
Abstract
This study explores a potential solution to the shortage of kidneys for transplantation in end-stage renal disease (ESRD). Currently, kidney transplantation stands as the optimal option, yet the scarcity of organs persists. Employing tissue engineering, researchers sought to assess the feasibility of generating kidneys for transplantation. Pig kidneys were utilized since they possess higher similarities to human kidneys. Cells were removed via decellularization, which maintains the organ’s microarchitecture. Subsequently, pig kidney cells and human red blood cells were perfused into the vacant kidney structure to reconstitute it. The methodologies employed showed promising results, suggesting a viable approach to increase the recellularization rate in whole pig kidneys. This proof-of-concept establishes a groundwork for potentially extending this technology to human kidneys, tackling the organ shortage, thus positively enhancing outcomes for ESRD patients by increasing the availability of transplantable organs.
Similar content being viewed by others
Introduction
End-stage renal disease (ESRD) is a condition in which the kidneys lose their ability to filter toxins and excess fluids from the bloodstream, leading to a dangerous accumulation of waste products and electrolytes in the body. This not only diminishes the patient’s quality of life but also results in premature mortality1. By 2021, the prevalence of ESRD in Canada had reached 48,000 cases, marking a 24% increase over the past decade. This surge in cases imposes a considerable annual healthcare burden of 40 billion dollars on the system2,3,4. Moreover, the prevalence of ESRD is expected to rise further due to its strong association with chronic diseases like diabetes mellitus and hypertension, both prevalent in the aging Canadian population5,6,7. Currently, there exist two primary treatments for ESRD: dialysis and kidney transplant4. Although a kidney transplant is considered the optimal treatment, most patients initially undergo dialysis. The demand for chronic dialysis has nearly doubled over the past two decades, costing the Canadian healthcare system over $100,000 per patient annually3,8,9. Kidney transplant remains the most cost-effective and survival-enhancing option, with a 41% increase in transplant cases in the last decade10. However, the shortage of available organs is a critical issue, with 77% of individuals on the organ waitlist in Canada awaiting a kidney and an average wait time of 3 years and 10 months4,10,11. In response to this organ shortage problem, tissue engineering emerges as a potential solution. Tissue engineering combines engineering principles with cell biology to develop biological substitutes capable of restoring or enhancing tissue and organ function. One technique in tissue engineering is decellularization, which removes cellular components from tissues or organs while preserving the extracellular matrix (ECM). The ECM provides structural support, native vascular networks, and bioactive molecules, thereby enhancing cell recellularization and differentiation12,13. Recellularization involves reseeding the decellularized organ with cells. While this process can be performed via perfusion through the renal artery, renal vein, or ureter, challenges such as oxygenating cells, recirculating media to preserve nutrients, and ensuring cell viability and migration persist. Current methods exhibit a low reseeding success rate and difficulty forming functional renal structures14,15,16,17,18.
This study aims to enhance kidney recellularization methods using primary porcine renal cells as a proof of principle. The research demonstrates that perfusion-based decellularization and recellularization can achieve a high cell density in the ECM, low cytotoxicity levels, and the presence of kidney cell markers throughout the kidney. Further optimization needs to be done, and a higher number of markers are required to identify all the cells inside the kidney. Nevertheless, this research could pave the way for similar techniques in human kidneys, offering hope for regenerative medicine in ESRD treatment and improving organ availability for transplantation.
Materials and methods
The reporting of this study meets the ARRIVE essential 10 criteria.
Experimental animals and tissue collection
Our team operates under an organ retrieval animal protocol (A22-0119) sanctioned by the Animal Care Committee of the University of British Columbia (UBC), confirming that all experiments were performed in strict accordance with guidelines and regulations established by the Institutional Animal Care and Use Committee (IACUC) of the University of British Columbia. This protocol ensures compliance with ethical standards and animal welfare principles while facilitating collaboration with existing research endeavours.
All kidneys utilized in this study were sourced from adult female Yorkshire pigs (n = 19). No animals were specifically procured for sacrifice; instead, the organs were retrieved from pigs involved in other ongoing research protocols upon the conclusion of their respective experiments. Prior to kidney retrieval, pigs were anesthetized by injecting Ketamine S (20 mg/kg i.m.), infusing 500 ml of lactated Ringer’s solution (1–4 ml/kg/h to prevent hypovolemia, followed by propofol (20–30 mg i.v. in increments). Pigs are then intubated with an endotracheal tube (5.5–6 mm internal diameter). Maintenance of anesthesia is performed by morphine (20 mg i.v.) and propagated by an infusion of propofol (6–8 ml/kg/h). For euthanasia, we inject morphine (1 mg/kg i.v.) followed 10 min later by propofol (10 mg/kg i.v), rocuronium (1 mg/kg i.v.) and subsequent injection of potassium chloride (40mmol) unless otherwise specified by approved animal research protocols. Before euthanizing the pigs, 50 ml of 10% 1000 USP units/mL sodium heparin (C504036 Fresenius Kabi) was perfused in bolus via IV. Both the renal artery and the renal vein were clamped before removing one kidney, leaving the blood vessels intact for cannulation. The other kidney was removed with blood vessels because it was used for tissue dissociation and cell retrieval. The fresh kidneys were transported in PBS and ice to the biosafety cabinet SterilGard III (GMI).
Decellularization
Following tissue collection, the kidneys underwent a thorough cleaning process. They were immersed three times for 3 s each in 300 mL of sterile ddH2O containing 0.1% hydrogen peroxide (product code 033323 from Fisher Scientific). Subsequently, the organs were rinsed with sterile PBS. To facilitate decellularization, catheterization of the renal vein and artery was performed using a sterile angiocath connected to an intravenous (IV) line (BAX2C8401 from Baxter). Male-to-male luer lock adapters were inserted at both ends of the IV line, and 2 − 0 silk sutures (J&J-685G Ethicon) were used to secure the tubes in place.
Decellularization was achieved through perfusion cycles that alternated between the renal artery and renal vein using a sterilized in vitro bioreactor treated with ethylene oxide. Male Luer lock adapters were connected to the bioreactor valves, which, in turn, were linked to a programmable peristaltic pump (BS-900 from Braintree Scientific Inc.) through a 3/16” ID hose. Seven different methods were explored to optimize kidney decellularization, and the details of each method and their respective results can be found in the supplemental material (Appendix 1).
The optimized decellularization protocol (method g) involved the following steps:
-
1.
Perfusion of sterile ddH2O at 10 ml/min for 30 min, followed by an increase to 25 ml/min for another 30 min.
-
2.
Two cycles of detergents: first, 1% Triton 100X (M143 from VWR) at 25 ml/min for 120 min and then at 10 ml/min overnight for 840 min.
-
3.
Perfusing 0.1% SDS (BP1311-1 from Fisher Bioreagents) at 25 ml/min for 360 min, totalling 45 h of decellularization. This process took place in a bioreactor with dimensions of 30 cm in height and 25.4 cm in diameter.
-
4.
Conditioning the matrix by perfusing sterile ddH2O at 10 ml/min for one week.
-
5.
A final wash with 1,800 mL of sterile water containing 5% antibiotics and antimycotics 100X (A/A) (#15240062 Gibco, NY USA).
Decellularized organs were preserved while awaiting reseeding, using HBSS (product #14025092, Gibco) buffer with 1% A/A (Gibco) at 4 °C.
To validate the decellularization process, five sections from specific locations of the decellularized kidney and five sections from the same locations of a native kidney were analyzed to quantify the remaining DNA. This analysis was conducted using the DNeasy Blood & Tissue Kit (product #69504, Qiagen, CA, USA), processing 20 mg of tissue and eluting in 80 µL of elution buffer.
Histology
Sections from the superior pole, inferior pole and close to the renal hilum were extracted in both the cortex and medulla regions to perform the histological analysis of the decellularized and recellularized kidneys. The tissue samples were fixed for 24 h in 10% neutral buffered formalin (Scigen CA, USA). Afterwards, they were dehydrated in 70% ethanol and embedded in paraffin. Sections of 5 μm of thickness were mounted on slides deparaffinized in the oven at 60 C for one hour and rehydrated in washes of xylene and ethanol to later stain with hematoxylin and eosin (H&E). The slides were examined using SCN400 Slide Scanner (Leica Biosystems, IL, USA) and analyzed with Aperio Digital Whole Slide Scanner (Leica Biosystems).
Immunohistochemistry
The samples of the recellularized organs were deparaffinized, rehydrated, antigen retrieved, permeabilized and blocked. Afterwards, the slides were incubated with 3% hydrogen peroxide (Sigma-Aldrich, MO, USA) for 10 min and washed with PBS. Followed by the incubation with Ki-67 rabbit monoclonal antibody (RM-9106, Thermo Scientific, MA, USA) or with Anti-Cleaved Capsase-3 (ab2302, Abcam, Ma, USA) for 90 min at room temperature. The samples were washed with PBS and incubated with a Vectastain ELITE ABC-Peroxidase kit (Vector Laboratories, CA, USA) for 30 min and washed to incubate later with DAB for 5 min. Finally, the slides were washed with ddH2O and dehydrated by immersion in ethanol and xylene. The staining intensity and distribution of positive cells were calculated by analyzing five sections of the recellularized kidney, using the image analysis software QuPath v0.4.3 Windows19 to quantify cellular proliferation and apoptosis within the recellularized tissue. The Movat Pentachrome Stain Kit (Scytek Laboratories, UT, USA) was used with deparaffinized sections of the recellularized kidney according to the manufacturer’s protocol to see the co-localization of the ECM components with the cells.
Proteomics
Samples of decellularized and fresh native kidneys were processed for protein extraction. Both groups were minced rigorously and 200 mg of tissue with 200uL of Ripa (R0278 Sigma-Aldrich, MO, USA) and cOmplete™ Protease Inhibitor Cocktail (Sigma-Aldrich) were homogenized in Tissue Grinding tubes with ceramic beads (P000922-LYSK0-A.0, Bertin Instruments, Montignyle-Bretonneux, France) using the Precellys® homogenizer (Bertin Instruments). Afterwards, the supernatant was sonicated (FB705, Fisher Scientific) for milliseconds at 50 of amplitude and centrifuged at max rpm for 4 min. The peptide concentration was determined using the Quick Start Bradford Assay (5000205, Bio Rad) according to the manufacturer’s instructions and a microplate reader (Epoch, BioTek). The single-pot, solid-phase-enhanced sample-preparation (SP3) method was used for the extraction and processing of the proteins; 100ug of each of the samples of decellularized and native kidney extracts were reduced with 4uL of 200mM 1, 4-Dithiothreitol (DTT, 1019777700, Sigma-Aldrich, 10197777001, Darmstadt, Germany) then alkylated by addition of 4uL of 400mM iodoacetamide (IAA) (90034, Thermo Fisher, MA, USA). SP3 protein clean up and digestion was performed using 1 mg (20 µl) SeraMag Speedbeads (GE Healthcare) per sample, adding acetonitrile to 70% then washed with 200ul 80% EtOH (x3), and finally suspending in 50mM HEPES pH 8, 2 µg trypsin/LycC. After removing the beads, the digests were acidified by adding 50 µl 0.2% FA. Finally, the samples were desalted by solid phase extraction with C18 Sep-Pak cartridges (Waters), conditioned with 1 ml acetonitrile/0.1%. The eluate was dried using a centrifugal evaporator (CentriVap) and the residue dissolved in a 20ul 0.1% FA. Prepared samples were analyzed using an Easy nLC 1200 connected to an Orbitrap Lumos. The Easy nLC was set up with a PepMap RSLC C18 75 μm x 50 cm, 2 μm column at 50⁰C using the following gradient: 0–5 min 2% ACN; 5–140 min, 2–28% ACN; 140–150 min, 28–40% ACN; 150–160 min, 40–95% ACN; 160–170 min, 95% ACN. Column equilibration was 8 µl and sample injects 2 µl with 6 µl loading volume with total run times about 3 h. The Easynano source was operated at 2400 V and m/z 375–1500 orbitrap (OT) MS1 survey scan data collected at 120,000 resolution (profile), RF lens 30%, AGC 4e5, 50ms max. Data dependant MS2 data was collected with peptide monoisotopic selection, 5.0e4 threshold, charge states 2–7, 60 s dynamic exclusion (10ppm), quad isolation m/z 0.8, HCD activation 30%, OT 15,000 resolution (centroid), auto scan range with first mass m/z110, 5.0e4 target with max inject time of 100ms and cycle time of 3 s.
Data was processed using Thermo Proteome Discoverer 2.5 with Sequest and the Sus Scrofa dataset (Sus Scrofa TaxID = 9823, 2022-03-02 and Homo SapiensTaxID = 9606) with the following parameters: precursor mass 350–5000 Da; enzyme, trypsin; missed cleavage, 2; min/max peptide length 6/144; precursor/fragment tolerance, 10ppm/0.02Da; all neutral losses; b,y ions only; static modification, carbamidomethyl; dynamic modifications, oxidation (K, M, P), protein terminus acetyl; Cn max 0.05; PSM, peptide and decoy DB target FDR’s, 0.01 strict, 0.05 relaxed, based on q-value. Filtering was set for peptides with at least medium confidence; however, only high-confidence data was selected with final post-process filtering with Discoverer. Protein and Protein Group were exported to Excel for further sorting and analysis.
Primary cell culture
The cells used for recellularization were obtained from fresh kidneys of pigs undergoing euthanasia in our Animal facility under our animal protocol (A22-0119) approved by the University of British Columbia. To obtain a population of cells labelled as whole kidney pig cells (WKPC), the organ was minced and weighed. ∼5.5 mg was dissociated with Collagenase A (10103586001, Sigma-Aldrich) at 2.25 mg/ml and Dispase II (17105-041, Gibco) 10U in a mixture of HBSS and DMEM and incubated at 37 C for two hours gently shaking the mixture continuously. The mixture was filtered through a 70 μm and 40 μm cell strainer and centrifuged to be later incubated for 10 min with TrypLE (12605028, Gibco). The culture was monitored and expanded until passage five at 37 °C and 5% CO2 in 175 cm flasks, using kidney mix media (KMM) supplemented with 5.1% of FBS (Hyclone, UT, USA) and 5% A/A, the composition consisted of one third of DMEM (11965092, Gibco), one third of REBM (CC-3190, Lonza, GA, USA) supplemented with 0.1% hEGF, 0.1% Insulin, 0.1% Hydrocortisone, 0.1% GA-1000, 0.1% Transferrin, 0.1% Triiodothyronine, 0.1% Epinephrine, and one-third of EGM (CC-3162, Lonza), supplemented with 0.04% Hydrocortisone, 0.4% hFGF-B, 0.1% VEGF, 0.1% R3-IGF-1, 0.1% Ascorbic Acid, 0.1% hEGF, 0.1% GA-1000, 0.1% Heparin. The passage was done when the cells were at 80% confluency with 0.25% Trypsin-EDTA (#25200056, Gibco, MD, USA) for 10 min at 37 °C was used for detachment and with later passages, a cell scrapper was needed. WKPC were frozen in liquid nitrogen with Bambanker (BB01, Cederlane) in cryovials for better preservation. The number of cells was determined with the Automated Cell Counter TC20 (Bio-Rad, WA, USA).
Flow cytometry
Primary porcine renal cells were used with and without culture directly after cell dissociation. Expanded WKPC cells were detached using Cell Stripper (VWR, Cat# CA4500-668), a non-enzymatic reagent washed with PBC and resuspended in FACS buffer (1X PBS, 2.5mM EDTA, 2% FBS, 0.05% sodium aside). To analyze for viability, The Zombie NIR™ dye (423107, BioLegend, CA, USA) in a 1:100 dilution was added to the cells and incubated for 15 min. For blocking, the cells were incubated on ice with Fc block (564220, BD biosciences) diluted in a1:300 for 5 min. For surface staining, the primary antibodies were added and incubated on ice in the dark for 45 min. Due to the lack of antibodies for pig samples, anti-human antibodies were used with predicted reactivity with pig. The following primary antibodies were used: anti-human CD14-AF700 (301822, BioLegend), anti-mouse/human E-Cadherin-BV421 (147319, Bio Legend), anti-mouse/human AQP1-AF594 (bs-1506R-A594, Bioss USA, MA, USA), anti-mouse/human AQP4-APC-Cy7 (bs-0634R-APC-Cy7, Bioss USA), anti-mouse/human Podocin-PE (bs-6597R-PE, Bioss USA), anti-human CD31-APC (303115, BioLegend). After washing, the cells underwent intracellular protein staining, involving fixation, permeabilization, and incubation in the dark with Anti-Human Vimentin-AF488 (562338, BD Biosciences) for 15 min. Finally, the cells were filtered, resuspended in FACS buffer, and analyzed by FACS Canto II Flow Cytometry System (BD Bioscience). The percentage of positively stained cell count was quantified using FlowJo_V10.
Quantitative real-time PCR (RT-qpcr)
WKPC from every passage (2 × 106 cells) were washed with 100 uL of PBS and then stored in 100uL of RNA at − 20 °C. The total RNA was extracted with the RNeasy kit (Qiagen) following the manufacturer’s instructions. The cDNA synthesis was performed using 1000ng of RNA and the High Capacity cDNA Reverse Transcription Kit (4368813 Applied Biosystems, CA, USA). The analysis of real-time PCR (RT-PCR) was done QuantStudio 7Pro (Applied Biosystems) using PowerTrack™ SYBR™ Green Master Mix (4309155 applied biosystem). The reaction was carried out with an initial denaturation of 2 min followed by 40 cycles of denaturation of 95 for 5 s and elongation at 60 for 35 s. Using 300nM of gene-specific designed primers for cell kidney markers and GADPH as a housekeeping gene for normalization and quantifying gene expression, which can be found in the supplemental material (Appendix 2).
Recellularization
The tubbing, Luer lock adaptors and the bioreactor were sterilized using ethylene oxide. The decellularized organ was connected through the renal vein and the renal artery to the valves of the bioreactor employing the same IV lines that were used for decellularization, connecting them with Luer lock adapters to the valves of the bioreactor; the assembling, sterilization and cell perfusion was done on the biosafety cabinet. For the sterilization of the organ, ddH2O with 1% A/A was perfused through the renal artery and renal vein at 10 ml/min using a peristaltic pump (BS-900 Braintree Scientific Inc.); after the continuous perfusion, 150 ml of ddH2O with 10% A/A were left inside the organ for 2 h.
Using a syringe 40 × 106 WKPC were infused into the renal artery resuspended in 45 ml of DMEM 10% FBS and 1% A/A, 40 × 106 WKPC perfused through the renal vein and 20 × 106 WKPC perfused through the ureter, previously catheterized, -17inHg of negative pressure was applied after 10 min in static state, only during the first day of cell perfusion. Five identical cycles of cell perfusion were done, having a total of 500 × 106WKPC. DMEM 10% FBS and 1% A/A, was perfused through the renal artery every 24 h for four days for 1 h at 10 ml/min. At the end of the fourth day, the bioreactor was set up in continuous recirculation of supplemented DMEM at 10 ml/min inside an incubator for eight days; the media was changed every day and oxygenated by agitation for one hour daily. On the ninth day, oxygenation was provided using red blood cells (RBC). The RBCs were isolated by mixing 60mL of human blood with 12mL of Ficoll-Paque PLUS (#17144003, Cytivia Life Sciences) and then centrifuged at 400 g for 30 min at 20 °C. The upper layers, including the granulocytes, were discarded using a sterile pipette. Approximately 30 billion RBCs were mixed with 400mL of media and oxygenated for 30 min using a stirring plate. The RBC, along with the media, were perfused at 30 ml/min into the kidney with daily oxygenation by agitation. The media was stored every day using cOmplete™ Protease Inhibitor Cocktail (Roche, Germany) at -80 C, later on it was analyzed to count and collect the number of cells that flowed out of the kidney, obtaining the retention of cells following the methodology proposed by Hochman-Mendez20. On the twelfth day, the kidney was sectioned and prepared for histology analysis.
LDH assay
The media obtained after the recellularization was thawed and analyzed using the LDH-Glo™ Cytotoxicity Assay (Promega, WI, USA) according to the manufacturer’s instructions, using 2 µl of 10% Triton X-100 per 100 µl of media as a negative control. The assay was done with 25uL of samples and 60 min of incubation time, and luminescence was recorded using a luminometer infinite m200 pro (Tecan, Switzerland).
Immunofluorescence
For the analysis of cells, WKPCs were cultured in Nunc™ Lab-Tek™ Flask on Slide (170920, Thermo Fisher, MA, USA)) and were allowed to grow for 72 h, washed with 1X filtered PBS and fixed with 4% paraformaldehyde (Sigma-Aldrich, MO, USA). Regarding the study of the recellularized kidney sections, five slides of the inferior pole with longitudinal cut were deparaffinized, once rehydrated they were treated with Diva Decloaker 10X (Cedarlane, ON, CA) for 30 min in boiling water for the antigen retrieval, then washed with dH2O. Both sections and cell slides were washed with PBS, permeabilized with 0.3% Triton X-100, incubated with Fc blocker and blocked for non-specific binding with 2.5% horse serum (Vector Laboratories, CA, USA) for 30 min.
Slides were stained with Mouse Anti-E-Cadherin at 1:500 (610181 BD Bioscience), mouse anti-Aquaporin-1 AT 1:400 (ab9566 ), mouse anti-CD31 at a1:100 (ab24590), rabbit anti-Vimentin at 1:200 (mAb 5741), rabbit anti-aquaporin-1 at 1:500(mAb59678 ), rabbit anti-podocin at 1:500 (ab216341) antibodies incubated overnight at 4 °C. Goat Anti-Mouse IgG (Alexa Fluor 569) and Goat Anti-Rabbit IgG (Alexa Fluor 488) were used as secondary antibodies for staining at a 1:200 dilution for two hours in the dark, the slides were mounted with DAPI (Vector Laboratories, CA, USA) and imaged with confocal microscope at 20X and 60X magnifications (Olympus FV3000RS).
Statistical analysis
R studio (2021.09.0) was used to analyze the statistical difference between the two groups using a paired t-test. It was considered statistically different when the probability value was P < 0.05. Data were presented as mean ± S.D.
Results
Decellularization
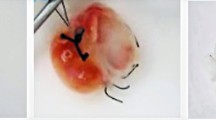
The primary objective was to minimize the exposure of the extracellular matrix (ECM) to detergents by adjusting the flow rates rather than altering the duration of detergent exposure in each cycle. We successfully established a decellularization protocol for pig kidneys using perfusion, achieving complete decellularization within 45 h (Fig. 1). Macroscopic evaluation demonstrated effective cell removal through detergent perfusion via the renal vein and renal artery, resulting in partially transparent decellularized organs that retained their structures and vascular networks.

Evaluation of decellularized pig kidney. (a) H&E showed a complete cell removal in the renal medulla (b) microscopic evaluation of the preservation of the cortex of a pig kidney. The arrows point to the preservation of the glomeruli. Slides scanned at 40X (Scale bar 100 μm & 100 μm, respectively). (c) DNA quantification of pig native and decellularized kidneys (Native 1113.17 ng/µl ± 100.02 ng/µl, Decellularized 12.24 ng/µl ± 4.07 ng/µl, n = 5). The DNA content was reduced 99% in decellularized pig kidneys. (d) Macroscopic progression of a decellularized pig kidney, after 45 h of decellularizing reagents perfusion through the renal artery and renal vein, we obtained a complete cell removal.
Consistent with the macroscopic assessment, microscopic evaluation of various sections of the decellularized organs using H&E staining revealed complete cell removal in the renal medulla and cortex of pig kidneys. This preservation of the organ’s microarchitecture indicated the production of a high-quality ECM suitable for recellularization. In the medulla, the characteristic honeycomb-like appearance was maintained (Fig. 1a), while in the cortex, the loops corresponding to the glomerulus, a crucial kidney component, were preserved (Fig. 1b).
Double-stranded DNA content was quantified and compared between decellularized kidneys (12.24 ng/µl ± 4.07 ng/µl) and native kidneys (1113.17 ng/µl ± 100.02 ng/µl), revealing a statistically significant difference with a p-value of < 0.0001 (1.578*10^-05). This difference signifies a reduction of over 99% in residual DNA in the decellularized pig kidneys, suggesting the elimination of the majority of cells and any remaining DNA (Fig. 1c). Figure 1d shows the macroscopic appearance of the kidney prior, during and after decellularization.
Proteomic analysis of the extracellular matrix
Out of the 4,610 proteins identified in native pig kidneys, 3,162 proteins were found in the decellularized pig kidneys. A comparison between the decellularized and native kidneys revealed an overlap of 2,715 proteins, with 447 proteins uniquely identified in the decellularized tissue and 1,895 proteins exclusively detected in the native kidney. The absence of certain proteins in the decellularized kidney can be attributed to the removal of cellular components, including enzymes and organelle-specific proteins. It is important to note that the samples were derived from different kidneys.
By identifying and quantifying proteins within the ECM, we aimed to understand its composition and tissue-specific differences. Table 1presents a total of 170 proteins, which include critical components of the extracellular matrix, selecting a lower number of proteins from the total pool allowed us to concentrate on the core matrisome and unique ECM regulators. These proteins were meticulously selected due to their indispensable roles in orchestrating essential cellular processes, particularly in facilitating changes in cell adhesion, which are crucial for cellular movement and tissue organization. Moreover, the selected proteins comprise the core matrisome of the ECM found in every tissue (collagen, elastin, laminin, fibronectin, integrins) and include specific proteins found in the kidney that regulate its function (nephronectin, uromodulin). These proteins are instrumental in stabilizing cell-matrix interactions, ensuring structural integrity, and facilitating proper signalling within the ECM21,22.
Notably, the decellularized organ exhibited the presence of seven different types of growth factors (Fig. 2a). The number of peptides corresponds to the unique peptides identified per protein by the mass spectrometer, while coverage indicates the proportion of amino acids covered by the identified peptides relative to the total protein length. Abundance reflects the combined intensity of all analyzed peptides, serving as a marker for quantifying protein relative abundance in the organ23,24.

Proteomic analysis of decellularized and native pig (Sus scrofa) kidney. (a) Fold change (Relative abundance of decellularized organ / Relative abundance of native organ) of critical extracellular matrix components. The fold change indicates alterations in relative protein concentration during the decellularization process. Removal of cellular components resulted in increased relative concentrations of certain proteins in the decellularized organ, reflected by fold changes higher than 1, particularly among key ECM components (elastin, fibronectin, laminin). (b) Fold change in different types of collagen within the ECM. Significant increases in collagen I and collagen V levels were observed, aligning with the predominant collagen composition of the decellularized organ’s ECM. Enhanced relative abundance was noted across most collagen types in the decellularized organ.
The decellularized kidney displayed high concentrations of key proteins such as fibronectin, elastin, glycoproteins, integrin, laminin, and fibrillin (Fig. 2a). An increased relative abundance of collagen was notably observed in the decellularized organ, with most collagen types exhibiting a fold change ≥ 1. This observation underscores the significance of collagen in maintaining structural integrity, which is well preserved in the decellularized organ (Fig. 2b). These proteins play a critical role in establishing a functional scaffold, preserving cell viability, and facilitating cellular recruitment12,25,26. The fold rate indicates changes in relative protein concentration during the decellularization process. Due to the removal of cellular components, some proteins showed increased relative concentrations in the decellularized organ, as indicated by fold rates greater than 1.
Notably, certain growth factors exhibited higher relative concentrations in the decellularized kidney, including acidic fibroblast growth factor (FGF), epidermal growth factor, and fibroblast growth factor (Fig. 2a). Furthermore, the decellularized scaffold retained 64% of VEGH compared to the native kidney. This growth factor is crucial for kidney bioengineering as it plays a role in angiogenesis and renal podocyte development(Fig. 2a)27.
Characterization of the primary porcine renal cells
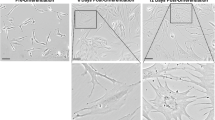
During the primary culture of WKPC with kidney mix media (IP under submission), we observed various morphologies, primarily during early passages. However, with successive passages, the heterogeneity of the cell population decreased, and fibroblast-shaped cells began to dominate the culture, reaching confluency earlier with each passage.
Analysis of WKPC revealed changes in marker expression over successive passages. RT-qPCR analysis demonstrated an upregulation of CDH1 and Vimentin as passages increased, compared to uncultured WKPC, indicating a higher presence of epithelial and fibroblast cells in the culture (Fig. 3a). In contrast, the expression of AQP1, AQP4, CD14, CD31, and Podocin decreased compared to the original uncultured WKPC. The most marked decrease was observed in immune cells expressing CD14. Notably, podocin exhibited a lower decrease in expression compared to the other markers (Fig. 3b). These findings suggest that WKPC should be utilized at earlier passages, preferably up to passage two, to ensure effective repopulation with a variety of cells as found in the native kidney.

Evaluation of the WKPC. (a) qPCR of whole kidney pig cells. The analysis showed the presence of CDH1 and Vimentin expression increasing with passages, as depicted in the top graph. (b) The markers AQP1, AQP4, CD14, CD31 and Podocin, were also present but exhibited a decreasing level of expression with the passages (n = 4). After the dissociation and culture of primary porcine renal cells it was found a heterogenous population with cells expressing kidney cell markers such as AQP1(c), and epithelial cells CDH1 (d), AQP4 (e), endothelial cells (f) and stroma cells with podocytes (g). The analysis was performed at 20X magnification, with a scale bar of 20 μm.
Both qPCR and immunofluorescence confirmed the presence of epithelial cells expressing CDH1 (E-cadherin), AQP1 indicative of cells found in proximal tubules, and AQP4 indicating the presence of cells conforming to the collecting ducts28. Additionally, cells expressing Vimentin, typically found in the renal capsule, glomeruli, and vasculature, were identified, suggesting the presence of renal tubular cells and fibroblasts29,30. The analysis also revealed the presence of endothelial cells expressing CD31 and podocytes expressing podocin, crucial for glomeruli formation, the site of the initial filtration step28,31,32. Furthermore, a lower expression of immune cells, particularly monocytes expressing CD14, was observed33(Fig. 3b).
These results were confirmed with immunofluorescence analysis. The majority of the cells showed positive staining for AQP1, AQP4, Vimentin, and CDH1 (E-cadherin), indicating their epithelial cell nature. A small percentage of the population demonstrated positive staining for CD31 and podocin, suggesting the presence of endothelial cells and podocytes, respectively (Fig. 3c-g).
These findings validate the efficacy of the method used to isolate WKPC, which involved dissociating kidney sections rather than utilizing the whole organ. Using this approach, we generated populations of cells expressing a diverse range of kidney-specific markers crucial for performing various kidney functions. This highlights the potential of WKPC as a valuable resource for further studies to optimize the recellularization techniques of a whole pig kidney and later test for functionality both in vivo and in vitro.
Flow cytometric analysis corroborated what was found in the RT-qPCR assay, showing that the WKPC population changes over passages. Table 2provides marker expression percentages for different renal cell antibodies in WKPC at various passages. Passage zero represents cells without culture, freshly isolated from a native kidney. The cell viability of WKPC remained constant through passages, suggesting that fluctuations in percentages over passages might be due to the faster duplication rates of stromal and fibroblast cells expressing vimentin rather than cell death34,35.
In cells obtained directly from the tissue (P0), a predominance of epithelial cells expressing AQP4 and AQP1 was observed, along with a noticeable percentage of endothelial cells. However, by passage 3, there was a notable shift in cell composition. Most cells expressed Vimentin, indicative of a stroma or fibroblast-like phenotype, while the percentages of AQP1 and CD31-positive cells decreased.
Results showed that Vimentin expression gradually increased from 15.20% in passage 0 to 66.10% in passage 3, indicating an enrichment of Vimentin-positive cells over successive passages, in agreement with RT-qPCR results. This change may be attributed to culture conditions favouring the growth of Vimentin-expressing cells. In contrast, Podocin expression increased from 13.70 to 47.90% in passage 2 and then slightly decreased to 37.60% in passage 3.
Moreover, the expression of CD14, a marker associated with immune cells, remained relatively low throughout the passages, ranging from 0.12 to 2.08%. The expression of AQP1 decreased from 65.30% in uncultured cells to 22.20% in passage 3, indicating a potential loss or reduced presence of proximal tubular cells. CD31, a marker for endothelial cells, decreased from 65.90 to 22.40% in passage 3. AQP4, a marker for collecting duct cells, showed a slight decrease from 69% in passage 0 to 50.80% in passage 3.
These findings highlight the heterogeneity and phenotypic changes within the WKPC population during culture, underscoring the importance of considering passage number when utilizing these cells for recellularization or other applications.
Evaluation of a whole recellularized pig kidney
In the panel of Fig. 4a it is shown six sections representing the histological examination of a native kidney, a decellularized kidney, and a recellularized kidney. Starting from the left, the decellularized kidney displays a significant (p < 0.05) absence of cellular material while keeping the ECM intact. In the middle, the native kidney section serves as a reference for comparison, it displays the characteristic histological features of a healthy kidney, including well-defined renal structures such as glomeruli, tubules, and blood vessels. On the right side, we have the recellularized kidney section, resembling every time more a native kidney. This section demonstrates a high percentage of repopulation of the decellularized ECM with new cells. The image reveals a reestablishment of cellular density and organization. Regions with clusters of cells, forming cell colonies, can be observed. Furthermore, the presence of various renal structures, including tubules and circular formations resembling glomeruli, suggests the recellularization of diverse renal cells forming different renal structures.

Recellularized pig kidney. (a) H&E staining of the reseeded pig kidney revealed a significant increase in cell presence and the formation of glomeruli-like structures compared to the decellularized kidney the samples were collected after 12 days of culture. The slide was scanned at 40X magnification. (b) Macroscopic evaluation of recellularized pig kidney. The recellularization process involved the perfusion of culture media, WKPC, and RBC through the renal artery, renal vein, and ureter for a culture time of 12 days.
The accompanying image showcases the macroscopic transformation of the kidney over the course of several days (Fig. 4b). Starting from a decellularized organ, the kidney progressively evolved to contain 500 million well-characterized WKPC and was initially perfused solely with media. Ultimately, the kidney contained an impressive population of 318 billion red blood cells (RBCs). It is worth noting that the proportion of WKPC within the total cell population accounted for only 0.15% of the perfused RBCs, leading to erythrocyte accumulation all over the matrix. However, prior to the perfusion of RBCs, on the fifth day, the kidney closely resembled a native kidney, without blood flow.
The histological evaluation revealed a robust presence of cells distributed throughout the matrix, indicating a high recellularization percentage and the formation of colonies by WKPCs. H&E staining was performed on various sections, including the superior pole, superior half, inferior half, and inferior pole, encompassing the parenchyma, collecting ducts, and medulla (Fig. 5). The sampling of both cortex and medulla from different sections validated the successful reach of cells to the organ’s periphery. Cells exhibited alignment within tubular structures, closely resembling a native kidney, in the superior half. Furthermore, in the inferior half, there is evidence of cells recovering the wall of blood vessels. The presence of diverse cell types indicated the establishment of different cell populations crucial for functional kidney regeneration. Notably, interconnected cell networks were observed within the scaffold, indicating cell-to-cell communication and potential migration within the matrix. The image displayed a well-organized arrangement of cells, with varying cell densities across different regions. There were no signs of cell aggregation. Circular structures were observed, suggesting the repopulation of glomeruli in the cortex, especially in the superior and inferior poles (Fig. 5). The higher number of RBCs observed could potentially obstruct the microarchitecture of the organ, posing a challenge for cell migration. Nevertheless, the protocol demonstrated effective cell perfusion in the cortex and medulla.

Histological evaluation of a recellularized pig kidney. After 12 days of culture different sections of the organ were collected and analyzed, including the superior pole, superior half, inferior half, and inferior pole. Sampling zones encompassed the parenchyma, collecting ducts, and the medulla. The distribution of cells was found to be uniform throughout the entire tissue, demonstrating a high percentage of cell reseeding and migration towards the periphery of the organ. The slide was scanned at 40X magnification.
Identification of primary porcine renal cells in recellularized pig kidney
All the immunofluorescence slides were compared with their closest respective H&E for analysis of cellularity and tissue structure. Fluorescence microscopy analysis revealed endothelial cells expressing CD31 were predominately aligned within vascular structures in both the medulla and the cortex (Fig. 6a), with enhanced expression in the half sections of the kidney. Furthermore, positive staining for AQP4 indicated site-specific adhesion of the extracellular matrix by epithelial cells in the tubular regions of the medulla(Fig. 6c), particularly in the collecting ducts. Vimentin expression was detected in the renal tubules (Fig. 6e), showing a high abundance in all sections. Additionally, the presence of aquaporin 1-expressing epithelial cells formed structures resembling the native proximal tubular epithelium observed in the cortex, primarily concentrated in the poles of the organ (Fig. 6g). The podocin staining disclosed glomeruli-like structures in the seeded kidney that were densely populated with podocytes in the cortex across all sampled sections (Fig. 6i), highlighting their successful integration. Podocin was co-stained with CD31 to analyze the relationship between the cells in the glomeruli-like structure of the kidney (Fig. 6k). Moreover, a Movat pentachrome staining showed the co-localization of the recellularized kidney cells with the components of the ECM, particularly the collagen recovering the glomerular basal membrane (Fig. 6l). Furthermore, recellularized pig kidney was stained with nephrin, confirming the presence of podocytes in the glomerular area (Appendix 3 supplementary material). These findings suggest that the seeded cells exhibit site-specific localization and the ability to form structures resembling the native renal components (Fig. 6a-l). H&E sections were attached to the immunofluorescence images to improve the understanding of cell distribution within different compartments of the recellularized kidney and aid in understanding the morphology of the tissue for the location of CD31 (Fig. 6b), AQP4 (Fig. 6d), Vimentin (Fig. 6f), AQP1 (Fig. 6h) and Podocin (Fig. 6j). Ki-67 staining demonstrated the presence of 58.1 ± 17.2% brown-labelled proliferating cells distributed throughout the recellularized kidney, indicating active cell division of the WKPC inside the organ. This suggests that the recellularization process has successfully supported cell proliferation within the tissue scaffold (Fig. 6m). In contrast, the Caspase-3 staining revealed 14.22 ± 4.46% of brown-labelled apoptotic cells in the recellularized pig kidney, indicating minimal cell death within the tissue. This indicates a favourable environment for cell survival and suggests that the recellularization protocol provides enough oxygen and nutrients to support the milieu for the maintenance and viability of the introduced WKPC (Fig. 6mm).

Staining of the reseeded pig kidney illustrates the presence of cell markers in various kidney sections. (a) Endothelial cells expressing CD31 (c) aquaporin 4 was detected in tubules of the recellularized kidney (e) vimentin (g) as well as proximal tubule markers aquaporin. Notably, (i) podocytes are stained with podocin-positive (k) podocin co-stained with CD31 cells are observed, forming glomeruli-like structures. The corresponding H&E staining’s (b, d, f, h and j) are shown next to the immunofluorescence staining to aid in understanding the morphology of the tissue. Note: tissue sections are not consecutive. The IF analysis was conducted at 20X magnification, with a scale bar of 20 μm. (l) Movat staining of a recellularized glomeruli showing presence of collagen in the glomeruli basal membrane stained in yellow. Elastin fibres are stained in black, nuclei in blue, collagen in yellow, mucin in bright blue, fibrin in bright red and muscle in red. Movat and H&E slides were scanned at 40X (m) The Ki-67 staining showed the presence of 58.1 ± 17.2% positive cells, indicating active proliferation. Conversely, Caspase-3 staining exhibited 14.22 ± 4.46% positive cells, suggesting low levels of apoptosis and cell death after recellularization. (n) The LDH values exhibited variation throughout the recellularization process, consistently measuring above 15 mU/mL. The media perfused through the recellularized kidney demonstrated low cytotoxicity, suggesting that the cells perfused remained viable within the organ.
The number of cells present in the culture media accounted for less than 1% of the initially seeded cells, indicating that the recellularization process was successful since the ECM retained 99% of the cells. Although LDH values gradually increased during the reseeding process, there was not a considerable production of LDH, consistently remaining below 16 U/L. The normal level of LDH in human blood is below 280 U/L, demonstrating that 16U/L does not express signs of cytotoxicity. The histological analysis and the assessment of LDH levels mutually reinforce each other, providing robust evidence for the successful retention and viability of the seeded cells within the recellularized kidney. The protocol of recellularization resulted in low tissue damage, low cell death and cell migration to the periphery of the organ within the recellularized kidney (Fig. 6n).
A preliminary experiment was conducted for 12 days without mechanical agitation of the media and RBCs. The proliferation, cell death and LDH values obtained are presented in Appendix 4 of the Supplemental Material. Initially, LDH levels were recorded at 10 U/L, with a considerable increase observed starting from day 6, reaching values exceeding 20 U/L by the final day. Concurrently, the proliferation rate, assessed by Ki-67 staining, was found to be 16.06 ± 0.19%, while the percentage of apoptotic cells, evaluated by caspase-3 staining, was 46.56 ± 3.72%. These findings suggest that the absence of mechanical agitation of the media contributed to elevated LDH values during the initial days of culture. Additionally, the oxygenation provided by RBC perfusion was associated with increased proliferation, decreased apoptotic cell percentage, and reduced LDH values toward the latter stages of organ culture. These observations underscore the importance of optimizing culture conditions, including mechanical agitation and oxygenation strategies, to enhance organ viability and functionality during ex vivo culture.
Discussion
The successful reseeding of entire pig kidneys represents a remarkable advancement in the fields of tissue engineering and transplantation. Given the intricate structure of the kidney, which includes a complex network of capillaries and thousands of nephrons with intricate functional structures, the decellularization process is the most suitable method for bioengineering a functional kidney. To further advance kidney bioengineering, it is imperative to establish an optimal protocol for recellularizing kidneys of clinically relevant sizes.
The success of the decellularization process was confirmed by the DNA content, which measured below 50 ng of DNA per milligram of tissue, meeting the criteria for organ decellularization. Preservation of the microarchitecture, including the integrity of the glomerular loops, as well as the retention of proteins within the extracellular matrix (ECM), further validated the efficacy of the decellularization process7. Zambon et al36. demonstrated that a combination of SDS and Triton X-100 is the optimal decellularization methodology, achieving comparable macroscopic and microscopic results while substantially reducing perfusion time from 72 to 45 h. This modification minimizes ECM damage while ensuring complete cell removal.
It’s worth noting that Sullivan et al. decellularized pig kidneys for 36 h using only 0.5% SDS, which might result in structural protein and growth factor damage. Zambon et al13. further evaluated this method and observed ECM damage when perfusing with 0.5% SDS, as confirmed by scanning electron microscopy (SEM). In contrast, Tan37found that decellularizing a rat kidney with 0.1% SDS and 1X Triton did not damage the ECM, as evidenced by SEM analysis. Additionally, canine and pig kidneys were decellularized in 8 and 12 h, respectively, using SDS 0.5% perfusion and cryoablation. However, this process did cause damage to the overall ECM structure38,39.
Histological analyses have consistently shown the preservation of vital ECM components such as collagen, laminin, fibronectin, elastin, and glycosaminoglycans (GAGs) after decellularization using 0.1% SDS and Triton X-1007,26,40,41. Nevertheless, there has been a lack of a proteomic profile of the ECM post-decellularization of pig kidneys. Leuning et al42. reported a proteomic analysis for rat and human kidneys, obtaining results similar to our findings in porcine decellularized kidneys. Notably, our study revealed a higher presence of growth factors, particularly VEGF and FGF, which are crucial for cell adherence27. This may be attributed to the decellularization protocol using Triton/SDS, as Caralt et al. demonstrated good retention of FGF and VEGF in the matrix after decellularizing a rat kidney40. In the proteomic analysis of decellularized pig kidneys, we observed an increase in the relative concentration of elastin and certain collagens, including collagen IV, collagen V, and collagen I, which are primary constituents of the ECM. This finding contradicts the results reported by Hussein et al43., who observed a drastic reduction in elastin but aligns with the statement that collagen is preserved in a decellularized pig kidney scaffold. Additionally, the level of VEGF retained in the decellularized kidney was 64%, compared to the 38% reported by Hussein et al43.. Similarly, these researchers reported the retention of 44% for FGF, whereas, in our study, there was a 2.98-fold increase in the relative concentration of this growth factor44. These findings support the preservation of critical ECM components, which play a pivotal role in the recellularization process by promoting cell migration and enhancing cell viability. Preserving the growth factors offers the opportunity to guide the phenotypic specification of cells toward their original location-specific types44.
It was found in this study that combining perfusion and diffusion techniques enabled better preservation of cell viability. Antegrade perfusion supplies cells to the vasculature and at the glomerular level, while retrograde perfusion repopulates the collecting system and the tubular lumen26. Negative pressure has been employed to enhance cell migration and extravasation. Previous studies have reported the benefits of using negative pressure during cell seeding; when regulated with controlled time and pressure, it does not damage the extracellular matrix (ECM) and promotes a more uniform cell distribution17,42,45,46. Poornejad et al46. specifically demonstrated a remarkable improvement in cell migration from the tubules to the cortex using negative pressure (-40mmHg) during ureteral perfusion, which aligns with the results obtained by our group. We can conclude that − 17inHg yielded more favourable results in terms of recellularization efficiency and distribution, indicating its potential as a key factor in optimizing the recellularization process. It is worth noting that our protocol differed from other studies by focusing on whole organ recellularization, perfusing various cell types through the vein, artery, and ureter, resulting in better outcomes with a higher recellularization percentage. This approach successfully demonstrated the presence of endothelial cells in the blood vessels, epithelial cells in the tubules, and podocytes in glomerular-like structures.
We successfully isolated, cultured, and expanded primary porcine renal cells, which exhibited the expression of renal cell markers. These findings are consistent with those of Abolbashari et al47., who also demonstrated the presence of AQP1, AQP4, and Podocin in cultured primary porcine renal cells. However, the percentages obtained during flow cytometry analysis differed between the two studies. The WKPC cells showed lower expression of AQP1 (65.3% vs. 72%) but higher expression of AQP4 and Podocin (69% vs. 20% and 13.7% vs. 2%, respectively). This indicates a heterogeneous population of vital kidney cells expressing markers crucial for the bioengineering of a functional kidney. Moreover, the researchers demonstrated that primary porcine renal cells maintain renal cell functions, such as sodium uptake, hydrolase activity, and erythropoietin production. Consequently, assessing the in vitro functionality of the whole pig kidney recellularization should be a focus of future trials. It is worth mentioning that for successful recellularization, early passages of the cells should be utilized to preserve the majority of the cells. Flow cytometry and RT-qPCR analysis revealed that, over passages, the number of cells expressing Vimentin surpassed the desired population. Furthermore, optimizing the culture media is necessary to maximize the preservation of endothelial cells and monocytes, as these cell types showed a higher decrease in abundance over passages.
Abolbashari et al47. developed a methodology for the formation of renal tubular structures; however, their seeding method involved cell injection, which may result in ECM damage and inefficient repopulation of the whole organ. In contrast, the use of perfusion allowed for a uniform distribution and migration of cells throughout the entire recellularized kidney. We observed a higher number of cells and a larger formation of colonies within the recellularized kidney at 12 days of culture, using − 17 inHg of negative pressure and oxygenation, compared to lower values of negative pressure, fewer days in culture and the control group with no oxygenation, indicating a higher recellularization percentage and increased formation of renal structures, including tubular-like and glomerular-like formations. The comparison and methodology of the recellularization trials can be found in Figure A4 in Appendix 5 of the supplementary material. Furthermore, the presence of cells expressing AQP1 and AQP4 in the appropriate structures further supports the hypothesis that ECM components and distribution facilitate the migration of cells to their respective locations.
Regarding cell viability, the analysis conducted through ki-67 and caspase-3 staining supported the effectiveness of the recellularization approach in promoting cell proliferation and reducing cell death, crucial factors for the successful development of functional renal tissue. The 58.1% proliferation rate within the recellularized kidney significantly (p< 0.05) surpasses the findings of Leuning et al42., who reported a proliferation rate of only 7.8%. Additionally, more stained cells were observed compared to the studies conducted by Hussein et al43. and Abolbashari et al47.. Conversely, similar results were obtained when compared to Remuzzi et al.‘s42findings in rat kidney recellularization. Bonandrini et al18. reported higher proliferation using PCNA in porcine kidney recellularization after 72 h in culture.
Concerning cell death during pig kidney reseeding, the percentage of cell death on days 3, 7, 14, and 28 was lower in Abolbashari et al47. study compared to the findings in our recellularization process, where 14.22% expressed caspase-3. This coincides with the results reported by Bonandrini et al45. using embryonic stem cells. Although the percentage of cells expressing caspase-3 is not as low as in other researchers’ findings, it still falls within an acceptable range. However, addressing this issue and further reducing cell death should be a focus of future investigations.
During the optimization process, it was observed that oxygenating the media through agitation was less effective than desired. Analysis of the culture media revealed poor retention and diffusive delivery of oxygen due to the low oxygen affinity of the culture media. Additionally, the organ lacked the constant gas exchange that occurs in 2D cultures, limiting effective oxygen transfer. To overcome these challenges, a natural oxygen carrier, hemoglobin found in red blood cells (RBCs), was utilized. The use of erythrocytes allowed for efficient oxygen delivery to the cells without relying on bubble generation and rupture, thus avoiding potential cell damage caused by shear forces. This approach also eliminated the limitations associated with gas liberation during agitation.
The increased presence of RBCs resulting from this approach may lead to a higher repopulation of RBCs, which could potentially obstruct the microarchitecture of the organ and hinder cell migration. However, this test demonstrated that the method used is helpful in perfusing cells along different sections of the kidney since we can detect a high number of RBCs. Nonetheless, we continue to observe the formation of circular structures along the ECM. To further optimize the protocol, it is necessary to increase the amount of perfusing WKPC by at least eightfold and explore using different oxygen carriers47.
Besides the oxygenation, some limitations of bioengineering organs are the homogeneous distribution of cells, the preservation of the cell viability, the migration of cells to the periphery and into the right compartments and the re-endothelialization of the vascular network of the decellularized organ. Ideally recellularized sections should be analyzed with co-localization of the components of the extracellular matrix, and cell markers, however there is a big limitation on the availability of antibodies for specific species, such as Sus Scrofa, used in this manuscript. In the context of transplanting bio-engineered kidneys, it is understood that porcine primary renal cells may not be suitable for clinical applications. Once the recellularization process achieves an optimal recellularization percentage with a uniform distribution, it will be important to explore alternative sources of autologous cells from the patients themselves, including stem cells that could differentiate into specific kidney cells based on their location due to the proteins and growth factors found in the extracellular matrix48. However, this study served as a proof-of-principle, aiming to optimize the recellularization technique. It was demonstrated that the use of WKPCs showed promising characteristics, particularly their rapid proliferation rate, making them potential candidates for recellularization when a large number of cells are required and for further evaluation of in vivo functionality of the bioengineered organs.
Notably, this study successfully obtained a remarkable number of cells from a small amount of tissue. From just 5.5 mg of tissue, approximately 160*10^6 cells were obtained. Extrapolating these findings to the whole tissue, it can be estimated that approximately 3.98*10^9(three billion nine hundred eighty-five million) cells could be obtained. These results align with previous findings reported in the literature47.
Conclusion
In conclusion, the increasing incidence of End-Stage Renal Disease (ESRD) exacerbates the shortage of available organs for transplantation. The limitations of dialysis emphasize the urgent need for advancements in organ bioengineering. Bioengineered kidneys represent a promising solution to bridge the gap between organ supply and demand, ultimately enhancing the quality of life for patients in need.
Efficient decellularization of clinically relevant-sized kidneys while preserving the structural and compositional integrity of the extracellular matrix (ECM) is a critical milestone in the development of bioengineered organs. This study has demonstrated that the use of perfusion, negative pressure, nutrient immersion, and oxygenation considerably enhances the recellularization process, promotes cell viability, and facilitates the formation of functional renal structures.
Future research endeavours should focus on optimizing the culture conditions and automating key processes to make these bioengineered kidneys suitable for clinical applications, thus laying the groundwork for their potential use in human transplantation. Ultimately, this ground-breaking research opens up new avenues for exploring and developing regenerative therapies, fundamentally transforming the landscape of organ transplantation and positively impacting the lives of countless individuals in need of life-saving treatments.
Data availability
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
References
Benjamin, O. & Lappin, S. L. End-stage renal disease. in StatPearls (StatPearls Publishing, Treasure Island (FL). Preprint at https://pubmed.ncbi.nlm.nihgov/29763036/ (2021).
Canadian Institute for Health Information. Treatment of End-Stage Organ Failure in Canada, Canadian Organ Replacement Register, 2012 to 2021: End-Stage Kidney Disease and Kidney Transplants. https://www.cihi.ca/en/annual-statistics-on-organ-replacement-in-canada-2012-to-2021 (2023).
Manns, B., McKenzie, S. Q., Au, F., Gignac, P. M. & Geller, L. I. The Financial impact of advanced kidney disease on Canada Pension Plan and private disability insurance costs. Can. J. Kidney Health Dis. 4, 2–8 (2017).
Canadian Institute for Health Information. Annual statistics on organ replacement in Canada, 2011 to 2021[report]. Ottowa, ON: CIHI. https://www.cihi.ca/en/annual-statistics-on-organ-replacement-in-canada-2012-to-2021 (2022).
Canadian Institute for Health Information. Treatment of End-Stage Organ Failure in Canada, Canadian Organ Replacement Register, 2011 to 2020: End‐Stage Kidney Disease and Kidney Transplants. Ottowa, ON: CIHI. https://www.cihi.ca/en/organ-replacement-in-canada-corr-annual-statistics-2020, supplemented with data collected by the Quebec Branch of The Kidney Foundation of Canada as provided by renal units. (2021)
Narres, M. et al. The incidence of end-stage renal disease in the Diabetic (compared to the non-diabetic) Population: a systematic review. PLoS One. 11, 18–24, e0147329 (2016).
Sullivan, D. C. et al. Decellularization methods of porcine kidneys for whole organ engineering using a high-throughput system. Biomaterials. 33, 7756–7764 (2012).
Trends in end-stage kidney disease in Canada,* 2019 | CIHI. https://www.cihi.ca/en/trends-in-end-stage-kidney-disease-in-canada-2019 (2019).
Canadian Institute for Health Information. Annual Statistics on Organ Replacement in Canada: Dialysis, Transplantation and Donation, 2010 to 2019. https://www.cihi.ca/sites/default/files/document/corr-snapshot-2019-en.pdf (2019).
Caddick, A. Kidney transplants – the gold standard treatment. Open. Access. Government. https://www.openaccessgovernment.org/kidney-transplants-treatment/7550/ (2014).
Canadian Institute for Health Information. e-Statistics Report on Transplant, Waiting List and Donor Statistics. https://www.cihi.ca/sites/default/files/document/corr-transplant-wait-list-donorstats-2020-en.xlsx(2020).
McInnes, A. D., Moser, M. A. J. & Chen, X. Preparation and Use of Decellularized Extracellular Matrix for tissue Engineering. J. Funct. Biomaterials. 13, 240 (2022).
Paulo Zambon, J., Atala, A. & Yoo, J. J. Methods to generate tissue-derived constructs for regenerative medicine applications. Methods. 171, 3–10 (2020).
Figliuzzi, M., Remuzzi, G. & Remuzzi, A. Chapter 63 - Recellularization of Kidney Scaffold With Stem Cells. in Kidney Transplantation, Bioengineering and Regeneration (eds. Orlando, G., Remuzzi, G. & Williams, D. F.) 877–886 Academic Press, https://doi.org/10.1016/B978-0-12-801734-0.00063-1 (2017).
Gaspar, D. A., Gomide, V. & Monteiro, F. J. The role of perfusion bioreactors in bone tissue engineering. Biomatter. 2, 167–175 (2012).
Salehi-Nik, N. et al. Engineering Parameters in Bioreactor’s Design: A Critical Aspect in Tissue Engineering. Biomed Research International 762132, https://doi.org/10.1155/2013/762132 (2013).
Song, J. J. & Ott, H. C. Organ engineering based on decellularized matrix scaffolds. Trends Mol. Med. 17, 424–432 (2011).
Bonandrini, B. et al. Recellularization of well-preserved acellular kidney scaffold using embryonic stem cells. Tissue Eng. Part. A. 20, 1486–1498 (2014).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878. https://doi.org/10.1038/s41598-017-17204-5 (2017).
Hochman-Mendez, C. et al. Restoring anatomical complexity of a left ventricle wall as a step toward bioengineering a human heart with human induced pluripotent stem cell-derived cardiac cells. Acta Biomater. 141, 48–58 (2022).
Alberts, B. et al. Garland Science,. The Extracellular Matrix of Animals. in Molecular Biology of the Cell. 4th edition. https://www.ncbi.nlm.nih.gov/books/NBK26810/ (2002).
Frantz, C., Stewart, K. M. & Weaver, V. M. The extracellular matrix at a glance. J. Cell Sci. 123, 4195–4200 (2010).
Zhang, B., Pirmoradian, M., Zubarev, R. & Käll, L. Covariation of peptide abundances accurately reflects protein concentration differences. Mol. Cell. Proteomics: MCP. 16, 936 (2017).
Han, X., Aslanian, A. & Yates, J. R. Mass Spectrometry for Proteomics. Curr. Opin. Chem. Biol. 12, 483–490 (2008).
Parihar, A. et al. 3D Printing: Advancement in Biogenerative Engineering to combat shortage of organs and Bioapplicable materials. Regen Eng. Transl Med. 8, 173–199 (2022).
Ross, E. A. et al. Embryonic stem cells proliferate and differentiate when seeded into kidney scaffolds. J. Am. Soc. Nephrol. 20, 2338–2347 (2009).
Kim, B. S. & Goligorsky, M. S. Role of VEGF in kidney development, Microvascular Maintenance and pathophysiology of Renal Disease. Korean J. Intern. Med. 18, 65–75 (2003).
Kriz, W. & Kaissling, B. Structural Organization of the mammalian kidney. Seldin Giebisch’s Kidney. 1, 479–563 (2008).
Rowan, C. J., Sheybani-Deloui, S. & Rosenblum, N. D. Origin and function of the renal stroma in Health and Disease. Results Probl. Cell. Differ. 60, 205–229 (2017).
Kidd, M. E., Shumaker, D. K. & Ridge, K. M. The role of Vimentin Intermediate filaments in the progression of Lung Cancer. Am. J. Respir Cell. Mol. Biol. 50, 1–6 (2014).
Bombelli, S. et al. PKHhigh/CD133+/CD24 – renal stem-like cells isolated from human nephrospheres exhibit in Vitro Multipotency. Cells. 9, 1805 (2020).
Guo, K. et al. Deubiquitylase OTUD6B stabilizes the mutated pVHL and suppresses cell migration in clear cell renal cell carcinoma. Cell. Death Dis. 13, 1–11 (2022).
Kapellos, T. S. et al. Human monocyte subsets and phenotypes in Major Chronic Inflammatory diseases. Front. Immunol. 10, 2–5 (2019).
Starzonek, C. et al. Enrichment of Human dermal stem cells from primary cell cultures through the elimination of fibroblasts. Cells. 12, 949 (2023).
Klemmt, P. A. B., Carver, J. G., Kennedy, S. H., Koninckx, P. R. & Mardon, H. J. Stromal cells from endometriotic lesions and endometrium from women with endometriosis have reduced decidualization capacity. Fertil. Steril. 85, 564–572 (2006).
Jp, Z. et al. Comparative analysis of two porcine kidney decellularization methods for maintenance of functional vascular architectures. Acta Biomater. 75, 226–234. https://doi.org/10.1016/j.actbio.2018.06.004 (2018).
Tan, Z. Organ decellularization used as a novel approach to engineer three-dimensional urogenital tumor models. (University Br. Columbia. https://doi.org/10.14288/1.0379729 (2019).
Tajima, K. et al. Decellularization of canine kidney for three-dimensional organ regeneration. Vet. World. 13, 452–457 (2020).
Poornejad, N. et al. Efficient decellularization of whole porcine kidneys improves reseeded cell behavior. Biomed. Mater. 11, 025003 (2016).
Caralt, M. et al. Optimization and critical evaluation of decellularization strategies to develop renal extracellular matrix scaffolds as biological templates for organ engineering and transplantation. Am. J. Transpl.15, 64–75 (2015).
Uzarski, J. S. et al. Sustained in vivo perfusion of a re-endothelialized tissue engineered kidney graft in a human-scale animal model. Front. Bioeng. Biotechnol. 11, 1184408 (2023).
Leuning, D. G. et al. Vascular bioengineering of scaffolds derived from human discarded transplant kidneys using human pluripotent stem cell–derived endothelium. Am. J. Transpl. 19, 1328–1343 (2019).
Hussein, K. H. et al. Biocompatibility and hemocompatibility of efficiently decellularized whole porcine kidney for tissue engineering. J. Biomedical Mater. Res. Part. A. 106, 2034–2047 (2018).
de Haan, M. J. A., Witjas, F. M. R., Engelse, M. A. & Rabelink, T. J. Have we hit a wall with whole kidney decellularization and recellularization: a review. Curr. Opin. Biomedical Eng. 20, 100335 (2021).
Remuzzi, A. et al. Experimental evaluation of kidney regeneration by Organ Scaffold recellularization. Sci. Rep. 7, 43502 (2017).
Poornejad, N. et al. Re-epithelialization of whole porcine kidneys with renal epithelial cells. J. Tissue Eng. 8, 2041731417718809 (2017).
Abolbashari, M. et al. Repopulation of porcine kidney scaffold using porcine primary renal cells. Acta Biomater. 29, 52–61 (2016).
Liu, C., Pei, M., Li, Q. & Zhang, Y. Decellularized extracellular matrix mediates tissue construction and regeneration. Front. Med. 16, 56–82 (2022).
Acknowledgements
This study received no funding. We would like to express our gratitude to Dr. Dirk Lange and Dr. David Harriman for his insights during the planning of the experiments.
Author information
Authors and Affiliations
Contributions
ACLB, AS and CCM conceived the study and designed the experiments. ACLB and CUMA carried out the organ harvesting, cell culture, immunofluorescence, and flow cytometry. ACLB was in charge of the decellularization and recellularization. HA and ACLB performed the proteomics analysis. ACLB and CUMA analyzed the data. ACLB, CCM and AS wrote the manuscript. All the authors read and approved the submitted manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Luque-Badillo, A.C., Monjaras-Avila, C.U., Adomat, H. et al. Evaluating different methods for kidney recellularization. Sci Rep 14, 23520 (2024). https://doi.org/10.1038/s41598-024-74543-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-74543-w
