 3H, CH3), 5.25 (s, 1H, CH), 6.95 (s, 2H, NH2), 7.07 (s, 4H, CH), 7.33 (d, 1H, CH), 7.39 -7.45 (m, 2H, CH), 7.83 (d, 1H, CH), 7.89 -7.93 (m, 2H, CH). 13C NMR (DMSO‐d6, 100 MHz): 20.97, 38.19, 58.48, 116.25, 117.23, 120.98, 124.12, 125.34, 127.35, 127.48, 128.90, 129.69, 129.86, 130.64, 131.27, 136.14, 143.28, 147.21, 160.05 ppm.
3-Amino-1-(4-methoxyphenyl)-1H-benzo[f]chromene-2-carbonitrile (6o)
1H NMR (DMSO‐d6, 400 MHz): δ 3.67 (s, 3H, CH3), 5.25 (s, 1H, CH), 6.82 (d, 2H, CH), 6.93 (s, 2H, NH2), 7.10 (d, 2H, CH), 7.33 (d, 1H, CH), 7.39 – 7.46 (m, 2H, CH), 7.85 (d, 1H, CH), 7.89–7.93 (m, 2H, CH). 13C NMR (DMSO‐d6, 100 MHz): 37.74, 55.42, 58.65, 114.50, 116.42, 117.24, 121.03, 124.15, 125.33, 127.46, 128.49, 128.90, 129.81, 130.63, 131.28, 138.37, 147.15, 158.29, 160.01 ppm.
Ethyl 2-amino-4-(4-cyanophenyl)-5-oxo-4H,5H-pyrano[3,2-c]chromene-3-carboxylate (6p)
1H NMR (DMSO‐d6, 400 MHz): δ 1.08 (t, 3H, CH3), 3.98 (q, 2H, CH2), 4.75 (s, 1H, CH), 7.45 (d, 2H, CH), 7.49 (td, 1H, CH), 7.68–7.73 (m, 3H, CH), 7.94 (s, 2H, NH2), 7.98 (dd, 1H, CH). 13C NMR (DMSO‐d6, 100 MHz): 14.59, 36.21, 59.60, 76.42, 106.03, 109.65, 113.50, 117.04, 119.32, 123.06, 125.15, 129.77, 132.43, 133.39, 150.99, 152.66, 153.97, 158.97, 160.32, 167.73 ppm.
2-Amino-4-(4-bromophenyl)-5-oxo-4H,5H -pyrano[3,2-c] chromene-3-carbonitrile (6q)
1H NMR (DMSO‐d6, 400 MHz): δ 4.48 (s, 1H, CH), 7.25 (d, 2H,), 7.45–7.49 (m, 4H, 2CH, NH2), 7.50 (d, 2H, CH), 7.72 (td, 1H, CH), 7.90 (dd, 1H, CH). 13C NMR (DMSO‐d6, 100 MHz): 36.93, 57.86, 103.88, 113.40, 117.05, 119.56, 120.69, 122.97, 125.17, 130.50, 131.82, 133.49, 143.22, 152.64, 154.02, 158.40, 160.00 ppm.
Results and discussion
As illustrated in Fig. 3, Fe3O4@g-3N4@@Thiamine was prepared by a simple synthetic method. At first, iron salts were added to g-C3N4 nanosheets resulting from the thermal polymerization of urea, the process of liquid exfoliation and ultrasonic treatment. Then, thiamine hydrochloride was added to it as a linker through cyanuric chloride and finally it was identified by several analyses such as FT-IR, EDX, elemental mapping, TGA, FE-SEM, XRD, CHNS, VSM, and BET.
One of the ways to identify the synthesized catalyst is to use FT-IR spectroscopy and follow its changes in each step (Fig. 8). The presence of a strong band at 597 cm−1 is relevant to the stretching vibration of the Fe–O bond. As a result of s-Triazine′s bending vibration, there is a strong peak at 804 cm−1. In this case, the C-N stretching vibration modes have absorption peaks at 1240–1480 cm−1. C=N stretching vibrations cause the strong band at 1660 cm−1. There are also peaks at 3000–3600 cm−1 as a result of stretching vibrations of NH and OH. The FT-IR spectrum showed that the characteristic peak of thiamine for the (C=N)+ and NH2 stretching frequency is in the ranges 1665 and 3424 cm−1, respectively. C–H stretching (symmetric and asymmetric) is also evident at about 2936 cm−1.

FT-IR spectroscopy steps of the synthetic catalyst.
The FE-SEM and TEM analyses to peruse the morphology of the catalyst surface, the uniform distribution, and the spherical shape of the particles are shown in Figs. 9 and 10. The FESEM images clearly show the layered structure with a relatively smooth surface functionalized with TCT and thiamine.

The FE-SEM images of Fe3O4@g-C3N4@Thiamine.

The TEM images of Fe3O4@g-C3N4@Thiamine.
The EDX elemental analysis reveals the attendance of O, C, N, S, Fe, and Cl elements, which confirms the functionalization of Fe3O4@g-C3N4 substrate with thiamine (Fig. 11). EDX mapping images of the synthesized nanocatalyst confirm the good dispersion of elements (Fig. 12).

EDS spectra of Fe3O4@g-C3N4@Thiamine.

Fe3O4@g-C3N4@Thiamine elemental mapping.
CHNS elemental analysis for Fe3O4@g-C3N4@Thiamine in the presence of 1 mmol cyanuric chloride and 3 mmol thiamine hydrochloride shows the percentages of (C = 31.93%), (H = 3.97%), (N = 46.80%), and (S = 0.18%). In another part of the research, in the presence of 2 mmol cyanuric chloride, and 3 mmol thiamine hydrochloride, the content of C, H, N, and S amazingly increased to (C = 37.30%), (H = 3.80%), (N = 30.25%), and (S = 3.12%) changed. According to the measured data, it can be concluded that about 33% of thiamine hydrochloride molecules are grafted on Fe3O4@g-C3N4@TCT. To clarify this issue, it can be stated that in the presence of 3 mmol thiamine hydrochloride, and 2 mmol of the linker, cyanuric chloride can react with -NH groups in addition to the NH2 group. Therefore, the ratio of reactive functional groups boosted and more thiamine hydrochloride will be connected to Fe3O4@g-C3N4@TCT with covalent bonds.
Figure 13 illustrates the XRD patterns of the individual steps in the synthesis of the Fe3O4@g C3N4@thiamine nanocatalyst. Comparing the XRD pattern of the catalyst with the Fe3O4 pattern exhibited that all the characteristic peaks of the Fe3O4 nanoparticle, 2θ = 30.17°, 35.57°, 43.18°, 53.82°, 57.21° and 62.90°, correspond to (220), (311), (400), (422), (511), and (440). g-C3N4 also has a strong peak at 28.6°, which corresponds to (002).

The XRD patterns of the individual steps in the synthesis of the Fe3O4@gC3N4@thiamine.
The amount of surface H2O absorbed in the nanocatalyst is lost below 150°CC. At 150–400 °C, the Fe3O4@g-C3N4@Thiamine curve shows an apparent weight loss of 41%, indicating that the sheet contains organic components (Thiamine bound to cyanuric chloride). In the final stage, the Fe3O4@g-C3N4 starts to decompose at temperatures > 450 °C (Fig. 14).

The TGA curve of A: Fe3O4@g-C3N4, and B: Fe3O4@gC3N4@Thiamine.
By utilizing VSM analysis, an evaluation of the magnetic properties of Fe3O4 and Fe3O4@g-C3N4@thiamine nanocatalyst was done. The value of 76.81 and 22.28 emug−1 was found respectively (Fig. 15). The decrease in the magnetization strength of Fe3O4@g-C3N4@Thiamine compared to Fe3O4 is owing to the presence of non-magnetic g-C3N4 sheets and the loading of ligands. The synthetic nanocatalyst can be well separated by an external magnetic field, which indicates that even with g-C3N4 coating by cyanuric chloride and thiamine, the magnetic property is still visible and preserved.

Magnetization curves of Fe3O4@gC3N4@Thiamine nanocatalyst.
For the purpose of calculating the specific surface area (SBET), the BET method was utilized. The results show that Fe3O4@g-C3N4 and Fe3O4@g-C3N4@Thiamine have specific surfaces (70 m2/g) and (46 m2/g), respectively. It is evident, that the specific surface area decreases with the functionalization of Fe3O4@g-C3N4 by cyanuric chloride (TCT) followed by Thiamine, which confirms the success of the modifications on the surface of Fe3O4@g-C3N4. In addition, pore size and distribution analysis confirmed the presence of micropores in g-C3N4 and Fe3O4@g-C3N4 @Thiamine surfaces (Fig. 16, and Table 2, 3).

The t-plot and BET diagrams of Fe3O4@g-C3N4, and Fe3O4@g-C3N4@Thiamin.
To evaluate the activity of Fe3O4@gC3N4@Thiamine nanocatalyst, the three-component reaction of, 1,3-dicarbonyl compounds, isatin derivatives, and malononitrile was applied for the synthesis of spirooxindole-pyrans.
To achieve this goal, we selected the reaction of 1 mmol malononitrile, 1 mmol barbituric acid, and 1 mmol isatin as a sample reaction. Then we evaluated multiple factors examples include temperature, the amount of catalyst, and, solvent. At the beginning of the process, the model reaction in the vicinity of Fe3O4@g-C3N4@Thiamine was examined in various solvents, such as toluene, CH2Cl2, THF, MeOH, CH3CL, H2O, MeCN, EtOH, and solvent-free conditions. It turned out that the solvent-free conditions produced the most profitable results (98% yield). (Table 4, entries 1–9). On the other hand, by increasing the temperature up to 70 °C, there was no noticeable difference in the product's yield (Table 4, entries 9–11). Furthermore, by increasing the amount of Fe3O4@gC3N4@Thiamine from 0.01 to 0.015 g, the yield grew from 85 to 98%, but further increase did not affect the reaction process (Table 4, entry 9 and, entries 12–15).
To increase the range of the reaction according to the optimal reaction conditions, we used the Fe3O4@g-C3N4@Thiamine in the multi-component reaction for the preparation of spirooxindoles-pyran derivatives. The consequences show that both electron-donating and electron-withdrawing groups have excellent efficiency in producing favorable products in a brief period of time (Table 5).
Fe3O4@g-C3N4@Thiamine begins and continues the synthesis of spirooxindole-pyrans derivatives by activating carbonyl groups. First, in the presence of a catalyst, compound A is obtained by conducting knoevenagel condensation between malonitrile and isatin. Then by attacking: 1,3‐dicarbonyl compounds activated by the catalyst, intermediate B is created. Finally, the desired products are synthesized by enolization and intramolecular cyclization followed by tautomerization (Fig. 17)52.

A proposed mechanism for synthesizing compounds 4a-q.
Also, the performance of Fe3O4@g-C3N4@Thiamine nanocatalyst in the condensation reaction of three components including aldehyde derivatives, different OH-acids (4-hydroxycoumarine, β-naphthol, and dimedone), and active methylenes (malononitrile and ethyl cyanoacetate) was investigated. For this purpose, the reaction of 4-chlorobenzaldehyde, malononitrile, and dimedone in the presence of a catalyst was proposed as a model reaction to bring up the optimal conditions. In the beginning, the sample reaction was experimented the different solvents, such as Et2O, n-hexane, THF, MeCN, CH3Cl, MeOH, H2O, EtOH, and solvent-free conditions (Table 6, entries 1–9). The best performance of the catalyst was related to the solvent-free conditions; therefore, the continuation of the reaction was examined in these conditions boosting the temperature from 25 to 80 °C did not have efficacy on the reaction time. For that reason, the temperature of 25 °C was selected as the optimal temperature (Table 6, entries 9–11). Increasing the loading of Fe3O4@gC3N4@thiamine from 15 to 20 mg resulted in a reaction efficiency of 98%, so the amount was chosen as the optimal amount (Table 6, entries 9, and 12–15).
Next, to show the efficiency of the nanocatalyst, various aldehydes were reacted with OH-acids and active methylenes under ideal conditions. The summarized results, presented in Table 7, indicate both electron-donating and electron-withdrawing aldehydes produced the related products in excellent yields.
The proposed synthesis mechanism for 2-amino-4H-chromene derivatives is illustrated in Fig. 18. It seems that in the presence of Fe3O4@g-C3N4@Thiamine catalyst the first condensation of malononitrile, and aldehyde leads to the formation of arylidene malononitrile. The next step, in the presence of a catalyst, is done by adding OH-acid compounds to intermediate I, and species II is formed. In the last step, species I is enolized to intermediate III and the purpose products are formed by intramolecular cyclization 58.

A proposed mechanism for synthesizing compounds 6a-w.
To gain a deeper understanding of the importance and effectiveness of the work, it is useful to compare the synthetic nanocatalyst with prior catalysts (Table 8). To reach this goal, we used model reactions specific to both syntheses. This study highlights the significant potential of using this efficient catalyst for synthesizing target compounds. The catalyst proves advantageous because it promotes short reaction times, operates at suitable temperatures, and maintains high-efficiency levels.
Due to the economic detriments as well as the environmental pollution of chemicals, the capability to easily separate them from the environment can be one of the advantages of catalysts. On the other hand, it has been proven that heterogeneous catalysts perform better than homogeneous catalysts in many applications. Among heterogeneous catalysts, magnetic nanostructures are more efficient due to their facile separation by external magnetic fields. To achieve this objective, Fe3O4@g-C3N4@Thiamine was offered as a heterogeneous magnetic nanocatalyst, and in addition to investigating its catalytic capability, its efficiency after recycling was also perused. After the reaction is completed (model reaction), magnetic separation of the catalyst from the reaction mixture was performed, it was washed with EtOH and dried, then it was put again in another fresh reaction mixture. The recycled catalyst shows stable catalytic activity and retains its potential for reuse cycles without significant degradation for up to seven cycles (Fig. 19). The XRD patterns and FT-IR spectrum after seven cycles (Fig. 20) demonstrated that Fe3O4@g-C3N4@Thiamine maintains its structure, properties, and activity throughout the reaction.

Investigating the reusability of Fe3O4@g-C3N4@Thiamine.

XRD patterns and FT-IR spectrum (A fresh nanocatalyst, and B recycled catalyst) after seven cycles.
Conclusion
Chemical modification of g-C3N4 two-dimensional magnetic nanosheet by stabilizing a safe ligand, as a recyclable green catalyst, can increase the solubility and stability of magnetic graphene nitride. For this purpose, a novel and simple synthetic strategy was introduced to modify g-C3N4 through covalent bonding between thiamine and cyanuric chloride as a surface-attached linker. The chemical nature and properties of the catalyst were determined by a wide range of analyses. On the other hand, due to the significance of synthesizing heterocyclic compounds and their applications in human life, we used this biocompatible catalyst in the synthesis of spirooxindole-pyrans, which evidence indicate its excellent performance in mild and completely green conditions. The above evidence demonstrates that the high catalytic activity of Fe3O4@g-C3N4@Thiamine may be a result of the synergistic effect of g-C3N4 and Thiamine. Also, owing to the magnetic nature of the Fe3O4@g-C3N4@Thiamine, segregating it from the reaction medium is simple and it was used in seven consecutive cycles without noticeable reduction. In addition, the ability of the sorbent to be used for MSPE application was also studied. In this study, the magnetic sorbent was synthesized and used for microextraction of selected pesticides in tomato, cucumber, and carrot samples by the MSPE-HPLC–UV method. After determining the optimal extraction condition, MSPE-HPLC was used to quantify the target analyte. The desired sorbent had good features such as simplicity, cheapness, portability, high precision, wide linear range, good reproducibility, and low detection limit. Ultimately, the method is employed to detect the trace pollutant in real samples.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Peñas-Sanjuán, A., Anderson, J. A., López-Garzón, R., Pérez-Mendoza, M. & Melguizo, M. Carbon-supported statistically distributed polyethyleneimine/palladium (II) complexes as efficient and sustainable Sonogashira catalysts. React. Funct. Polym. 190, 105643 (2023).
Azizi-Amiri, M. et al. Efficient catalytic synthesis of xanthenes with copper immobilized on amine-modified NaY. Chem. Methodol. 8, 1–22. https://doi.org/10.48309/chemm.2024.424058.1737 (2023).
Climent, M. J., Corma, A. & Iborra, S. Homogeneous and heterogeneous catalysts for multicomponent reactions. RSC Adv. 2, 16–58 (2012).
Jiang, B., Rajale, T., Wever, W., Tu, S. J. & Li, G. Multicomponent reactions for the synthesis of heterocycles. Chem. Asian J. 5, 2318–2335 (2010).
Peiman, S., Maleki, B. & Ghani, M. Fe3O4@SiO2@Mel-Rh-Cu: A high-performance, green catalyst for efficient xanthene synthesis and its application for magnetic solid phase extraction of diazinon followed by its determination through HPLC-UV. Chem. Methodol. 8, 257–278. https://doi.org/10.48309/chemm.2024.442693.1767 (2024).
Farajpour, M., Vahdat, S. M., Baghbanian, S. M. & Hatami, M. Ag-SiO2 nanoparticles: Benign, expedient, and facile nano catalyst in synthesis of decahydroacridines. Chem. Methodol. 7, 540–551. https://doi.org/10.22034/chemm.2023.386678.1653 (2023).
Olveira, S., Forster, S. P. & Seeger, S. Nanocatalysis: academic discipline and industrial realities. J. Nanotechnol. 2014, 324089 (2014).
Zhu, Q.-L. & Xu, Q. Immobilization of ultrafine metal nanoparticles to high-surface-area materials and their catalytic applications. Chem 1, 220–245 (2016).
Hakimi, F., Sharifi-Zarchi, A. & Golrasan, E. Bifunctional polyethylene glycol/ethylenediamine nanomagnetic phase-transfer catalyst: Preparation, characterization, and application in knoevenagel condensation. Chem. Methodol. 7, 489–498. https://doi.org/10.22034/chemm.2023.392041.1667 (2023).
Chaturvedi, S., Dave, P. N. & Shah, N. K. Applications of nano-catalyst in new era. J. Saudi Chem. Soc. 16, 307–325 (2012).
Singh, S. B. & Tandon, P. K. Catalysis: A brief review on nano-catalyst. J. Energy Chem. Eng 2, 106–115 (2014).
Naderi, S., Sandaroos, R., Peiman, S. & Maleki, B. Synthesis and characterization of a novel crowned schiff base ligand linked to ionic liquid and application of its Mn(III) complex in the epoxidation of olefins. Chem. Methodol. 7, 392–404. https://doi.org/10.22034/chemm.2023.385248.1651 (2023).
Wang, T., Song, B. & Wang, L. A new filler for epoxy resin: Study on the properties of graphite carbon nitride (g-c3n4) reinforced epoxy resin composites. Polymers 12, 76 (2020).
Chidhambaram, N. & Ravichandran, K. Single step transformation of urea into metal-free g-C3N4 nanoflakes for visible light photocatalytic applications. Materials Letters 207, 44–48 (2017).
Chen, X. et al. Superior antibacterial activity of sulfur-doped g-C3N4 nanosheets dispersed by Tetrastigma hemsleyanum Diels & Gilg’s polysaccharides-3 solution. Int. J. Biol. Macromol. 168, 453–463 (2021).
Zhu, B., Xia, P., Ho, W. & Yu, J. Isoelectric point and adsorption activity of porous g-C3N4. Appl. Surface Sci. 344, 188–195 (2015).
Ghafuri, H., Tajik, Z., Ghanbari, N. & Hanifehnejad, P. Preparation and characterization of graphitic carbon nitride-supported l-arginine as a highly efficient and recyclable catalyst for the one-pot synthesis of condensation reactions. Sci. Rep. 11, 19792 (2021).
Shao, Y. et al. Piezocatalytic performance difference of graphitic carbon nitride (g-C3N4) derived from different precursors. Chem. Phys. Lett. 801, 139748 (2022).
Fidan, T. et al. Functionalized graphitic carbon nitrides for environmental and sensing applications. Adv. Energy Sustain. Res. 2, 2000073 (2021).
Peiman, S., Baharfar, R. & Maleki, B. Immobilization of trypsin onto polyamidoamine dendrimer functionalized iron oxide nanoparticles and its catalytic behavior towards spirooxindole-pyran derivatives in aqueous media. Mater. Today Commun. 26, 101759 (2021).
Maleki, B., Jamshidi, A., Peiman, S. & Housaindokht, M. R. Tri-vanadium substituted dawson-type heteropolytungstate nanocomposite (g-C3N4/Fe3O4@ P2W15V3) as a novel, green, and recyclable nanomagnetic catalyst in the synthesis of tetrahydrobenzo [b] pyrans. Polycycl. Arom. Compd. 44, 994–1010 (2023).
Mhaibes, R. M., Arzehgar, Z., Heydari, M. M. & Fatolahi, L. ZnO nanoparticles: A highly efficient and recyclable catalyst for tandem Knoevenagel-Michael-cyclocondensation reaction. Asian J. Green Chem. 7, 1–8 (2023).
Hsu, C.-Y. et al. Nano titanium oxide (nano-TiO2): A review of synthesis methods, properties, and applications. Case Stud. Chem. Environ. Eng. 100626 (2024).
Fazeli-Nasab, B., Shahraki-Mojahed, L., Beigomi, Z., Beigomi, M. & Pahlavan, A. Rapid detection methods of pesticides residues in vegetable foods. Chem. Methodol. 6, 24–40 (2022).
Suo, F. et al. Mesoporous activated carbon from starch for superior rapid pesticides removal. Int. J. Biol. Macromol. 121, 806–813 (2019).
El-Naggar, M. E. et al. Hydroxyethyl cellulose/bacterial cellulose cryogel dopped silver@ titanium oxide nanoparticles: Antimicrobial activity and controlled release of Tebuconazole fungicide. Int. J. Biol. Macromol. 165, 1010–1021 (2020).
Farhadi, B., Ebrahimi, M. & Morsali, A. Microextraction and determination trace amount of propranolol in aqueous and pharmaceutical samples with oxidized multiwalled carbon nanotubes. Chem. Methodol. 5, 227–233 (2021).
Sahebnasagh, S., Fadaee-Kakhki, J., Ebrahimi, M., Bozorgmehr, M. & Abedi, M. Pre-concentration and determination of fluoxetine in hospital wastewater and human hair samples using solid-phase µ-extraction by Silver nanoparticles followed by spectro-fluorimetric. Chem. Methodol. 5, 211–218 (2021).
Farhadi, B., Ebrahimi, M. & Morsali, A. Pre-concentration and sensitive determination of propranolol and metoprolol using dispersive solid-phase microextraction and high-performance liquid chromatography in biological, wastewater, and pharmaceutical samples. Chem. Methodol. 6, 750–761 (2022).
Sargazi-Karbasaki, S. et al. Fabrication of sulfamic acid functionalized magnetic nanoparticles with denderimeric linkers and its application for microextraction purposes, one-pot preparation of pyrans pigments and removal of malachite green. J. Taiwan Inst. Chem. Eng. 118, 342–354. https://doi.org/10.1016/j.jtice.2020.12.025 (2021).
Palasak, J. et al. Preconcentration of triazole fungicides using effervescent assisted switchable hydrophilicity solvent-based microextraction prior to high-performance liquid chromatographic analysis. Microchem. J. 182, 107882 (2022).
Kachangoon, R., Vichapong, J., Santaladchaiyakit, Y. & Srijaranai, S. Green fabrication of Moringa oleifera seed as efficient biosorbent for selective enrichment of triazole fungicides in environmental water, honey and fruit juice samples. Microchem. J. 175, 107194 (2022).
Han, X., Chen, J., Li, Z., Quan, K. & Qiu, H. Magnetic solid-phase extraction of triazole fungicides based on magnetic porous carbon prepared by combustion combined with solvothermal method. Anal. Chimica Acta 1129, 85–97 (2020).
Wang, Y., He, M., Chen, B. & Hu, B. Hydroxyl-containing porous organic framework coated stir bar sorption extraction combined with high performance liquid chromatography-diode array detector for analysis of triazole fungicides in grape and cabbage samples. J. Chromatograph. A 1633, 461628 (2020).
Boroumand, H., Alinezhad, H., Maleki, B. & Peiman, S. Triethylenetetramine-grafted magnetic graphene oxide (Fe3O4@ GO-NH2) as a reusable heterogeneous catalyst for the one-pot synthesis of 2-amino-4 H-benzopyran derivatives. Poly. Aromat. Compd. 43, 7853–7869 (2023).
Naderi, S., Sandaroos, R., Peiman, S. & Maleki, B. Novel crowned cobalt (II) complex containing an ionic liquid: A green and efficient catalyst for the one-pot synthesis of chromene and xanthene derivatives starting from benzylic alcohols. J. Phys. Chem. Solids 180, 111459 (2023).
Maleki, B., Sandaroos, R., Naderi, S. & Peiman, S. A crowned manganese-based Schiff complex supported on nanocellulose as an efficient and sustainable heterogeneous catalyst for the oxidation of benzyl alcohols. J. Organomet. Chem. 990, 122666 (2023).
Darvishy, S., Alinezhad, H., Vafaeezadeh, M., Peiman, S. & Maleki, B. S-(+) Camphorsulfonic acid glycine (csag) as surfactant-likes brønsted acidic ionic liquid for one-pot synthesis of ß-amino carbonyl. Polycycl. Arom. Comps. 1–13 (2022).
Peiman, S., Baharfar, R. & Hosseinzadeh, R. CuI NPs immobilized on a ternary hybrid system of magnetic nanosilica, PAMAM dendrimer and trypsin, as an efficient catalyst for A3-coupling reaction. Res. Chem. Intermed. 48, 1365–1382 (2022).
Baharfar, R., Peiman, S. & Maleki, B. Fe3O4@ SiO2@ D-NHCS-Tr as an efficient and reusable catalyst for the synthesis of indol-3-yl-4 H-chromene via a multi-component reaction under solvent-free conditions. J. Heterocycl. Chem. 58, 1302–1310 (2021).
Yang, J. et al. Synthesis of Fe3O4/g-C3N4 nanocomposites and their application in the photodegradation of 2,4,6-trichlorophenol under visible light. Mater. Lett. 164, 183–189 (2016).
Mohammadi, P., Heravi, M. M. & Sadjadi, S. Green synthesis of Ag NPs on magnetic polyallylamine decorated g-C3N4 by Heracleum persicum extract: Efficient catalyst for reduction of dyes. Sci. Rep. 10, 6579 (2020).
Zhang, M. et al. Catalyst-free, visible-light promoted one-pot synthesis of spirooxindole-pyran derivatives in aqueous ethyl lactate. ACS Sustain. Chem. Eng. 5, 6175–6182 (2017).
Maghsoodlou, M. T., Heydari, R., Mohamadpour, F. & Lashkari, M. Fe2O3 as an environmentally benign natural catalyst for one-pot and solvent-free synthesis of spiro-4H-pyran derivatives. Iran. J. Chem. Chem. Eng. 36, 31–38 (2017).
Guo, R.-Y. et al. Meglumine: A novel and efficient catalyst for one-pot, three-component combinatorial synthesis of functionalized 2-amino-4 H-pyrans. ACS Comb. Sci. 15, 557–563 (2013).
Umar-Basha, K. N. & Gnanamani, S. An efficient and easy method for the one-pot synthesis of spirooxindoles in the presence of Na2CO3. J. Heterocycl. Chem. 56, 2008–2016 (2019).
Nagaraju, S., Paplal, B., Sathish, K., Giri, S. & Kashinath, D. Synthesis of functionalized chromene and spirochromenes using l-proline-melamine as highly efficient and recyclable homogeneous catalyst at room temperature. Tetrahed. Lett. 58, 4200–4204 (2017).
Goli-Jolodar, O., Shirini, F. & Seddighi, M. Introduction of a novel basic ionic liquid containing dual basic functional groups for the efficient synthesis of spiro-4H-pyrans. J. Mol. Liquids 224, 1092–1101 (2016).
Li, Y., Chen, H., Shi, C., Shi, D. & Ji, S. Efficient one-pot synthesis of spirooxindole derivatives catalyzed by L-proline in aqueous medium. J. Comb. Chem. 12, 231–237 (2010).
Chandam, D. R., Mulik, A. G., Patil, D. R. & Deshmukh, M. B. Oxalic acid dihydrate: proline as a new recyclable designer solvent: A sustainable, green avenue for the synthesis of spirooxindole. Res. Chem. Intermed. 42, 1411–1423 (2016).
Mohamadpour, F., Maghsoodlou, M. T., Lashkari, M., Heydari, R. & Hazeri, N. Synthesis of quinolines, spiro [4 H-pyran-oxindoles] and xanthenes under solvent-free conditions. Org. Preparat. Proced. Int. 51, 456–476 (2019).
Tufail, F. et al. A practical green approach to diversified spirochromene/spiropyran scaffolds via a glucose–water synergy driven organocatalytic system. New J. Chem. 42, 17279–17290 (2018).
Hanim, S. A. M., Malek, N. A. N. N. & Ibrahim, Z. Analyses of surface area, porosity, silver release and antibacterial activity of amine-functionalized, silver-exchanged zeolite NaY. Vacuum 143, 344–347 (2017).
Mohammadi, A. A., Asghariganjeh, M. R. & Hadadzahmatkesh, A. Synthesis of tetrahydrobenzo [b] pyran under catalysis of NH4Al (SO4) 2· 12H2O (Alum). Arab. J. Chem. 10, S2213–S2216 (2017).
Lu, J., Fu, X.-W., Zhang, G. & Wang, C. β-Cyclodextrin as an efficient catalyst for the one-pot synthesis of tetrahydrobenzo [b] pyran derivatives in water. Res. Chem. Intermed. 42, 417–424 (2016).
Chaskar, A. Catalyst free one-pot three-component synthesis of 2-Amino-4Hchromene derivatives in aqueous deep eutectic mixture at room temperature. Lett. Org. Chem. 11, 480–486 (2014).
Hu, H., Qiu, F., Ying, A., Yang, J. & Meng, H. An environmentally benign protocol for aqueous synthesis of tetrahydrobenzo [b] pyrans catalyzed by cost-effective ionic liquid. Int. J. Mol. Sci. 15, 6897–6909 (2014).
Kalhor, M., Bigdeli, M., Mirshokraie, S. A. & Moghanian, H. ZnO@ zeolite-Y mesoporous as an eco-friendly and efficient nanocatalyst in the three-component synthesis of 2-amino-benzochromenes. Org. Chem. Res. 6, 219–232 (2020).
Maleki, B., Tayebee, R., Khoshsima, A. & Ahmadpoor, F. Facile protocol for the synthesis of 2-amino-4H-chromene derivatives using choline chloride/urea. Org. Prep. Proced. Int. 53, 34–41 (2020).
Javadi, S. & Habibi, D. The new sulfonic-acid-based Ce-Mn-Layered Doble Hydroxide as a capable nano-catalyst for the green-synthesis of spiro [indoline-pyran] ones and spiro [acenaphthylene-pyran] ones. Res. Chem. Intermed. 49, 2005–2024 (2023).
Javanshir, S., Saghiran-Pourshiri, N., Dolatkhah, Z. & Farhadnia, M. Caspian Isinglass, a versatile and sustainable biocatalyst for domino synthesis of spirooxindoles and spiroacenaphthylenes in water. Monatshefte für Chemie-Chem. Month. 148, 703–710 (2017).
Kamali, F. & Shirini, F. An efficient one-pot multi-component synthesis of spirooxindoles using Fe3O4/g-C3N4 nanocomposite as a green and reusable catalyst in aqueous media. J. Mol. Struct. 1227, 129654 (2021).
Singh, P. P. Supported ionic liquids and their applications in organic transformations. Curr. Org. Synth. 19, 905–922 (2022).
Mobinikhaledi, A., Foroughifar, N. & Fard, M. A. B. Simple and efficient method for three-component synthesis of spirooxindoles in aqueous and solvent-free media. Synth. Commun. 41, 441–450 (2011).
Molla, A. & Hussain, S. Base free synthesis of iron oxide supported on boron nitride for the construction of highly functionalized pyrans and spirooxindoles. RSC Adv. 6, 5491–5502 (2016).
Hojati, S. F., Amiri, A. & Mahamed, M. Polystyrene@ graphene oxide-Fe3O4 as a novel and magnetically recyclable nanocatalyst for the efficient multi-component synthesis of spiro indene derivatives. Res. Chem. Intermed. 46, 1091–1107 (2020).
Taib, L. A., Keshavarz, M. & Parhami, A. Click approach to the novel 1, 2, 3-triazolium phosphotungstate organic–inorganic hybrids for the highly promoted synthesis of spirooxindoles. Res. Chem. Intermed. 1–21 (2022).
Azath, I. A., Puthiaraj, P. & Pitchumani, K. One-pot multicomponent solvent-free synthesis of 2-amino-4 H-benzo [b] pyrans catalyzed by per-6-amino-β-cyclodextrin. ACS Sustain. Chem. Eng. 1, 174–179 (2013).
Sayyahi, S., Fallah-Mehrjardi, M. & Saghanezhad, S. J. Synthesis of heterocyclic compounds by catalysts supported on nano-magnetite (Fe3O4)-an update. Mini-Rev. Org Chem. 18, 11–26 (2021).
Hosseini-Mohtasham, N. & Gholizadeh, M. Horsetail plant (Equisetum arvense) and horsetail plant ash: Application and comparison of their catalytic activities as novel and natural porous lewis acid catalysts for the one-pot green synthesis of 2-amino-4 H-chromene derivatives under solvent-free conditions. J. Iran. Chem. Soc. 17, 397–409 (2020).
Maleki, A., Jafari, A. A. & Yousefi, S. Green cellulose-based nanocomposite catalyst: Design and facile performance in aqueous synthesis of pyranopyrimidines and pyrazolopyranopyrimidines. Carbohyd. Polym. 175, 409–416 (2017).
Aghaei-Hashjin, M., Yahyazadeh, A. & Abbaspour-Gilandeh, E. Zr@ IL-Fe3O4 MNPs as an efficient and green heterogeneous magnetic nanocatalyst for the one-pot three-component synthesis of highly substituted pyran derivatives under solvent-free conditions. RSC Adv. 11, 23491–23505 (2021).
Acknowledgements
A research grant from the University of Mazandaran (number 33/38554) supported this study.
Author information
Authors and Affiliations
Contributions
Writing: S.P.; B.M.; M.Gh Conceptualization: S.P.; B.M.; M. Gh. Data curation: S.P.; B.M.; M.Gh. Formal analysis: S.P.; B.M.; M.Gh. Project administration: S.P.; B.M.; M.Gh. Methodology: S.P.; B.M.; M.Gh. Validation: S.P.; B.M.; M.Gh. Review and editing: S.P.; B.M.; M.Gh. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This work does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Peiman, S., Maleki, B. & Ghani, M. Fe3O4@gC3N4@Thiamine: a novel heterogeneous catalyst for the synthesis of heterocyclic compounds and microextraction of tebuconazole in food samples. Sci Rep 14, 21488 (2024). https://doi.org/10.1038/s41598-024-72212-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-72212-6
