
Abstract
Coronary artery disease (CAD) is a predominant cardiovascular condition influenced by risk factors, with an emphasis on major depressive disorder (MDD). However, the shared mechanisms and therapeutic targets for CAD and MDD remain incompletely comprehended. Functional enrichment analyses were conducted to investigate the pathways associated with the differentially expressed genes (DEGs) in the CAD and MDD datasets. Hub genes were identified utilizing the Protein-Protein Interaction network and Cytoscape software. The single sample gene set variation analysis was applied to assess immune cell infiltration in the CAD and MDD datasets. Weighted gene co-expression network analysis and molecular biological experiments were executed to evaluate these hub genes. Molecular docking was conducted to identify drug candidates targeting these hub genes. The overlapping DEGs between the CAD and MDD datasets were mainly enriched in the Herpes simplex virus 1 infection and the NF-kappa B signaling pathways. CDC42, NDUFB3, and TXN were validated within the eigengenes of the blue module, which exhibited a significant association with the CAD phenotype. The drug candidate GS-9620 was identified as a potential protective agent against both disorders. In conclusion, CDC42, NDUFB3, and TXN held potential as molecular biomarkers and therapeutic targets for the simultaneous treatment of CAD and MDD.
Similar content being viewed by others

Introduction
Coronary artery disease (CAD) significantly contributes to high mortality rates and is a disorder resulting from atherosclerosis, which can manifest as stable angina, unstable angina, myocardial infarction (MI), or even sudden cardiac death1. The risk factors of CAD include environmental factors, lifestyle, genetic factors, and psychosocial stress2. Despite the availability of advanced medications, the morbidity and mortality of CAD remain high3. Major depressive disorder (MDD), the most prevalent psychiatric disorder, impacts more than 300 million individuals globally and severely limits psychosocial function and quality of life. Although various treatments, including psychological and medical therapies, are currently available, rates of intervention for depression persist at low levels4.
Both CAD and MDD are prevalent conditions that imposing a substantial economic strain on society. In recent years, over 100 research studies and a minimum of 6 meta-analyses have demonstrated strong evidence that depression independently contributes to the risk of developing CAD and worsening prognosis5,6,7,8,9,10. For example, Friedman et al. establish a strong association between depression and CAD through a meta-analysis5. Reiner et al. report that the incidence of CAD in individuals is predicted by depression in a dose-dependent manner through a meta-analysis6. Amanda et al. demonstrate that depression serves as both an etiologic and prognostic factor in CAD through a meta-analysis7. Kone et al. indicate that a depressed mood moderately elevates the risk for MI, cerebrovascular diseases, and other forms of CAD through a meta-analysis8. Wulsin et al. reveal that symptoms of depression independently increase the risk of developing CAD more than exposure to passive smoking9. Gan et al. suggest that there is an independent association between depression and a notably heightened risk of CAD through a meta-analysis10. The high occurrence of comorbidity between CAD and MDD indicates underlying common pathological mechanisms, such as biological mechanisms (activation of the immune system11, dysregulation of the hypothalamic-pituitary-adrenocortical axis12, disruption in autonomic tone of the heart13, irregularities in blood clotting processes14, and dysfunction of the endothelium in coronary arteries15) and behavioral mechanisms1 (poor adherence to medication, exercise, diet, smoking cessation, and sedentary behavior). However, the precise genetic mechanisms between MDD and CAD are not yet fully understood. Therefore, it is crucial to comprehend the shared pathological mechanism and identify therapeutic targets for both conditions, as well as to explore effective pharmacological and psychological treatments.
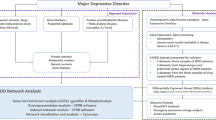
In this research, the utilization of gene microarray technologies offered a robust method for identifying possible therapeutic targets shared by both CAD and MDD. We performed functional enrichment analysis of differentially expressed genes (DEGs) derived from datasets related to CAD and MDD obtained from the NCBI Gene Expression Omnibus database (GEO, https://www.ncbi.nlm.nih.gov/geo/), which allowed us to identify pivotal molecular targets and pathways involved in the common pathogenesis of these 2 conditions. Hub genes were identified among the common DEGs between CAD and MDD datasets by calculating scores using 5 algorithms: Degree, Stress, Density of Maximum Neighborhood Component (DMNC), EcCentricity, and Radiality, which were components of the CytoHubba plugin in Cytoscape software. Furthermore, we validated the expression of these hub genes in CAD and MDD datasets, and established Receiver Operating Characteristic (ROC) curves to assess the possible diagnostic relevance of the hub genes associated with these disorders. We also examined the extent of immune cell infiltration and evaluated the relationship between hub genes and immune cell infiltration across both disorders. Additionally, we evaluated the importance of the hub genes through a weighted gene co-expression network analysis (WGCNA) and a real-time polymerase chain reaction analysis (RT-PCR). Ultimately, the binding energy and targeted chemical interactions between candidate drugs and hub genes were calculated and identified utilizing molecular docking analysis. The workflow diagram of this research was depicted in Fig. 1.

The workflow chart of the study.
This comprehensive investigation served as an innovative study in revealing shared pathological mechanisms associated with the co-occurrence of CAD and MDD, providing significant perspectives on the genetic basis and possible therapeutic approaches for both disorders.
Materials and methods
Data collection and sources
Datasets for this research were derived from the GEO database. The GSE71226 dataset included a total of 6 samples of peripheral blood: 3 normal and 3 CAD samples. The GSE76826 dataset included a total of 32 samples of peripheral blood: 12 normal and 20 MDD samples. These datasets were considered as the sets for the primary analysis. The GSE20680 dataset, comprising 52 normal and 87 CAD samples of peripheral blood, along with the GSE38206 dataset, which included 18 control and 18 MDD samples of peripheral blood, was utilized to confirm the expression of the identified marker genes. In addition, the GSE12288 dataset, comprising 112 non-CAD and 110 CAD samples of peripheral blood, was gathered for WGCNA evaluation purposes.
Identification of DEGs
Differential expression analyses of gene expression data from the GSE71226 and GSE76826 datasets were conducted utilizing the “limma” (version 3.50.1) R package16. DEGs were determined based on the following criteria: |Log2FC| > 0 and adj. P < 0.05 in CAD and MDD dataset. Additionally, volcano plots were generated utilizing the “ggplot2” (version 3.3.6) R package to visualize differences in gene expression across the datasets17.
Functional enrichment analysis
Metascape software (version 3.5.20240101, http://metascape.org/) was utilized for analyzing potential functions associated with DEGs. This analysis encompassed Gene Ontology (GO) terms and Reactome pathway enrichment. To explore the possible biological functions and mechanisms associated with the genes, we utilized the “clusterProfiler” (version 4.2.2) R package to evaluate the enrichment of Kyoto Encyclopedia of Genes and Genomes (KEGG) terms for the DEGs18.
Determination of hub genes for CAD and MDD
A protein-protein interactions network (PPI) of DEGs was created utilizing the STRING database (version 10.5, https://cn.string-db.org/) with the “minimum required interaction score” parameter set at 0.419. The PPI network was subsequently loaded into Cytoscape software (version 3.10.2, https://cytoscape.org/). The CytoHubba plugin (version 0.1, https://apps.cytoscape.org/apps/cytohubba) was employed alongside the Degree, Radiality, DMNC, EcCentricity, and Stress algorithms to identify the top 10 genes with highest scores20. A Venn diagram was generated to visualize the overlapping genes identified by the 5 algorithms. Finally, the GeneMANIA database (version 2.1, http://genemania.org/) was utilized to display co-expressed genes associated with the hub genes.
ROC curve analysis
Based on the GSE20680 and GSE38206 datasets, we conducted a ROC curve analysis to elucidate the sensitivity and specificity of hub genes in diagnosing CAD and MDD. The area under the curves (AUCs) were calculated using the “pROC” (version 1.18.0) R package21,22. with an AUC greater than 0.5 considered indicative of diagnostic capability23.
Expression analysis
In the GSE20680 and GSE38206 datasets, we conducted an expression analysis for each selected hub gene, comparing non-CAD and CAD samples, as well as non-MDD and MDD samples, to validate its accuracy. The gene expression data for the control and disease groups were organized into a data frame that included gene names, expression levels, and group information. Subsequently, the average expression levels of each gene in both the control and disease groups were calculated for both the control and disease groups. Finally, bar plots and other visualizations were created using “ggplot2” (version 3.3.6) R package to illustrate the gene expression levels across the different groups24.
Immune infiltration analysis
Single sample gene set variation analysis (ssGSVA) for immune cell infiltration was conducted utilizing the “GSVA” (version1.40.1) R package25. Following the acquisition of the LM22 signature matrix, the Wilcoxon test was conducted to assess variations in immune cell infiltration between patients with CAD or MDD and healthy controls. A P-value of less than 0.05 was deemed statistically significant. The heatmaps were generated with the “pheatmap” (version 1.0.12) R package26.
Spearman’s rank correlation analysis of hub genes and infiltrating immune cells
Spearman’s rank correlation analysis was performed to examine the relationships between hub genes and the proportions of infiltrating immune cells in the CAD and MDD datasets, utilizing “ggplot2” (version 3.3.6) and “reshape2” (version 1.4.4) R packages27. Finally, lollipop plots were utilized to visually represent the association between hub genes and infiltrating immune cells.
WGCNA analysis
The “WGCNA” (version 1.70-3) R package was employed to compute the Pearson correlation coefficients among gene pairs and create a gene co-expression matrix28. An unsigned co-expression network was then constructed sequentially in accordance with the principles of scale-free networks, utilizing a soft threshold. The resulting adjacency matrix was converted into a topological overlap matrix. Clustering analysis was conducted next to uncover gene modules. Hierarchical clustering was utilized to construct a dendrogram, and module eigengenes were computed to assess their correlation with CAD phenotype. The module with the high correlation coefficient and the low P-value was designated as the CAD-associated module. After identifying the candidate module containing hub genes, criteria were established to filter significant genes within this module by ensuring that |module membership| exceeded 0.8 and |gene significance| surpassed 0.229,30. The heatmaps were generated with the “pheatmap” (version 1.0.12) R package26.
Potential drug evaluation and molecular docking
The file in SDF format of the primary active components of drugs was retrieved from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). Additionally, the protein structures corresponding to the hub genes were sourced from the Protein Data Bank database (version 3.48.0, https://www.rcsb.org/). Optimization treatments, such as the removal of water molecules and small molecular ligands, were performed on the hub genes using PyMOL software (version 3.0, https://pymol.org/). Hydrogenation and charge treatments were conducted using the AutoDock tool (version 4.2.6, https://autodock.scripps.edu/download-autodock4/). The molecular docking was carried out with the hub genes as the receptor and its corresponding active component as the ligand using the AutoDock Vina (version 1.1.2, https://vina.scripps.edu/) within PyRx software (version 1.1, https://sourceforge.net/projects/pyrx/) to calculate the binding energy31. Finally, the results were visualized using PyMOL software.
Mice and MI surgery
The 10-week-old male C57BL/6 mice were purchased from Shanghai Model Organisms Center, Inc (Shanghai, China). Sham (n = 4) or ligation of the left anterior descending artery (LAD) (n = 4) surgery was carried out on mice in accordance with a previously established protocol32. Initially, the mice were temporarily anesthetized using 2% isoflurane through inhalation. Subsequently, a minor incision was created in the skin of the left thorax to access the heart, where permanent ligation of the LAD was achieved with 6 − 0 silk sutures. For those subjected to sham surgery, the same procedure was performed, although the ligation of the LAD was omitted. All experiments were conducted in compliance with the United Kingdom Animals (Scientific Procedures) Act 1986 and the American Veterinary Medical Association Guidelines for the Euthanasia of Animals (2020) and the ARRIVE guidelines (https://arriveguidelines.org).
Real-time polymerase chain reaction (RT-PCR)
RT-PCR was carried out in accordance with a previously established protocol33. RNA was isolated from the myocardial tissues utilizing TRIzol reagent (Thermo Fisher Scientific, USA, #15596026CN). cDNA synthesis was conducted with the Prime Script RT Kit (Takara, Japan, #RR047A). Quantitative PCR was employed to assess the mRNA expression levels of the genes of interest, using SYBR1 Premix Ex Taq II (Takara, Japan, #RR430B). The results were normalized against the housekeeping gene GAPDH. The primer sequences used in this study were as follows:
CDC42: Forward: 5’-CCCATCGGAATATGTACCAACTG-3’; Reverse: 5’-CCAAGAGTGTATGGCTCTCCAC-3’.
NDUFB3: Forward: 5’- ACAGACAGTGGAAAATTGAAGGG-3’; Reverse: 5’- GCCCATGTATCTCCAAGCCT-3’.
TXN: Forward: 5’-ACCTGGTCCTCCGCCTGAG-3’; Reverse: 5’- ATGCCCTCTTTATCCTGGATCTTGG-3’.
GAPDH: Forward: 5’-AGGTCGGTGTGAACGGATTTG-3’; Reverse: 5’- GGGGTCGTTGATGGCAACA-3’.
Statistical analysis
Statistical analyses in bioinformatics were performed utilizing R software (version 4.4.1, http://www.r-project.org/). RT-PCR results were presented as the mean ± standard error of the mean (SEM) from 4 independent experiments. All statistical analyses for the RT-PCR experiments were carried out using GraphPad Prism 8 (version 8.0, https://www.graphpad-prism.cn/). The normality of the RT-PCR data was assessed using the Shapiro-Wilk test. An unpaired Student’s t-test was employed to evaluate the differences between the 2 groups. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Result
Data acquisition and processing
Datasets for humans, including the GSE71226, GSE76826, GSE20680, and GSE38206 datasets, were collected from the NCBI GEO database. Our analysis demonstrated a linear distribution trend in the expression matrices of these datasets, indicating no significant batch effects of the data (Supplemental Fig. 1a-d). To further ensure the high quality of datasets, we conducted the Principal Component Analysis (PCA) on GSE71226 and GSE76826. Notably, distinct separation of all samples within the CAD and MDD datasets was observed, indicating the good reproducibility of the data (Fig. 2a, b).

Identification of DEGs. (a, b) The PCA analysis of GSE71226 and GSE76826 datasets. (c, d) Volcan plots displaying the DEGs in the GSE71226 and GSE76826 datasets. (e, f) Venn diagrams displaying the number of up- and down-regulated DEGs in the GSE71226 and GSE76826 datasets.
Determination of DEGs in CAD and MDD datasets
We retrieved a total of 22,188 and 38,152 genes from the GSE71226 and GSE76826 datasets, respectively. To pinpoint key genes within the datasets, we utilized a screening threshold of adj. P < 0.05 and |log2FC| > 0. Subsequently, we created 2 volcano plots for both the CAD and MDD datasets. Specifically, the GSE71226 dataset exhibited a total of 2,293 DEGs, which encompassed 684 genes that were up-regulated and 1,609 genes that were down-regulated. The GSE76826 dataset exhibited a total of 4,485 DEGs, with 3,086 of these genes being up-regulated and 1,799 down-regulated (Fig. 2c, d). Venn diagrams highlighted a collective overlap of 120 up-regulated genes and 209 down-regulated genes existing between the GSE71226 and GSE76826 datasets (Fig. 2e, f).
Functional enrichment analysis of DEGs
Functional enrichment analysis was performed using Metascape software to investigate the biological functions and pathways related to DEGs in the CAD and MDD datasets. With a significance threshold of adj. P < 0.05, A total of 20 GO terms and Reactome pathways showing most significant enrichment of DEGs in the CAD or MDD dataset were identified. The analysis of cell component revealed that DEGs in the CAD dataset were primarily enriched in endoplasmic reticulum lumen and extracellular matrix. The analysis of biological process and molecular function revealed that DEGs in the CAD dataset were primarily enriched in pathways associated with primary immunodeficiency, IgM immunoglobulin complex, immunoglobulin binding, inflammation response, Wnt signaling pathway, positive regulation of MAPK cascade, regulation of hormone levels, regulation of cell activation, and activin receptor signaling pathways (Fig. 3a, b and Supplemental Table 1), suggesting that the DEGs involved in the pathogenesis of CAD was closely related to immune dysregulation and inflammation response34,35. In the MDD dataset, the analysis of cell component revealed a significant enrichment of DEGs in nuclear speck, centrosome, and Golgi membrane. The analysis of biological process and molecular function indicated that DEGs in the MDD dataset showed significant enrichment in pathways related to chromatin organization, DNA metabolic process, protein serine kinase activity, DNA-templated transcription, mRNA metabolic process, regulation of RNA splicing, transcription factor binding, and regulation of DNA metabolic process (Fig. 3c, d and Supplemental Table 2), suggesting that the DEGs involved in the pathogenesis of MDD was closely related to transcription regulation36. Moreover, GO analysis of the overlapping DEGs between CAD and MDD datasets exhibited predominant enrichment in cell component of endosome membrane and nuclear speck, as well as pathways linked to Herpes simplex virus 1 infection, regulation of adenylate cyclase-activating G protein-couples receptor signaling, regulation of macro-autophagy, NF-kappa B signaling, Herpes simplex virus 1 infection, regulation of hormone secretion, phosphatidylinositol signaling system, and regulation of T cell receptor signaling (Fig. 3e and Supplemental Table 3). Concurrently, KEGG analysis of the common DEGs between CAD and MDD datasets displayed predominant enrichment in 2 pathways related to Herpes simplex virus 1 infection and NF-kappa B signaling, indicating that immune system dysregulation and inflammatory processes might act as pathogenic mechanisms for both CAD and MDD37,38,39,40 (Fig. 3f and Supplemental Table 3).

Functional enrichment analysis of DEGs. (a, b) Functional enrichment analysis of DEGs in the GSE71226 dataset. (c, d) Functional Enrichment analysis of DEGs in the GSE76826 dataset. (e, f) Functional enrichment analysis of overlapping DEGs between the CAD and MDD datasets.
Determination of the shared hub genes between CAD and MDD datasets
The 329 shared DEGs from the CAD and MDD datasets were uploaded to the STRING database to create a PPI network which was subsequently uploaded to the Cytoscape software. Finally, the top 10 DEGs with the highest scores in Degree, Stress, DMNC, EcCentricity, and Radiality algorithms were determined from a total of 329 DEGs using the CytoHubba plugins in Cytoscape software. Other algorithms, such as Diameter, Average Distance, Betweenness, Closeness, Eigenvector Centrality, Bridging Centrality, Centroid value, and Edge Betweenness, were not included in the analysis due to the lack of intersecting hub genes. Moreover, 4 hub genes, which included NADH: ubiquinone oxidoreductase subunit B3 (NDUFB3), cell division cycle 42 (CDC42), cytochrome c oxidase subunit 7A2 (COX7A2), and thioredoxin (TXN), were determined through a Venn diagram (Fig. 4a) and visualized in the PPI network. Protein molecules that potentially interacted with hub genes included interleukin 18 (IL18), caspase 1 (CASP1), MDM2 proto-oncogene (MDM2), tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta (YWHAZ), proteasome 20 S subunit alpha 6 (PSMA6), proteasome 20 S subunit alpha 6 (SLIRP), LSM3 Homolog, U6 Small Nuclear RNA And MRNA Degradation Associated (LSM3), cytochrome c oxidase subunit 6 C (COX6C), ubiquinol-cytochrome c reductase complex III subunit VII (UQCRQ), proteasome 20 S subunit alpha 4 (PSMA4), cytochrome c oxidase subunit 7 C (COX7C), NDUFA4 mitochondrial complex associated (NDUFA4), and TNF receptor superfamily member 13 C (TNFRSF13C) (Fig. 4b). Furthermore, the co-expressed genes linked to these hub genes were further examined using GeneMANIA database41. Among them, the most significant genes associated with these hub genes were TXN interacting protein (TXNIP), thioredoxin reductase 1 (TXNRD1), Rho GDP dissociation inhibitor alpha (ARHGDIA), ribonucleotide reductase regulatory TP53 inducible subunit M2B (RRM2B), IQ motif containing GTPase activating protein 3 (IQGAP3), peroxiredoxin 2 (PRDX2), cytochrome c oxidase subunit 7A2 like (COX7A2L), and mitogen-activated protein kinase kinase kinase 5 (MAP3K5) (Fig. 4c).

Identification of hub genes. (a) Venn diagrams showing the overlapping DEGs based on Degree, Stress, DMNC, EcCentricity, and Radiality algorithms. (b) PPI network of 4 hub genes (TXN, CDC42, COX7A2, and NDUFB3) with other proteins. (c) Co-expressed gene network of 4 identified hub genes (TXN, CDC42, COX7A2, and NDUFB3).
The expression and ROC curve analysis of hub genes
In the following analysis, we verified the expression levels of the 4 hub genes among individuals with CAD and MDD, utilizing the human CAD (GSE71226 and GSE20680) and MDD (GSE76826 and GSE38206) datasets, respectively. The bar and volcano plots showed an increase in CDC42, TXN, NDUFB3, and COX7A2 expression in patients with CAD compared to normal individuals (Fig. 5a and Supplemental Fig. 2a). Similarly, the bar and volcano plots indicated an increase in CDC42, NDUFB3, and TXN expression and a decrease in COX7A2 expression in MDD patients compared to normal individuals (Fig. 5b and Supplemental Fig. 2b). It can be seen that COX7A2 was expressed inconsistently in the CAD and MDD datasets. Therefore, we excluded COX7A2 from the list of hub genes in the subsequent analysis. These findings suggested that these 3 hub genes might serve as common pathogenic targets for patients with both CAD and MDD. Next, based on the GSE20680 and GSE38206 datasets, we developed a diagnostic prediction model utilizing ROC curves to assess the specificity and sensitivity as well as the potential diagnostic significance of these 3 hub genes in individuals with CAD and MDD. The findings indicated that the 3 hub genes possessed promising diagnostic values for both conditions. Specifically, the AUCs for NDUFB3, TXN, and CDC42 were 0.573, 0.583, and 0.520 in the CAD dataset (Fig. 5c), and 0.685, 0.676, and 0.701 in the MDD dataset (Fig. 5d), respectively23. These findings implied that these 3 hub genes could act as potential biomarkers for the accurate diagnosis of both CAD and MDD.

Validation and prognostic capability of hub genes. (a, b) Bar plots of hub genes in the GSE20680 (a) and GSE38206 (b) datasets. (c, d) ROC curve analysis of hub genes in the GSE20680 (c) and GSE38206 (d) datasets. P* < 0.05, P** < 0.01, P*** < 0.001.
Immune infiltration analysis in CAD and MDD datasets
CAD is a complex immune disease that immune infiltration is crucial in the onset and progression of atherosclerosis42,43,44. Moreover, immune cell infiltration is also closely linked to MDD, and treatment with immune modulators may alleviate depressive symptoms45,46,47. Thus, we applied the ssGSVA analysis to assess the levels and proportions of infiltrating immune cells in both CAD and MDD, using the GSE71226 and GSE76826 datasets. Our findings demonstrated that the CAD dataset exhibited a notable increase in dendritic cells (DCs), mast cells, eosinophils, neutrophils, and NK CD56bright cells, alongside a notable reduction in central Memory T cell (Tcm), macrophages, B cells, follicular helper T cells (TFH), T cells, and T helper cells in CAD patients when compared with the control group (Fig. 6a, b). Similarly, the MDD dataset showed an elevation in neutrophils and a reduction in B cells, CD8 T cells, NK CD56bright cells, T helper cells, and Tcm (Fig. 7a, b), which was in line with the CAD dataset results concerning B cells, neutrophils, Tcm, and T helper cells. These observations pointed towards a possible shared mechanism underlying immune cell infiltration between CAD and MDD, especially regarding neutrophils, B cells, Tcm, and T helper cells.

Immune infiltration analysis in the GSE71226 dataset. P* < 0.05, P** < 0.01, P**** < 0.0001.

Immune infiltration analysis in the GSE76826 dataset. P* < 0.05, P** < 0.01, P**** < 0.0001.
Correlation between the hub genes and infiltrating immune cells
The similarity in the composition of infiltrating immune cell represents merely one facet of the shared pathogenesis between CAD and MDD. It is crucial to confirm the connection between these 3 hub genes and infiltrating immune cells. To address this, we conducted the Spearman’s rank correlation analysis to explore the relationship between these 3 hub genes and infiltrating immune cells utilizing GSE71226 and GSE76826 datasets. The findings revealed that NDUFB3, CDC42, and TXN exhibited positive correlations with cytotoxic cells, T gamma delta cells (Tgd), Tcm, regulatory T cell (Treg), T helper type 1 cells (Th1), NK cells, T cells, and DC, and negative correlations with B cells, interdigitating DC (iDC), mast cells, macrophages, and eosinophils in the CAD dataset (Fig. 8a-c). In the MDD dataset, NDUFB3 and TXN showed positive correlations with Th1 cells, B cells, Tcm, iDC, NK CD56dim cells, Treg, and T helper cells, and negative correlations with mast cells, effective Memory T Cell (Tem), activated dendritic cells (aDC), CD8 T cells, DC, eosinophils, and macrophages. CDC42 were positively correlated with T helper type 17 cells (Th17), neutrophils, macrophages, and mast cells, and negatively correlated with Treg, TFH, eosinophils, Th1 cells, Th2 cells, cytotoxic cells, Tem, T helper cells, iDC, aDC, NK CD65bright cells, NK CD65dim cells, CD8 T cells, Tcm, and B cells (Fig. 9a-c). Overall, a consistent correlation was observed between these 3 hub genes and infiltrating immune cells in both the CAD and MDD datasets.

The correlation between hub genes and infiltrating immune cells in the GSE71226 dataset. Correlation of NDUFB3 (a), CDC42 (b), and TXN (c), with infiltrating immune cells in the GSE71226 dataset.

The correlation between hub genes and infiltrating immune cells in the GSE76826 dataset. Correlation of NDUFB3 (a), TXN (b), and CDC42 (c) with infiltrating immune cells in the GSE76826 dataset.
Evaluation of 3 hub genes by WGCNA
To further evaluate the importance of hub genes in CAD and MDD datasets, we first constructed a gene co-expression network utilizing the “WGCNA” R package, based on the expression levels of genes from the GSE12288 dataset. Following this, we conducted cluster analysis on all samples in the dataset, excluding 1 identified outlier (Fig. 10a and Supplemental Fig. 3a). We successfully established a scale-free network using a soft threshold power of β = 4 (Fig. 10b and Supplemental Fig. 3b). Subsequently, we calculated the average linkage and Spearman correlation coefficients to develop a hierarchical clustering tree, where each leaf represented a specific gene and each branch was associated with a module comprising genes that exhibited similar expression patterns (Fig. 10c, d). Finally, we merged functionally equivalent modules into a single large module, resulting in a total of 11 modules. The hub genes belonged to the eigengenes within the blue module that showed high correlation with CAD phenotype (Fig. 10e, f). Next, in order to investigate the correlation between module correlation (MM) and trait correlation (GS) in the blue module, eigengenes within the blue module were selected for scatter plot analysis. The results revealed a positive correlation between MM and GS, demonstrating that genes highly correlated with CAD traits also played an important role in the key modules (Fig. 10g). Additionally, the genes situated in the blue module were uploaded into Cytoscape software to generate a protein interaction map. The result found that these 3 hub genes were the core genes within the blue module, further validating their important role in the development of CAD (Fig. 10h).


Evaluation of hub genes in the GSE12288 dataset using WGCNA. (a) Hierarchical clustering and heatmap of samples. (b) Histogram of connectivity distribution. (c) Gene dendrogram of hierarchical clustering (d) Heatmap of adjacency matrix between different modules. (e) Heatmap of Pearson correlation analysis. (f) Heatmap displaying the relationship between various modules and CAD functionality. (g) Scatter plot between Module Membership (MM) and Gene Significance (GS) in the blue module. (h) PPI network of hub genes with other molecules.
Evaluation of hub genes by molecular biology experiments
To further enhance the evaluation of hub gene expression in infarcted hearts compared to non-infarcted hearts, mice were subjected to either sham (n = 4) or MI (n = 4) surgery for a duration of 4 weeks. The results from RT-PCR indicated that the levels of CDC42 (Fig. 11a), NDUFB3 (Fig. 11b), and TXN (Fig. 11c) were increased in the myocardial tissues of MI mice relative to those of sham-operated mice.

Evaluation of hub genes using RT-PCR. The mRNA levels of CDC42 (a), NDUFB3 (b), and TXN (c) in myocardial tissues of MI mice compared to those of sham-operated mice. P*** < 0.001.
Identification of candidate drugs and molecular docking for hub genes in CAD and MDD
AutoDock Vina within PyRx software is a crucial tool for identifying core drug components48. The 3 hub genes linked to CAD and MDD were employed to determine potential drug candidates using the PubChem and Protein Data Bank databases. From this analysis, the top 10 drugs with the most favorable P-value were chosen as potential candidates, including GS-9620, RN-1734, Cimetidine, SB-674,042, Ceritinib, 3,5-Dichlorosalicylic acid, Octreotide, LY344864, Buspirone, and Migalastat (Fig. 12a). In general, these candidates had the potential to treat both CAD and MDD by binding to hub genes. Among these, GS-9620 stood out with the highest binding energy, prompting molecular docking analysis to visualize its interactions with the 3 hub genes (Fig. 12b-d). Specifically, GS-9620 formed hydrogen bonds with GLY-47 and LEU-377 of CDC42, established additional hydrogen bonds with TYR-277 of NDUFB3, and interacted through hydrogen bonds with GLU-68, GLY-83, and LYS-85 of TXN. GS-9620 exhibited strong binding affinities with the 3 target proteins, with calculated binding energies of -7 kcal/mol (CDC42), -7.3 kcal/mol (NDUFB3), and − 5.8 kcal/mol (TXN), respectively. These results indicated the potential of GS-9620 for treating CAD in conjunction with MDD.

Identification of candidate drugs interacted with hub genes using molecular docking analysis. (a) The top 10 potential candidate drugs were identified based on their binding energy. (b–d) The molecular docking interactions between CDC42 (b), NDUFB3 (c), TXN (d), and GS-9620.
Discussion
CAD is a prevalent cardiovascular condition and a leading cause of mortality worldwide. Various risk factors contribute to the development of CAD, including smoking, hypertension, diabetes, obesity, and psychosocial stress2. Recent research has identified several types of psychosocial stress that play a role in the onset and progression of atherosclerosis and heart disease. These stresses encompass depression, anxiety disorders, and temperament traits like anger and hostility, among which depression has been extensively studied in relation to CAD13. MDD is a disorder that is highly heterogeneous, known for its significant levels of morbidity and mortality, and is recognized as the leading cause of disability on a global scale45. Compelling evidence suggests that depression serves as an independent risk factor for the development of CAD and can also exacerbate the prognosis post-diagnosis6,7,8,10,13. However, the specific underlying pathogenesis between CAD and MDD remains unclear, investigating the shared pathophysiological mechanism of CAD and MDD using bioinformatics approaches may yield significant insights.
In this study, by conducting an extensive bioinformatics analysis, we investigated the underlying genetic mechanisms connecting CAD and MDD. Our methodology involved the identification of DEGs in CAD and MDD utilizing the GSE71226 and GSE76826 datasets, respectively. Subsequently, we carried out functional enrichment analyses on the DEGs specific to CAD and MDD, as well as the overlapping DEGs between the 2 conditions. To identify hub genes among the common DEGs, we employed 5 algorithms from the CytoHubba plugin in Cytoscape software and pinpointed NDUFB3, CDC42, and TXN as hub genes. These findings were verified using human GSE20680 and GSE38206 datasets. Moreover, we explored the immune cell infiltration in both conditions and investigated the relationship between the hub genes and infiltrating immune cell utilizing the ssGSVA algorithm. Remarkably, our analysis revealed a strong association between the hub genes and infiltrating immune cell in the CAD and MDD datasets. In addition, we identified the blue module, which exhibited a significant correlation with CAD phenotype, and confirmed that the hub genes were part of the eigengenes within the blue module utilizing WGCNA analysis. Furthermore, we validated the expression of these hub genes in myocardial tissues of mice undergoing MI surgery and conducted a screening for candidate drugs and chemical interactions targeting the hub genes associated with CAD and MDD using molecular docking.
COX7A2, the seventh subunit of cytochrome c oxidase in the respiratory chain of mitochondria, is encoded by nuclear genes and forms polypeptide chains49,50. Alterations in the respiratory chain complex have been linked to the development of CAD51,52. Additionally, disruption in respiratory chain function, reduced expression of genes related to mitochondrial, impairment of mitochondrial lipids and membrane proteins, and increased oxidative stress, can all be observed in depression models53,54. Yet minimal research has identified COX7A2 as a valuable indicator for diagnosing or predicting CAD or MDD. Our study uncovered inconsistent alterations in COX7A2 levels among individuals suffering from CAD and MDD, indicating the failure of COX7A2 to act as a potential connection between CAD and MDD and to provide a promising therapeutic target for patients with both disorders.
CDC42, a crucial member of the Rho-GTPase family, plays a vital role in the development of CAD by modulating atherosclerosis, blood lipids, endothelial barrier function, inflammation55. The research conducted by Li et al. reveals that CDC42 assists in reducing atherosclerosis through enhancing reendothelialization of damaged arteries56. Khalid et al. propose that CDC42 inhibits inflammation through the maintenance of Treg homeostasis57. Lv et al. demonstrate that CDC42 promotes the regeneration of endothelial cells and aids in vascular recovery58. Moreover, CDC42 in the bloodstream regulates Th17 cell differentiation and is linked to an elevated risk of depression, anxiety, and cognitive impairment, potentially impacting cognitive function59. Geula et al. find that blocking CDC42 activity leads to increased anxiety levels in mice60. While the upregulation of CDC42 improved cognitive function in mice61. Our investigation revealed that CDC42 expression was increased in CAD and MDD patients compared to the control group. The significant correlation between CDC42 and the CAD phenotype was confirmed using WGCNA and RT-PCR. Additionally, molecular docking analysis identified chemical interactions between CDC42 and the potential drug candidate GS-9620, highlighting CDC42 as a potential link between CAD and MDD and proposing a promising therapeutic target for patients with both disorders.
NDUFB3, a critical subunit of complex I in the mitochondrial electron transport chain, is indispensable for the assembly and regulation of this complex, as well as for controlling the production of reactive oxygen species (ROS)62. Oxidative stress is a key factor in the onset and progression of CAD, impacting endothelial function and the formation and stability of coronary plaques63,64. Furthermore, oxidative stress plays a crucial role in the pathophysiology of MDD by involving free radicals and ROS, which are fundamental in predicting and measuring depressive conditions, as well as in evaluating the effectiveness of antidepressants65,66,67. Yet minimal research has identified NDUFB3 as a promising biomarker for diagnosing and predicting both CAD and MDD. Our study revealed increased NDUFB3 levels in individuals with CAD and MDD, with WGCNA and RT-PCR confirming a significant association between NDUFB3 and the CAD phenotype. Moreover, molecular docking analysis identified potential chemical interactions between NDUFB3 and the investigational drug GS-9620, indicating NDUFB3 as a potential link between CAD and MDD and proposing a therapeutic target for patients diagnosed with both conditions.
TXN, a 12 kDa redox protein from the oxidoreductase superfamily, is crucial for maintaining redox homeostasis68. Studies have observed decreased TXN levels in CAD patients, with these reduced levels closely linked to disease severity69. The endogenous inhibitor of TXN, TXNIP, plays a significant role in cellular redox regulation. An imbalance between TXNIP and TXN activates the NLRP3 inflammasome, leading to atherosclerotic plaque formation and IL-1β production70. Furthermore, TXN has also been identified as a biomarker for psychiatric disorders such as schizophrenia, bipolar disorder, and autism71. Chronic stress can reduce TXN activity, leading to increased oxidative protein cysteine modifications and activation of the apoptosis signal-regulating kinase 1 pathway72. Our research found higher TXN levels in individuals with both CAD and MDD. Notably, WGCNA and RT-PCR analyses confirmed a strong association between TXN and CAD phenotype. Furthermore, molecular docking analysis indicated potential chemical interactions between TXN and the drug GS-9620, supporting the hypothesis that TXN could serve as a crucial link between CAD and MDD, highlighting its potential as a therapeutic target for patients diagnosed with both conditions.
Previous researches have demonstrated the significant impacts of immune cell infiltration on the progression of both CAD and MDD42,43,45,46. A study has shown a strong association between the expression of CDC42 and immune infiltration, immune checkpoints, and Treg cell markers73. Analysis of the immune microenvironment landscape has revealed a strong correlation between NDUFB3 and 22 types of immune cells74. TXN has been found to promote cancer growth and virus infection by inhibiting an effective immune response75. Our analysis indicated that these 3 hub genes were closely linked to immune cell infiltration, suggesting their potential role in the shared pathogenesis of CAD and MDD through the regulation of immune cell infiltration.
Numerous approaches have been studied in clinical trials to address depression in individuals diagnosed with CAD, including antidepressants like fluoxetine, citalopram, and sertraline, interpersonal psychotherapy, cognitive-behavioral therapy, and exercise76,77,78. However, there is a lack of strategies to manage both conditions simultaneously76,79. Our investigation identified the compound GS-9620 as a potential drug targeting key genes involved in both CAD and MDD. GS-9620, known as Vesatolimod, is a small molecule drug developed by Gilead Sciences, Inc (California, United States), and classified as a toll-like receptor 7 (TLR7) agonist. Currently, the drug is in phase II clinical trials, where it is being investigated for the treatment of HIV infection and acquired immune deficiency syndrome80,81. TLR7 activation can trigger the release of anti-inflammatory cytokines and its presence in plaques has been associated with improved outcomes, suggesting TLR7 as a promising target for preventing atherosclerosis complications82,83. Furthermore, the peripheral administration of etanercept and fluoxetine has been found to alleviate depressive symptoms in rats by indirectly affecting the TLR7 pathway84. These findings underscore the significant role of TLR7 in the pathogenesis of CAD and MDD, with GS-9620 potentially providing protection against both conditions through TLR7 modulation.
Our study does have some limitations. Firstly, the CAD dataset (GSE71226) has a relatively small sample size. To enhance the reliability of our findings, obtaining larger CAD-related datasets in future studies will be beneficial. Secondly, more research is necessary to clarify the specific mechanisms linking immune cell infiltration to the 3 hub genes. Thirdly, there is a lack of additional molecular biology experiments to validate our findings. Fourthly, there is an absence of techniques such as molecular dynamics simulation, crystallography, and nuclear magnetic resonance, which can offer more precise insights into binding patterns and affinities.
Conclusion
The study not only introduced a novel method for detecting the 3 hub genes (CDC42, NDUFB3, and TXN) in samples from individuals with CAD and MDD, but also offered theoretical approaches and new insights into shared pathogenesis and potential treatment options for individuals with both CAD and MDD.
Data availability
The article contained the data that could be accessed in the public domain through the NCBI GEO database (GEO, https://www.ncbi.nlm.nih.gov/geo/), including GSE71226, GSE20680, GSE76826, GSE38206, and GSE12288 datasets.
References
Carney, R. M. & Freedland, K. E. Depression and coronary heart disease. Nat. Rev. Cardiol. 14, 145–155 (2016).
Malakar, A. K. et al. A review on coronary artery disease, its risk factors, and therapeutics. J. Cell. Physiol. 234, 16812–16823 (2019).
Libby, P. & Theroux, P. Pathophysiology of coronary artery disease. Circulation 111, 3481–3488 (2005).
Trivedi, M. H. Major depressive disorder in primary care: strategies for identification. J. Clin. Psychiatry 81, UT17042BR17041C (2020).
Booth-Kewley, S. & Friedman, H. S. Psychological predictors of heart disease: a quantitative review. Psychol. Bull. 101, 343–362 (1987).
R, R. Depression as a predictor for coronary heart disease. A review and meta-analysis. Am. J. Prev. Med. 23, 51–61 (2002).
Nicholson, A., Kuper, H. & Hemingway, H. Depression as an aetiologic and prognostic factor in coronary heart disease: a meta-analysis of 6362 events among 146 538 participants in 54 observational studies. Eur. Heart J. 27, 2763–2774 (2006).
Van der Kooy, K. et al. Depression and the risk for cardiovascular diseases: systematic review and meta analysis. Int. J. Geriatr. Psychiatry. 22, 613–626 (2007).
Lawson, R. W. & Singal, B. M. Do depressive symptoms increase the risk for the onset of coronary disease? A systematic quantitative review. Psychosom. Med. 65, 201–210 (2003).
Gan, Y. et al. Depression and the risk of coronary heart disease: a meta-analysis of prospective cohort studies. BMC Psychiatry. 14, 371 (2014).
Suarez, E. C., Krishnan, R. R. & Lewis, J. G. The relation of severity of depressive symptoms to Monocyte-Associated Proinflammatory cytokines and chemokines in apparently healthy men. Psychosom. Med. 65, 362–368 (2003).
Brown, E. S., Varghese, F. P. & McEwen, B. S. Association of depression with medical illness: does cortisol play a role? Biol. Psychiatry. 55, 1–9 (2004).
Goldston, K. & Baillie, A. Depression and coronary heart disease: a review of the epidemiological evidence, explanatory mechanisms and management approaches. Clin. Psychol. Rev. 28, 288–306 (2008).
Annique Schins, A. et al. Increased coronary events in depressed cardiovascular patients: 5-HT2A receptor as missing link? Psychosom. Med. 65, 729–737 (2003).
Broadley, A. J. M. et al. Arterial endothelial function is impaired in treated depression. Heart 85, 521–523 (2002).
Cao, Z. et al. Bioinformatics-led discovery of liver-specific genes and macrophage infiltration in acute liver injury. Front. Immunol. 14, 1287136 (2023).
Gustavsson, E. K. et al. Ggtranscript: an R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics 38, 3844–3846 (2022).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Shen, M. et al. Identification of key interferon-stimulated genes for indicating the condition of patients with systemic lupus erythematosus. Front. Immunol. 13, 962393 (2022).
Chin, C. H. et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8, S11 (2014).
Hu, Y. et al. Identification of ferroptosis-associated biomarkers for the potential diagnosis and treatment of postmenopausal osteoporosis. Front. Endocrinol. 13, 986384 (2022).
Fayyad-Kazan, H. et al. Circulating miR-150 and miR-342 in plasma are novel potential biomarkers for acute myeloid leukemia. J. Transl Med. 7, 31 (2013).
Obuchowski, N. A. & Bullen, J. A. Receiver operating characteristic (ROC) curves: review of methods with applications in diagnostic medicine. Phys. Med. Biol. 63, 07tr01 (2018).
Shi, Y. et al. Crosstalk of ferroptosis regulators and tumor immunity in pancreatic adenocarcinoma: novel perspective to mRNA vaccines and personalized immunotherapy. Apoptosis 28, 1423–1435 (2023).
Wang, S. et al. Suppression of P53 pathway is an important factor inducing Acute Graft-versus-host disease through T cell activation based on Bioinformatics Analysis. J. Inflamm. Res. 17, 4093–4104 (2024).
Zheng, X. et al. Identification and validation of immunotherapy for four novel clusters of colorectal cancer based on the tumor microenvironment. Front. Immunol. 13, 984480 (2022).
Eden, S. K., Li, C. & Shepherd, B. E. Nonparametric estimation of Spearman’s rank correlation with bivariate survival data. Biometrics 78, 421–434 (2021).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Wang, Y. et al. The shared biomarkers and pathways of systemic lupus erythematosus and metabolic syndrome analyzed by bioinformatics combining machine learning algorithm and single-cell sequencing analysis. Front. Immunol. 13, 1015882 (2022).
Li, M. et al. Novel Diagnostic Biomarkers Related to Oxidative Stress and Macrophage Ferroptosis in Atherosclerosis. Oxid Med Cell Longev 1–18 (2022). (2022).
Pinzi, L. & Rastelli, G. Molecular Docking: shifting paradigms in Drug Discovery. Int. J. Mol. Sci. 20, 4331 (2019).
Dutta, P. & Johny, E. Left coronary artery ligation: a Surgical Murine Model of myocardial infarction. J. Vis. Exp. 9 https://doi.org/10.3791/64387 (2022).
Nolan, T., Hands, R. E. & Bustin, S. A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 1, 1559–1582 (2006).
Malayala, S. V. & Raza, A. A. Comparative analysis of Cardiovascular Risk in Human Immunodeficiency Virus-Seropositive and -seronegative pre-menopausal women. J. Clin. Med. Res. 12, 409–414 (2020).
Zhang, J. et al. Secreted frizzled-related protein 4 exerts anti-atherosclerotic effects by reducing inflammation and oxidative stress. Eur. J. Pharmacol. 923, 174901 (2022).
de Klerk, E. & ‘t Hoen, P. A. C. Alternative mRNA transcription, processing, and translation: insights from RNA sequencing. Trends Genet. 31, 128–139 (2015).
Zhang, J., Liu, H. & Wei, B. Immune response of T cells during herpes simplex virus type 1 (HSV-1) infection. J. Zhejiang Univ. Sci. B. 18, 277–288 (2017).
Barnabei, L. et al. NF-κB: at the Borders of autoimmunity and inflammation. Front. Immunol. 12, 716469 (2021).
Li, Q. & Verma, I. M. NF-κB regulation in the immune system. Nat. Rev. Immunol. 2, 725–734 (2002).
Zhu, S. & Viejo-Borbolla, A. Pathogenesis and virulence of herpes simplex virus. Virulence 12, 2670–2702 (2021).
Franz, M. et al. GeneMANIA update 2018. Nucleic Acids Res. 46, W60–W64 (2018).
He, T. et al. Immune Cell Infiltration Analysis based on Bioinformatics reveals novel biomarkers of coronary artery disease. J. Inflamm. Res. 16, 3169–3184 (2023).
Jiang, X. et al. Identification of diagnostic biomarkers and immune cell infiltration in coronary artery disease by machine learning, nomogram, and molecular docking. Front. Immunol. 15, 1368904 (2024).
Feng, X. et al. Identification of diagnostic biomarkers and therapeutic targets in peripheral immune landscape from coronary artery disease. J. Transl Med. 20, 399 (2022).
Zhang, J. et al. Identification of mitophagy-related biomarkers and immune infiltration in major depressive disorder. BMC Genom. 24, 216 (2023).
Chang, L. et al. Identification of novel endoplasmic reticulum-related genes and their association with immune cell infiltration in major depressive disorder. J. Affect. Disord. 356, 190–203 (2024).
Liu, X. & Wu, Y. Identification of 7 mitochondria-related genes as diagnostic biomarkers of MDD and their correlation with immune infiltration: new insights from bioinformatics analysis. J. Affect. Disord. 349, 86–100 (2024).
Tessema, F. B. et al. Flavonoids and phenolic acids from Aerial Part of Ajuga Integrifolia (Buch.-Ham. Ex D. Don): Anti-shigellosis Activity and in Silico Molecular Docking studies. Molecules 28, 1111 (2023).
Chen, L. et al. Cox7a2 mediates steroidogenesis in TM3 mouse ley dig cells. Asian J. Androl. 8, 589–594 (2006).
Deng, S. et al. Overexpression of COX7A2 is associated with a good prognosis in patients with glioma. J. Neurooncol. 136, 41–50 (2017).
Maurer, I. & Zierz, S. Myocardial respiratory chain enzyme activities in idiopathic dilated cardiomyopathy, and comparison with those in atherosclerotic coronary artery disease and valvular aortic stenosis. Am. J. Cardiol. 72, 428–433 (1993).
Zierz, S. & I Maurer & Myocardial respiratory chain enzyme activities in anomalous origin of the left main coronary artery from the pulmonary trunk (bland-White-Garland syndrome) and comparison with atherosclerotic coronary artery disease. Am. J. Cardiol. 70, 1228–1231 (2019).
Khan, M. & Baussan, Y. Hebert-Chatelain, E. Connecting dots between mitochondrial dysfunction and depression. Biomolecules 13, 695 (2023).
Fernström, J. et al. Blood-based mitochondrial respiratory chain function in major depression. Transl Psychiatry. 11, 593 (2021).
Wu, L. et al. The clinical utility of circulating cell division control 42 in small-vessel coronary artery disease patients undergoing drug-coated balloon treatment. BMC Cardiovasc. Disord. 23, 496 (2023).
Li, J. et al. Eva1a ameliorates atherosclerosis by promoting re-endothelialization of injured arteries via Rac1/Cdc42/Arpc1b. Cardiovasc. Res. 117, 450–461 (2021).
Kalim, K. W. et al. Reciprocal Regulation of Glycolysis-Driven Th17 Pathogenicity and Regulatory T Cell Stability by Cdc42. J. Immunol. 200, 2313–2326 (2018).
Lv, J. et al. Endothelial Cdc42 deficiency impairs endothelial regeneration and vascular repair after inflammatory vascular injury. Respir Res. 19, 27 (2018).
Ma, H. et al. Linkage of blood cell division cycle 42 with T helper cells, and their correlation with anxiety, depression, and cognitive impairment in stroke patients. Braz J. Med. Biol. Res. 56, e12855 (2023).
Hanin, G. et al. Competing targets of microRNA-608 affect anxiety and hypertension. Hum. Mol. Genet. 23, 4569–4580 (2014).
Fang, X. et al. Overexpression cdc42 attenuates isoflurane-induced neurotoxicity in developmental brain of rats. Biochem. Biophys. Res. Commun. 490, 719–725 (2017).
Zhang, Z. et al. Hepatocellular carcinoma cells downregulate NADH:Ubiquinone Oxidoreductase Subunit B3 to maintain reactive oxygen species homeostasis. Hepatol. Commun. 8, e0395 (2024).
Casper, E. The crosstalk between Nrf2 and NF-κB pathways in coronary artery disease: can it be regulated by SIRT6? Life Sci. 330, 122007 (2023).
Spyridon Simantiris, C. et al. Oxidative stress biomarkers in coronary artery disease. Curr. Top. Med. Chem. 23, 2158–2171 (2023).
Bhatt, S., Nagappa, A. N. & Patil, C. R. Role of oxidative stress in depression. Drug Discov Today. 25, 1270–1276 (2020).
Vaváková, M., Ďuračková, Z. & Trebatická, J. Markers of Oxidative Stress and Neuroprogression in Depression Disorder. Oxid Med Cell Longev 1–12 (2015). (2015).
Hande Sipahi, A. F. et al. The interrelation between oxidative stress, Depression and inflammation through the Kynurenine Pathway. Curr. Top. Med. Chem. 23, 415–425 (2023).
Ghareeb, H. & Metanis, N. The Thioredoxin System: a Promising Target for Cancer Drug Development. Chemistry 26, 10175–10184 (2020).
Yunfei Wu, L., Zhong, L. & Yang & Decreased serum levels of thioredoxin in patients with coronary artery disease plus hyperhomocysteinemia is strongly associated with the disease severity. Atherosclerosis 212, 351–355 (2010).
Shateri, H. et al. Imbalance in thioredoxin system activates NLRP3 inflammasome pathway in epicardial adipose tissue of patients with coronary artery disease. Mol. Biol. Rep. 48, 1181–1191 (2021).
Aydın, E. P. et al. Thioredoxin is not a marker for treatment-resistance depression but associated with cognitive function: an rTMS study. Prog Neuropsychopharmacol. Biol. Psychiatry. 80, 322–328 (2018).
Zhou, H. et al. Increased thioredoxin-interacting protein in brain of mice exposed to chronic stress. Prog Neuropsychopharmacol. Biol. Psychiatry. 88, 320–326 (2019).
Jiang, T. et al. CDC42-A promising immune-related target in glioma. Front. Neurosci. 17, 1192766 (2023).
Shu, Q. et al. Identification and experimental validation of mitochondria-related genes biomarkers associated with immune infiltration for sepsis. Front. Immunol. 14, 1184126 (2023).
Benhar, M. et al. Dual targeting of the thioredoxin and glutathione systems in cancer and HIV. J. Clin. Invest. 126, 1630–1639 (2016).
Lisa, F. et al. Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the enhancing recovery in Coronary Heart Disease patients (ENRICHD) Randomized Trial. JAMA 289, 3106–3116 (2003).
Freedland, K. E. et al. Treatment of Depression after coronary artery bypass surgery. Arch. Gen. Psychiatry. 66, 387 (2009).
Glassman, A. H. et al. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 288, 701–709 (2002).
van Melle, J. P. et al. Effects of antidepressant treatment following myocardial infarction. Br. J. Psychiatry. 190, 460–466 (2018).
Devi SenGupta, C. et al. The TLR7 agonist vesatolimod induced a modest delay in viral rebound in HIV controllers after cessation of antiretroviral therapy. Sci. Transl Med. 13, eabg3071 (2021).
Usero, L. et al. The combination of an mRNA Immunogen, a TLR7 agonist and a PD1 blocking Agent enhances In-Vitro HIV T-Cell Immune responses. Vaccines 11, 286 (2023).
Karadimou, G. et al. Low TLR7 gene expression in atherosclerotic plaques is associated with major adverse cardio- and cerebrovascular events. Cardiovasc. Res. 113, 30–39 (2017).
Shafeghat, M. et al. Toll-like receptor 7 regulates cardiovascular diseases. Int. Immunopharmacol. 113, 109390 (2022).
Homberg, J. et al. Assessing the role of toll-like receptor in isolated, standard and enriched housing conditions. Plos One. 14, e0222818 (2019).
Acknowledgements
All authors thanked the Union Hospital affiliated to Huazhong University of Science for providing a platform to conduct this study.
Funding
This work was supported by the 2018 General Project of Hubei Provincial Health and Family Planning Commission (WJ2018H0120).
Author information
Authors and Affiliations
Contributions
Mengyun Hu, Rong Tan, Tao Liu, and Qin Wang designed the study. Caihong Lu and Ting Zhou conducted the bioinformatics analysis, while Mengyun Hu and Rong Tan focused on the molecular biological experiments. All authors contributed to writing and revising the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All animal experiments were approved by the Medical Ethics Committee of Union Hospital Affiliated to Huazhong University of Science and Technology. All experiments were conducted in compliance with the United Kingdom Animals (Scientific Procedures) Act 1986 and the American Veterinary Medical Association Guidelines for the Euthanasia of Animals (2020) and the ARRIVE guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, M., Tan, R., Lu, C. et al. Unveiling shared therapeutic targets and pathological pathways between coronary artery disease and major depressive disorder through bioinformatics analysis. Sci Rep 14, 29365 (2024). https://doi.org/10.1038/s41598-024-80920-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-80920-2
