
Abstract
Ischemic stroke (IS) is a leading global cause of mortality and disability, particularly prominent in patients with end-stage renal disease (ESRD). Despite clinical evidence of their comorbidity, the molecular mechanisms underlying their interaction remain elusive. This study aims to identify shared biomarkers, gene regulatory networks, and therapeutic targets through integrative bioinformatics analyses. Gene expression datasets for IS (GSE16561, GSE22255) and ESRD (GSE37171, GSE142153) were obtained from gene expression omnibus (GEO). Weighted gene co-expression network analysis (WGCNA) and differential expression genes (DEGs) analysis identified shared genes and enriched pathways. Protein-protein interaction networks were constructed using STRING with clustering algorithms. Immune cell infiltration analysis was performed via CIBERSORT. Transcriptional regulatory networks were predicted using RcisTarget and miRcode. Key gene expressions were validated by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) in clinical samples. We identified 417 intersecting genes through WGCNA and 1712 shared differentially expressed genes. Among these, seven key genes (MRPL49, MRPS2, MRPS9, MRPS10, MRPS11, MRPS27, TFB1M) demonstrated central roles in mitochondrial function. Immune infiltration analysis revealed significant correlations with T cells and neutrophils. Pathway enrichment implicated these genes in transforming growth factor beta (TGF-β) signaling, p53 pathway, and G2/M checkpoint. Clinical validation confirmed significant downregulation of MRPS9, MRPS10, MRPS11, MRPS27 and TFB1M in comorbid patients. This study systematically elucidates the mitochondrial-immune interaction mechanisms in IS-ESRD comorbidity, highlighting the pivotal role of mitochondrial ribosomal protein (MRP) family genes in regulating cellular energetics and inflammatory responses. These findings provide new foundations for targeted therapies.
Similar content being viewed by others
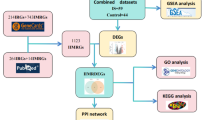


Introduction
Ischemic stroke (IS) is a condition characterized by sudden focal neurological deficits resulting from cerebral vascular infarction. Globally, it has emerged as one of the leading causes of long-term disability and mortality1,2. The etiology of IS is complex and includes multiple risk factors such as inflammation, calcium overload, and oxidative stress3. The high incidence and mortality associated with IS place a significant burden on patients, their families, and society at large. Similarly, end-stage renal disease (ESRD), the terminal stage of chronic kidney disease, is characterized by severely impaired renal function that is insufficient to maintain normal metabolic homeostasis in the body4. As with IS, the high prevalence and mortality of ESRD lead to profound social and economic consequences, further compounding the challenges faced by affected individuals and healthcare systems.
IS and ESRD are significant global public health challenges that share overlapping pathophysiologic processes and epidemiological links. Recent studies indicate a marked increase in the incidence of stroke among ESRD patients. Patients undergoing maintenance dialysis have a 4- to 10-fold increased risk of stroke compared to the general population5. The incidence and mortality of stroke are notably elevated in dialysis patients with chronic kidney disease, with mortality rates often being as much as 9 times higher than in the general population6,7. A longitudinal observational study conducted in Japan by Ueki et al. found that lower estimated glomerular filtration rate (eGFR) and higher proteinuria were independently associated with increased risks of recurrent stroke and all-cause mortality in ESRD patients8. In addition to traditional stroke risk factors such as dyslipidemia, hypertension, and diabetes, patients with ESRD face dialysis-specific risk factors. These include accelerated calcific arteriosclerosis, the effects of uremic toxins, dialysis techniques, vascular access complications, and the use of anticoagulants to maintain blood flow during extracorporeal circulation9. Furthermore, post-renal failure conditions, including inflammation, oxidative stress, anemia, reduced nitric oxide levels, platelet aggregation, and elevated homocysteine levels, contribute to vascular endothelial dysfunction. These changes exacerbate the risk of stroke10,11. Altered immune cell proportions in ESRD patients has been implicated in exacerbating stroke risk, primarily through mechanisms involving inflammation and vascular injury. However, the roles of altered immune cell profiles and inflammatory factors in directly damaging neurons and cerebral vasculature in ESRD patients remain underexplored in research12,13. Although evidence supports an association between IS and ESRD, the precise mechanisms underlying their comorbidity remain poorly understood14,15. A comprehensive understanding of the interplay between these two diseases is crucial for developing personalized and effective management strategies for affected patients.
With the rapid advancement of bioinformatics and the increasing adoption of gene chip technologies in recent years, bioinformatics analysis has become an indispensable tool in biomedical research. Shared transcriptional patterns in IS and ESRD provide new insights into their underlying etiologies. In this study, we utilized four microarray datasets (GSE16561, GSE22255, GSE373171, and GSE142153) from the Gene Expression Omnibus (GEO) database to analyze gene expression patterns in IS and ESRD patients. Differential expression genes (DEGs) in IS and ESRD were identified through comprehensive bioinformatics approaches, including functional enrichment analyses to elucidate their biological significance. Additionally, Weighted gene co-expression network analysis (WGCNA) was employed to identify co-expression modules and common differential genes shared between the two diseases. Core regulatory networks were further analyzed using STRING and Cytoscape, enabling an in-depth exploration of their structural and functional characteristics. Furthermore, the ImmuCellAI tool was used to quantitatively analyze 22 immune cell subtypes and assess their associations with IS and ESRD progression. Key signaling pathways implicated in the progression of these diseases were identified through gene set variation analysis (GSVA) and gene set enrichment analysis (GSEA). Potential microRNA (miRNA) regulatory factors involved in these pathways were predicted through the miRcode database. Finally, the expression levels and correlations of the identified comorbid genes in IS and ESRD were validated through clinical samples and data, providing further insight into their roles in disease progression. This study employed comprehensive bioinformatics methodologies, integrating differential gene expression analysis, key gene identification, and immune correlation validation, to systematically uncover shared molecular pathways and potential therapeutic targets in IS and ESRD. These findings offer novel insights for developing targeted interventions, with the ultimate goal of improving patient outcomes and quality of life.
Methods
Data download
The gene expression data in this study were obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/), a public repository curated by the National Center for Biotechnology Information (NCBI). The datasets include two IS cohorts, GSE16561 (Illumina platform GPL6883) and GSE22255 (Affymetrix platform GPL570), along with two ESRD cohorts, GSE37171 (Affymetrix platform GPL570) and GSE142153 (Agilent platform GPL6480). All raw data were retrieved from GEO Series Matrix Files, and gene identifiers were mapped using platform-specific probe annotation files. Comprehensive dataset details (e.g., sample size, experimental groups) are provided in Supplementary File 1.
Differential expression analysis
Limma package is an R software package for differential expression analysis of expression profiles to identify significantly differentially expressed genes between group comparisons16. Use the R package “Limma” to analyze the differences in the molecular mechanisms of IS and ESRD comorbidity data, and identify differentially expressed genes between control samples and disease samples. The screening criteria for significant differential expression were defined as log2|fold change (FC)|>1 and p value < 0.05, and Plot differential gene volcano plots and heat maps.
Functional enrichment analysis
Functional annotation of differentially expressed genes was performed using ClusterProfiler to comprehensively explore the functional correlation of these differentially expressed genes17. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) were used to evaluate relevant functional categories18,19. GO and KEGG enriched pathways with both p-value and q-value less than 0.05 were considered as significant categories.
WGCNA analysis
By constructing a weighted gene co-expression network, we can find cooperatively expressed gene modules and explore the correlation between gene networks and diseases, as well as the core genes in the network. The WGCNA-R package was used to construct a co-expression network of all genes in the data set20. The weighted adjacency matrix is converted into a topological overlap matrix (TOM) to estimate the network connectivity, and the hierarchical clustering method is used to construct the clustering tree structure of the TOM matrix. Different branches of the clustering tree represent different gene modules, and different colors represent different modules. Based on the weighted correlation coefficient of genes, genes are classified according to expression patterns, genes with similar patterns are grouped into one module, and all genes are divided into multiple modules based on gene expression patterns.
Immune cell infiltration analysis
The CIBERSORT method is a widely used method to evaluate immune cell types in the microenvironment21. This method is based on the principle of support vector regression and performs deconvolution analysis on the expression matrix of immune cell subtypes. It contains 547 biomarkers that distinguish 22 human immune cell phenotypes, including T cells, B cells, plasma cells, and myeloid cell subsets. This study uses the CIBERSORT algorithm to analyze patient data to infer the relative proportions of 22 types of immune infiltrating cells, and conduct correlation analysis on gene expression and immune cell content.
Transcriptional regulation analysis of key genes
This study used the R package “RcisTarget” to predict transcription factors. All calculations performed by RcisTarget are based on motifs22. The normalized enrichment score (NES) of a motif depends on the total number of motifs in the database. In addition to the motifs annotated by the source data, we inferred further annotation files based on motif similarity and gene sequence. The first step in estimating the overexpression of each motif on a gene set is to calculate the area under the curve (AUC) for each motif-motif set pair. This was performed based on recovery curve calculations of the gene set against the ordering of the motifs. The NES of each motif is calculated based on the AUC distribution of all motifs in the gene set.
MiRNA network construction
miRNAs are small non-coding RNAs (ncRNAs) that have been shown to regulate gene expression by promoting the degradation of messenger RNAs (mRNAs) or inhibiting the translation of mRNAs. Therefore, we further analyzed whether there are some miRNAs in key genes that regulate the transcription or degradation of some dangerous genes. We obtained key gene-related miRNAs through the miRcode database and visualized the gene’s miRNA network through Cytoscape software23.
GSVA analysis
GSVA is a non-parametric and unsupervised method to evaluate transcriptome gene set enrichment24. GSVA converts gene-level changes into pathway-level changes by comprehensively scoring the gene set of interest, and then determines the biological function of the sample. This study will download gene sets from the Molecular signatures database (version 7.0), and use the GSVA algorithm to comprehensively score each gene set to evaluate potential biological function changes in different samples.
GSEA analysis
GSEA analysis uses a predefined gene set to rank genes according to their differential expression levels in two types of samples, and then tests whether the preset gene set is enriched at the top or bottom of this ranking list25. This study uses GSEA to compare the differences in signaling pathways between the high expression group and the low expression group, and explore the molecular mechanism of the core genes of the two groups of patients. The number of substitutions is set to 1000, and the substitution type is set to phenotype.
Analysis by reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
From July 2023 to April 2024, we enrolled ten adult patients with both ESRD and IS in this study. Diagnosis of ESRD followed the KDIGO (Kidney Disease: Improving Global Outcomes) guidelines26, with inclusion criteria such as a glomerular filtration rate (GFR) consistently below 15 mL/min/1.73 m2, or proteinuria, hematuria, or structural abnormalities observed over three months. IS diagnosis adhered to the American Heart Association (AHA) and American Stroke Association (ASA) guidelines27, which required evidence of acute focal neurological deficits and ischemic areas confirmed by imaging. AND ten healthy individuals were recruited as controls. Blood samples (1 mL) were collected from each participant. This study adhered to the Declaration of Helsinki, and all procedures received approval from our hospital’s ethics committee, with informed consent obtained from all participants. Total RNA from whole blood was isolated using RNAiso Plus (Takara Bio, Japan) and and reverse transcribed Hifair® II 1st Strand cDNA Synthesis Kit (Yeasen Biotechnology, Shanghai). The PCR analysis was done on an ABI ViiA 7 system using Hieff® qPCR SYBR Green Master Mix(Low Rox Plus) (Yeasen Biotechnology, Shanghai) in triplicate. The 2–△△Ct method was used tocalculate the relative expressions of the target genes, with GAPDH as the housekeeping gene. All the sequences of the primers, melting curves, and amplification curves are listed in Supplementary File 2.
Statistical analysis
All statistical analyses were conducted using R (version 4.2.2) and GraphPad Prism 9. Two-sided tests were applied, with p < 0.05 deemed statistically significant.
Results
-
1.
We downloaded the GSE16561 and GSE22255 data sets from the NCBI GEO public database, a total of 103 IS samples, including 44 cases in the control group and 59 cases in the disease group. We used the Combat algorithm to correct the chip, and used the PCA chart to show the results before and after correction. discrepancy situation. The results show that the inter-chip batch effect is reduced after Combat algorithm correction (Fig. 1A, B). Use the limma package to calculate the differential genes between the two groups of patients. The differential gene screening conditions are: p < 0.05, and 3657 differential genes were obtained, of which 1420 were up-regulated and 2237 were down-regulated (Fig. 1C, D) .

PCA correction and differential expression analysis. (A) PCA plot of IS datasets (GSE16561 and GSE22255) before Combat correction. (B) PCA plot of IS datasets after Combat correction. (C) Volcano plot of IS differential expression analysis. 3657 DEGs identified (1420 upregulated, 2237 downregulated. (D) Heatmap of top 50 DEGs in IS. Yellow: high expression; green: low expression. (E) PCA plot of ESRD datasets (GSE37171 and GSE142153) before Combat correction. (F) PCA plot of ESRD datasets after Combat correction. (G) Volcano plot of ESRD differential expression analysis. 12659 DEGs identified (7058 upregulated, 5601 downregulated). (H) Heatmap of top 50 DEGs in ESRD. (I) Venn diagram of shared upregulated DEGs between IS and ESRD (594 genes). (J) Venn diagram of shared downregulated DEGs between IS and ESRD (1118 genes).
-
2.
Download the GSE37171 and GSE142153 data sets from the NCBI GEO public database, a total of 132 ESRD samples, including 50 cases in the control group and 82 cases in the disease group. Use the Combat algorithm to correct the chip, and use the PCA chart to show the difference before and after correction. The results show that the inter-chip batch effect is reduced after Combat algorithm correction (Fig. 1E, F). Use the limma package to calculate the differential genes between the two groups of patients. The differential gene screening conditions are: p < 0.05, and 12,659 differential genes were obtained, of which 7058 were up-regulated and 5601 were down-regulated (Fig. 1G, H). Next, we took the intersection of the differential genes of the two diseases obtained in the previous step. There were 594 up-regulated intersections (Fig. 1I) and 1118 down-regulated intersections (Fig. 1J). We further conducted pathway analysis on the intersection genes. The GO results showed that, Genes are mainly enriched in pathways such as ncRNA processing, tRNA processing, and tRNA metabolic process (Fig. 2A). KEGG results show that genes are mainly enriched in pathways such as DNA replication (Fig. 2B).

Functional enrichment of shared DEGs (A) GO analysis reveals significant enrichment in biological processes such as ncRNA processing and tRNA metabolism. (B) KEGG pathways including DNA replication.
-
3.
In order to confirm the key genes in the IS and ESRD cohorts, we further constructed the WGCNA network based on the expression profile data. In the IS data set, the soft threshold β is set to 18 (Fig. 3A), and then the gene module is detected based on the tom matrix. A total of 4 gene modules were detected in this analysis (Fig. 3B, C). They are black (3520), blue (1789), gray (6962), and pink (933), among which the blue module has the highest correlation with disease (cor = 0.43, p = 6e-6). In the ESRD data set, the soft threshold β was set to 11 (Fig. 3D), and then the gene modules were detected based on the tom matrix. A total of 5 gene modules were detected in this analysis (Fig. 3E, F), respectively blue (5005), gray (4110), read (369), turquoise (6824), yellow (477), among which the blue module has the highest correlation with disease (cor = 0.72, p = 3e-22).

WGCNA (A) Scale independence and mean connectivity analysis for IS WGCNA. (B) Hierarchical clustering dendrogram of IS genes. Four modules identified: black, blue, gray and pink. (C) Module-trait correlation heatmap for IS. Blue module shows the highest correlation with disease (cor = 0.43, p = 6e-6). (D) Scale independence and mean connectivity analysis for ESRD WGCNA. (E) Hierarchical clustering dendrogram of ESRD genes. Five modules detected: blue, gray, red, turquoise and yellow. (F) Module-trait correlation heatmap for ESRD. Blue module shows the strongest disease association (cor = 0.72, p = 3e-22). (G) Venn diagram of shared genes between IS and ESRD blue modules. Overlapping region contains 417 genes. (H) Intersection of blue module genes and DEGs identifies 237 key genes.
-
4.
To gain a deeper understanding of the molecular interactions among the genes within these modules and their pivotal roles in disease progression, we constructed a protein-protein interaction (PPI) network. Clustering analysis was subsequently performed to identify key genes, providing insights into their potential contributions to the disease’s underlying mechanisms. We intersected the module genes with the highest correlation between the two data sets and obtained 417 intersection genes (Fig. 3G). Finally, we intersected these 417 intersection genes with the previous 1712 differential intersection genes, and obtained 237 intersection genes (Fig. 3H). In order to further find out the key genes among these 237 genes, we used the STRING database (http://cn.string-db.org) construct a protein interaction network of common targets and use Cytoscape to visualize it (Fig. 4A). Through MCODE cluster analysis, we get 5 modules, and the scores in the modules The top seven genes are our key genes (Fig. 4B), which are: MRPL49, MRPS2, MRPS9, MRPS10, MRPS11, MRPS27, and TFB1M.

PPI network and key gene identification (A) PPI network of 237 shared genes constructed via STRING database and visualized in Cytoscape. Node size reflects connectivity, red nodes indicate hub genes. (B) Top-scoring module from MCODE clustering. Key genes (MRPL49, MRPS2, MRPS9, MRPS10, MRPS11, MRPS27 and TFB1M) all belonging to the MRP family.
-
5.
The microenvironment is mainly composed of immune cells, extracellular matrix, various growth factors, inflammatory factors and special physical and chemical characteristics, which significantly affects the diagnosis of disease and the sensitivity of clinical treatment. We further explored the potential molecular mechanisms by which key genes influence the progression of IS by analyzing the relationship between key genes and immune infiltration in the IS data set. In the IS data set, the proportion of immune cell content in each patient and the correlation between immune cells (Fig. 5A, B). In addition, the results showed that compared with control patients, T cells gamma delta, Neutrophils, Macrophages M0, Mast cells activated, etc. in the disease group were significantly higher than those in the control group (Fig. 5C). We further explored the relationship between key genes and immune cells, and the results showed that MRPL49 had a significant positive correlation with T cells CD8, B cells naive, and T cells follicular helper, and a significant negative correlation with Neutrophils, Macrophages M0, and Monocytes (Fig. 5D); MRPS2 has a significant positive correlation with T cells CD8, B cells naive, T cells regulatory (Tregs), etc., and Neutrophils, Macrophages M0 (Fig. 5E); MRPS9 has a significant positive correlation with T cells CD8, B cells naive, etc., and a significant negative correlation with Neutrophils, Mast cells activated, etc. (Fig. 5F); MRPS10 has a significant positive correlation with T cells CD8, B cells naive, etc., and Mast cells activated, Neutrophils, etc. showed a significant negative correlation (Fig. 5G); MRPS11 had a significant positive correlation with T cells CD8, Macrophages M2, etc., and a significant negative correlation with Neutrophils, Mast cells activated, etc. (Fig. 5H); MRPS27 There is a significant positive correlation with T cells CD8, T cells CD4 memory resting, etc., and a significant negative correlation with Neutrophils, Macrophages M0, etc. (Fig. 5I); TFB1M has a significant positive correlation with T cells CD8, T cells regulatory (Tregs), etc. Correlated, and significantly negatively correlated with Mast cells activated, Macrophages M0, etc. (Fig. 5J). We obtained the correlation between these key genes and different immune factors from the tumor-immune system interaction database (TISIDB) (http://cis.hku.hk/TISIDB/), including chemokines, immunoinhibitors, immunostimulators, major histocompatibility complexes (MHC) and receptors (Fig. 6). These analyzes suggest that key genes are closely related to the level of immune cell infiltration and play an important role in the immune microenvironment.

Immune cell infiltration in IS (A) The relative proportions of 22 immune cells in the control and disease groups. (B) Pearson correlation between the 22 immune cells, with blue indicating a negative correlation and red indicating a positive correlation. (C) Differences in immune cell counts between the control and disease groups, with blue representing control patients and yellow representing disease patients. (D–J) Correlation between key genes (MRPL49, MRPS2, MRPS9, MRPS10, MRPS11, MRPS27 and TFB1M) and immune cell counts.
-
6.
In the ESRD data set, the proportion of immune cells in each patient and the correlation between immune cells are shown in the figure (Fig. 7A, B). In addition, the research results show that compared with the control Compared with the patients in the disease group, B cell memory, Monocytes, and Neutrophils in the disease group were significantly higher than those in the control group (Fig. 7C). We further explored the relationship between key genes and immune cells, and the results showed that MRPL49 had a significant positive correlation with T cells CD8, B cells naive, etc., and a significant negative correlation with Macrophages M0, B cells memory, etc. (Fig. 7D); MRPS2 has a significant positive correlation with T cells CD8, B cells naive, etc., and Neutrophils, B cells memory (Fig. 7E); MRPS9 has a significant positive correlation with T cells CD8, B cells naive, etc., and Neutrophils, Mast cells activated, etc. There was a significant negative correlation (Fig. 7F); MRPS10 had a significant positive correlation with B cells naive, NK cells resting, etc., and a significant negative correlation with Macrophages M1, Monocytes, etc. (Fig. 7G); MRPS11 had a significant positive correlation with T cells CD8, There is a significant positive correlation with B cells naive, etc., and a significant negative correlation with Neutrophils, B cells memory, etc. (Fig. 7H); MRPS27 has a significant positive correlation with B cells naive, T cells CD8, etc., and a significant negative correlation with Neutrophils, Macrophages M1, etc. (Fig. 7I); TFB1M has a significant positive correlation with B cells naive, NK cells resting, etc., and has a significant negative correlation with T cells gamma delta, B cells naive, etc. showed significant negative correlation (Fig. 7G). We obtained the correlation between these key genes and different immune factors from the TISIDB database, including chemokines, immunoinhibitors, immunostimulators, MHC and receptors (Fig. 8). These analyzes suggest that key genes are closely related to the level of immune cell infiltration and play an important role in the immune microenvironment.

The correlation between key genes and immune factors in IS (A–E) The correlation between key genes (MRPL49, MRPS2, MRPS9, MRPS10, MRPS11, MRPS27 and TFB1M) and chemokines, immunoinhibitors, immunostimulators, MHC and receptors.

Immune cell infiltration in ESRD (A) The relative proportions of 22 immune cells in control and disease groups. (B) Pearson correlation among the 22 immune cells, with blue indicating a negative correlation and red indicating a positive correlation. (C) Differences in immune cell counts between control and disease groups, with blue representing control patients and yellow representing disease patients. (D–J) Correlation between key genes (MRPL49, MRPS2, MRPS9, MRPS10, MRPS11, MRPS27 and TFB1M) and immune cell counts.

The correlation between key genes and immune factors in ESRD (A–E) The correlation between key genes (MRPL49, MRPS2, MRPS9, MRPS10, MRPS11, MRPS27 and TFB1M) and chemokines, immunoinhibitors, immunostimulators, MHC and receptors.
-
7.
We next study the specific signaling pathways enriched in 7 key genes and explore the potential molecular mechanisms by which key genes affect the progression of IS and ESRD. The GSVA results in IS show that highly expressed MRPL49 is enriched in signaling pathways such as DNA repair and Myc targets (Fig. 9A); highly expressed MRPS2 is enriched in signaling pathways such as fatty acid metabolism and mTORC1 signaling (Fig. 9B); highly expressed MRPS9 is enriched in concentrated in Wnt/β-catenin signaling, peroxisome and other signaling pathways (Fig. 9C); Highly expressed MRPS10 is enriched in p53 pathway, adipogenesis and other signaling pathways (Fig. 9D); Highly expressed MRPS11 is enriched in Wnt/β-catenin signaling, interferon-gamma response and other signaling pathways (Fig. 9E); Highly expressed MRPS27 is enriched in signaling pathways such as G2/M checkpoint and protein secretion (Fig. 9F); highly expressed TFB1M is enriched in signaling pathways such as transforming growth factor beta (TGF-β) signaling pathway and peroxisome (Fig. 9G). This suggests that key genes may influence the progression of IS through these pathways. In addition, GSEA results show that the pathways enriched by MRPL49 include Base excision repair, RNA polymerase, Spliceosome and other pathways (Fig. 10A); the pathways enriched by MRPS2 include DNA replication, Nucleotide excision repair, RNA degradation, etc. Pathway (Fig. 10B); The pathways enriched by MRPS9 include Mismatch repair, Nucleotide metabolism, Proteasome and other pathways (Fig. 10C); the pathways enriched by MRPS10 include mRNA surveillance pathway, Nucleotide excision repair, Propanoate metabolism and other pathways (Fig. 10D); The pathways enriched by MRPS11 include aminoacyl − tRNA biosynthesis, focal adhesion, propanoate metabolism and other pathways (Fig. 10E); The pathways enriched by MRPS27 include Bile secretion, ECM − receptor interaction, Nitrogen metabolism and other pathways (Fig. 10F); the pathways enriched by TFB1M include Nucleocytoplasmic transport, Nucleotide metabolism, Oxidative phosphorylatio and other pathways (Fig. 10G).

The GSVA of key genes in IS (A–G) GSVA of key genes (MRPL49, MRPS2, MRPS9, MRPS10, MRPS11, MRPS27 and TFB1M) with blue representing pathways linked to high expression genes and green representing pathways linked to low expression genes.

The GSEA of key genes in IS (A–G) KEGG pathways correlated with key genes (MRPL49, MRPS2, MRPS9, MRPS10, MRPS11, MRPS27 and TFB1M).
-
8.
In ESRD, GSVA results show that highly expressed MRPL49 is enriched in signaling pathways such as TGF-β signaling and G2/M checkpoint (Fig. 11A); highly expressed MRPS2 is enriched in signaling pathways such as adipogenesi and MYC targets V2 pathways (Fig. 11B); Highly expressed MRPS9 is enriched in signaling pathways such as IL2-STAT5 signaling and TGF-β signaling pathways (Fig. 11C); Highly expressed MRPS10 is enriched in signaling pathways such as PI3K-Akt-mTOR signaling and p53 pathways (Fig. 11D); Highly expressed MRPS11 is enriched in signaling pathways such as G2/M checkpoint and MYC targets V2 pathways (Fig. 11E); Highly expressed MRPS27 is enriched in signaling pathways such as Notch signaling and IL2-STAT5 signaling pathways (Fig. 11F); Highly expressed TFB1M is enriched in signaling pathways such as mTORC1 signaling and DNA repair pathways (Fig. 11G). This suggests that key genes may influence the progression of ESRD through these pathways. In addition, GSEA results show that the pathways enriched by MRPL49 include Apoptosis, TNF signaling pathway, Toll-like receptor signaling pathway and other pathways (Fig. 12A); The pathways enriched by MRPS2 include Cell adhesion molecules, Cell cycle, Th17 cell differentiation and other pathways (Fig. 12B); the pathways enriched by MRPS9 include GnRH signaling pathway, Necroptosis, Oxytocin signaling pathway and other pathways (Fig. 12C); pathways enriched by MRPS10 include Calcium signaling pathway, Synaptic vesicle cycle, TNF signaling pathway (Fig. 12D); pathways enriched by MRPS11 include Complement and coagulation cascades, GABAergic synapse, Glutamatergic synapse and other pathways (Fig. 12E); MRPS27 enriched pathways include autophagy-animal, Circadian rhythm, Ribosome biogenesis in eukaryotes and other pathways (Fig. 12F); TFB1M enriched The pathways include cAMP signaling pathway, Circadian entrainment, ECM-receptor interaction and other pathways (Fig. 12G).

The GSVA of key genes in ESRD (A–G) GSVA of key genes (MRPL49, MRPS2, MRPS9, MRPS10, MRPS11, MRPS27 and TFB1M), with blue representing pathways linked to high expression genes and green representing pathways linked to low expression genes.

The GSEA of key genes in ESRD (A–G) KEGG pathways correlated with key genes (MRPL49, MRPS2, MRPS9, MRPS10, MRPS11, MRPS27 and TFB1M).
9. We used these 7 key genes as the gene set for this analysis and found that they are regulated by common mechanisms such as multiple transcription factors. Therefore, enrichment analysis of these transcription factors was performed using cumulative recovery curves. The results of motif-TF annotation and selection analysis of important genes show that the motif with the highest normalized enrichment score (NES: 11.7) is cisbp__M4151. We display all enriched motifs and corresponding transcription factors of key genes (Fig. 13A, B). In addition, we performed reverse prediction on 7 key genes through the mircode database and obtained 85 miRNAs, a total of 191 pairs of mRNA-miRNA relationships, and visualized them using Cytoscape (Fig. 13C).

The transcriptional regulation and miRNA network of key genes (A) Transcriptional regulation of key genes, with red nodes indicating key genes and green nodes indicating transcription factors. (B) Display of all motifs enriched for key genes and their corresponding transcription factors. (C) The miRNA network of key genes, with orange circles representing mRNAs and blue arrows representing miRNAs.
10. To further validate the actual expression levels and potential molecular functions of these genes in IS and ESRD patients, we conducted RT-qPCR. The mRNA expression of key genes in whole blood samples from IS-ESRD patients was analyzed using RT-qPCR. The results demonstrated that five genes (MRPS9, MRPS10, MRPS11, MRPS27, and TFB1M) were significantly down-regulated in IS-ESRD patients when compared with healthy controls (Fig. 14), the analysis results are detailed in Supplementary File 3. These findings indicate the diagnostic potential and significance of these genes in IS-ESRD patients. In summary, through multi-level bioinformatics analyses and experimental validation, we revealed the comorbid molecular mechanisms of IS and ESRD, identified key genes and their immune regulatory functions, and provided important insights for further exploration of therapeutic targets.

RT-qPCR validation of key genes. Transcription levels of MRPL49, MRPS2, MRPS9, MRPS10, MRPS11, MRPS27, TFB1M mRNA were analyzed in IS-ESRD patients (n = 10) and healthy controls (n = 10) (*p < 0.05, **p < 0.01, ***p < 0.001; ns not significant). Error bars: SEM; GAPDH as housekeeping gene.
Discussion
The increasing prevalence of IS and ESRD represented significant global public health challenges, severely impacting patients’ quality of life. Epidemiological evidence showed that ESRD patients, particularly those undergoing dialysis, were at an elevated risk of stroke compared to the general population, with both stroke incidence and mortality rates being markedly higher in this group28,29,30. This increased risk was primarily attributed to the complex metabolic disturbances and pathophysiological alterations commonly observed in ESRD patients. Emerging research had revealed substantial molecular overlaps between IS and ESRD, particularly in the context of metabolic abnormalities. One of the key shared features in both conditions was oxidative stress, alongside inflammation, which underlies many of the pathophysiological changes observed in these diseases31,32. In IS, hypoxia resulting from disrupted cerebral blood flow induced energy metabolism dysfunction, exacerbating oxidative stress, promoting apoptosis, and amplifying neurological damage33,34. Similarly, in ESRD, the accumulation of metabolic wastes and electrolyte imbalances due to renal failure severely disrupted normal cellular metabolic processes, leading to cellular dysfunction and tissue injury35,36. The overproduction of reactive oxygen species (ROS) and other free radicals in both diseases played a critical role in these processes. ROS-induced lipid peroxidation, protein denaturation, and DNA damage contributed to cell death via apoptosis or necrosis. These mechanisms not only enhanced neurological damage and brain tissue injury in IS but also promoted chronic inflammation and systemic metabolic dysregulation in ESRD. Thus, cellular dysfunction driven by oxidative stress is increasingly recognized as a shared and pivotal mechanism underlying both IS and ESRD.
In this study, we employed bioinformatics techniques to systematically screen 237 DEGs shared by IS and ESRD and identified seven key genes: MRPL49, MRPS2, MRPS9, MRPS10, MRPS11, MRPS27, and TFB1M. These genes were all members of the mitochondrial ribosomal protein (MRP) family, which played critical roles in maintaining mitochondrial protein synthesis and overall mitochondrial function. Previous studies have demonstrated that mutations or dysregulated expression of MRP family genes can lead to mitochondrial dysfunction, disrupting cellular energy metabolism and apoptosis regulation37,38,39. Validation through RT-qPCR revealed that MRPS9, MRPS10, MRPS11, MRPS27, and TFB1M were significantly downregulated in both IS and ESRD patients. These findings suggest that the downregulation of these genes might contribute to disease progression by impairing mitochondrial function and energy metabolism. In particular, MRPS9 had been shown to regulate cell proliferation, apoptosis, and mitochondrial function. It was closely associated with neuroinflammation and neuroprotection, specifically regulating the PI3K/Akt/mTOR pathway, which was crucial for neuronal survival following ischemia. MRPS9 inhibited caspase-3 activation and Bax translocation, thereby protecting neurons from ischemic injury40. In the context of ESRD, reduced MRPS9 expression was associated with renal fibrosis and decreased renal function, indicating its role in driving disease progression by impairing mitochondrial function and cellular energy metabolism41. Similarly, abnormal expression of MRPS10, a constituent protein of the large subunit of the mitochondrial ribosome, was implicated in both IS and ESRD. In kidney tissues from IS and ESRD patients, MRPS10 was significantly downregulated, which likely interfered with mitochondrial translation, resulting in defective assembly of respiratory chain complexes and exacerbating energy metabolism collapse. MRPS11, which was involved in regulating the mitochondrial stress response and apoptotic processes, also played a significant role in both diseases. A decrease in MRPS11 expression following ischemic brain injury suggested that its protective role in promoting neuronal survival after stroke was diminished42. In ESRD, downregulation of MRPS11 impairs mitochondrial function and elevated oxidative stress levels, further exacerbating renal injury and inflammation43. MRPS27, an essential component of the small subunit of mitochondrial ribosomes, was particularly critical under cellular stress conditions, especially in maintaining mitochondrial function44. Research showed that MRPS27 was significantly downregulated after ischemia, leading to mitochondrial dysfunction and promoting apoptosis45,46. In ESRD, decreased MRPS27 expression was closely associated with pathological processes such as renal fibrosis and inflammation, particularly in the development of chronic kidney disease47. Our RT-qPCR results aligned with these findings, suggesting that the downregulation of MRPS9, MRPS10, MRPS11, and MRPS27 exacerbated mitochondrial dysfunction and ATP depletion in patients with both IS and ESRD, amplifying neuronal and renal damage. Finally, TFB1M, a mitochondrial transcription factor, played a key role in regulating mitochondrial gene transcription, particularly under ischemic conditions. Dysfunction of TFB1M impairs the electron transport chain (ETC) complex, which disrupts energy metabolism and exacerbated oxidative stress48. In IS, downregulation of TFB1M impaired neuronal recovery from hypoxia, while in ESRD, abnormal TFB1M activity was linked to mitochondrial metabolic imbalance in renal cells, accelerating the deterioration of renal function48,49.In summary, the findings of this study emphasized the critical role of MRPS9, MRPS10, MRPS11, MRPS27, and TFB1M in the pathological mechanisms shared by IS and ESRD, particularly in their effects on mitochondrial function, oxidative stress, and apoptosis regulation. The dysregulation of these genes provided valuable insights into the molecular basis of both diseases and identified potential therapeutic targets for future clinical interventions, which might lead to breakthroughs in treatment.
Through GSVA and GSEA pathway enrichment analyses, several key pathways closely associated with the identified genes were identified, including the TGF-β signaling pathway, the p53 pathway, and the G2/M checkpoint. These pathways played pivotal roles in the pathophysiological mechanisms underlying both IS and ESRD. The TGF-β signaling pathway was central to the progression of both diseases, primarily through its regulation of fibrosis and inflammatory responses. In IS, TGF-β signaling influenced stroke progression by regulating neuroinflammation and the permeability of the blood-brain barrier (BBB). Overactivation of TGF-β triggered the release of pro-inflammatory cytokines such as Interleukin-1β (IL-1β), Tumor Necrosis Factor-alpha (TNF-α), and Interleukin-6 (IL-6) from microglia and astrocytes, thereby exacerbating brain tissue damage50. Moreover, TGF-β induced extracellular matrix remodeling, leading to cerebral fibrosis and limiting stroke recovery. In ESRD, TGF-β was the key regulatory factor responsible for tubulointerstitial fibrosis. Chronic activation of TGF-β upregulates the expression of α-smooth muscle actin and type I and III collagen through both SMAD-dependent and SMAD-independent pathways, promoting the transdifferentiation of renal tubular epithelial cells (TECs) into fibroblasts. This process culminates in renal parenchymal fibrosis and functional failure51. The p53 pathway exhibits a dynamic, dual role in both IS and ESRD. In IS, p53 limits the expansion of ischemic infarction by clearing irreversibly damaged neurons in the penumbra through activation of mitochondrial apoptotic pathways. However, its overexpression inhibits neural stem cell proliferation and angiogenesis while stimulating microglia to release pro-inflammatory cytokines such as IL-1β and TNF-α, which disrupt the BBB and exacerbate cerebral edema and secondary injury52. In ESRD, p53 alleviates the inflammatory microenvironment in the short term by inducing apoptosis of renal TECs to remove damaged cells. However, long-term activation of p53 leads to TEC senescence and fibrotic transformation, driving interstitial fibrosis and accelerating renal function deterioration53. This dual role highlights the delicate balance p53 maintains between cell clearance and tissue repair, with its dysregulation potentially resulting in either insufficient repair or excessive damage. The G2/M checkpoint, a critical regulator of the cell cycle, contributes to disease progression in both conditions through genomic instability. In IS, ischemia-induced neuronal entry into the G2/M phase results in apoptosis due to failed cell division, thus expanding the infarct core. Concurrently, G2/M phase arrest in astrocytes upregulates Matrix Metalloproteinase-9, disrupting the BBB and promoting the release of ROS and IL-6 by microglia, which aggravates neuroinflammation and cerebral fibrosis54. In ESRD, DNA damage or oxidative stress in renal TECs induces G2/M phase arrest, leading to dysregulation of the Cyclin B1/CDK1 complex and forcing cells into senescence. This process activates surrounding fibroblasts through the secretion of the senescence-associated secretory phenotype (SASP) and other inflammatory factors, creating a pro-fibrotic positive feedback loop55. These mechanisms illustrate that G2/M checkpoint dysregulation not only directly induces genomic instability but also amplifies inflammation and fibrosis through paracrine signaling, serving as a key driver of disease progression. The identification of these pathways further underscored the critical roles of the key genes in the shared pathophysiology of IS and ESRD. Additionally, these findings offer valuable insights for developing therapeutic and diagnostic strategies. Targeting these pathways holds promise for novel interventions aimed at improving patient outcomes in both diseases. For example, small-molecule inhibitors targeting the TGF-β pathway, such as galunisertib, have shown potential in preclinical studies for addressing both fibrosis in ESRD and neuroinflammation in IS56. Similarly, enhancing mitochondrial biogenesis with AMPK activators may help restore energy homeostasis in both diseases57. Furthermore, MRPS9 and MRPS11 may serve as potential biomarkers for early disease detection, with circulating mitochondrial DNA (mtDNA) levels serving as indicators of disease severity57. Thus, interventions targeting these pathways not only offer the potential for improved therapeutic outcomes but also lay the foundation for early diagnosis and personalized treatment strategies.Immune cell infiltration plays a pivotal role in the pathophysiology of both IS and ESRD. Studies have demonstrated that immune cells infiltrate extensively into brain tissue in IS patients, exacerbating neuronal damage through the release of inflammatory mediators58,59. Similarly, significant immune cell infiltration has been observed in renal tissues of ESRD patients60,61. In this study, the CIBERSORT algorithm was employed to analyze the proportion and distribution of immune cells in both IS and ESRD patients. The results revealed that candidate genes such as MRPL49 and MRPS27 were closely associated with immune cell infiltration, particularly with neutrophils and T cells. Specifically, MRPL49 expression showed a positive correlation with CD8 + T cells, suggesting its potential role in regulating adaptive immune responses. In contrast, MRPS27 expression was negatively correlated with M0 macrophages and neutrophils, indicating its involvement in promoting inflammatory responses and tissue damage. These findings suggest that MRPL49 and MRPS27 may act as important regulators within the immune microenvironment of both IS and ESRD. Consistent with these observations, previous research has shown that the accumulation of T cells and neutrophils in brain tissue after stroke correlates strongly with the severity of neuronal injury62,63. These immune cells exacerbate neuroinflammation and tissue damage by secreting inflammatory cytokines, such as TNF-αand IL-664,65. Similarly, immune cell infiltration is particularly pronounced in dialysis patients with ESRD, where it is closely associated with chronic inflammation and fibrosis66,67. The TGF-β signaling pathway plays a central role in regulating fibrosis and modulating immune responses in both IS and ESRD68,69. Furthermore, abnormalities in the p53 pathway and the G2/M checkpoint contribute to dysregulated immune cell proliferation and apoptosis, exacerbating immune dysfunction and promoting tissue damage70,71. In summary, these findings emphasize the critical role of immune cell infiltration in the progression of both IS and ESRD. The close association of candidate genes, such as MRPL49 and MRPS27, among others, with immune cell dynamics underscores their potential as key mediators in the immune microenvironment, offering valuable insights for novel therapeutic strategies targeting these diseases.
In addition to immune regulation, epigenetic mechanisms, particularly ncRNAs, may also contribute to disease progression. NcRNAs, including miRNAs and long non-coding RNAs (lncRNAs), play a significant role in regulating gene expression, especially in complex diseases such as IS and ESRD. Research has demonstrated that ncRNAs interact with mRNAs to modulate the expression of target genes, either by inhibiting or activating their transcription72. In this study, the miRcode database was utilized to predict potential miRNA regulators of the key genes identified. Several miRNAs were found to be potentially involved in the progression of IS and ESRD by regulating genes such as MRPL49 and MRPS2. For instance, miR-186 was shown to regulate MRPL49 expression, influencing oxidative stress responses and energy metabolism, which are critical in both diseases73. Similarly, miR-21 has been implicated in stroke through its regulation of phosphatase and tensin homolog (PTEN), playing a significant role in mediating inflammatory responses and apoptosis74. In the context of ESRD, miR-223 targets the NLRP3 inflammasome, thereby modulating inflammatory responses and renal tubular cell fibrosis, processes strongly associated with the decline of renal function75. These findings suggested that these miRNAs are part of a complex regulatory network that influences both the progression and prognosis of IS and ESRD. Overall, the identification of ncRNA-mediated mechanisms provides deeper insights into the molecular underpinnings of both diseases and highlights the potential of targeting specific miRNAs for diagnostic and therapeutic strategies.
In conclusion, this study systematically revealed the complex network of interactions between mitochondria and immune system in IS-ESRD co-morbidity through multi-omics analysis, and identified the central regulatory role of MRP family genes. These findings provide new perspectives for understanding the mechanism of multi-organ interaction and developing dual-targeted therapies. At the clinical translational level, a multimodal diagnostic model can be constructed for the early identification of high-risk groups by detecting the expression levels of MRP family genes (e.g., MRPS9, MRPS10) and mitochondrial function indicators (e.g., ROS content, ATP metabolites) in the peripheral blood. Therapeutic development should focus on intervention strategies targeting MRP genes, such as designing small molecule agonists to enhance mitochondrial translation function, using gene editing technology to repair mutated sites, or developing TGF-β/p53 dual-pathway inhibitor combination therapies to synergise fibrosis and cell cycle disorders. In addition, personalised therapies focusing on the repair of mitochondrial function, such as selective activation of the NRF2 pathway or modulation of mitochondrial autophagy activity, could be tailored based on immune cell infiltration characteristics (e.g. CD8 + T cell/M0 macrophage ratio) and epigenetic regulatory status. However, the present study has several limitations. Firstly, the sample size for clinical validation was relatively limited, and although key gene expression trends were validated in Asian samples by RT-qPCR, an expanded cohort is still needed to validate race-specific variants. Second, the dataset is mainly from European and American populations, and the genetic variation of MRP family genes in different populations may affect their clinical applicability. For example, the minor allele frequency of the rs11156878 locus of the MRPP3 gene is significantly lower in Asian populations than in European populations, which may explain the relatively higher stroke risk in Asian ESRD patients76. Therefore, future development of multigene risk scores based on MRP genes needs to integrate multi-ethnic data to optimise race-specific prediction models. Nevertheless, the results of this study provide important insights into the molecular mechanisms of IS-ESRD, and the MRP family of genes and pathway networks it reveals lays the foundation for potential therapeutic target development, and further validation of the gene functions and exploration of their dynamic regulatory mechanisms in multiorgan injury are needed.
Data availability
This study analyzed publicly available datasets. These datasets can be found in the GEO database (https://www.ncbi.nlm.nih.gov/geo/), with accession numbers GSE16561, GSE22255, GSE37171, and GSE142153. The codes are available in the Supplementary file, and the raw data are available from the corresponding author (shsh0844@163.com) on reasonable request.
References
Soltanpour, M., Greiner, R., Boulanger, P. & Buck, B. Improvement of automatic ischemic stroke lesion segmentation in CT perfusion maps using a learned deep neural network. Comput Biol. Med. 137, 104849 (2021).
Xu, Y. et al. Inhibition of 15-hydroxyprostaglandin dehydrogenase protects neurons from ferroptosis in ischemic stroke. MedComm 5(1) (2024).
Kim, S. J. & Bang, O. Y. Antiplatelet therapy for preventing stroke in patients with chronic kidney disease. Contrib. Nephrol. 179, 119–129 (2013).
Eirin, A. & Lerman, L. O. Mesenchymal stem/stromal cell-derived extracellular vesicles for chronic kidney disease: are we there yet? Hypertension 78 (2), 261–269 (2021).
Zhu, X., Han, Q., Xia, L., Shang, J. & Yan, X. Efficacy of two hemodialyses in patients with chronic renal failure complicated by massive intracerebral hemorrhage. Ann. Clin. Transl Neurol. 10 (7), 1186–1199 (2023).
Egashira, S., Koga, M. & Toyoda, K. Intravenous thrombolysis for acute ischemic stroke in patients with end-stage renal disease on hemodialysis: A narrative review. J. Cardiovasc. Dev. Dis. 9 (12), 446 (2022).
Bobot, M. et al. Kidney disease and stroke: epidemiology and potential mechanisms of susceptibility. Nephrol. Dial Transpl. 38 (9), 1940–1951 (2023).
Ueki, K. et al. Decreased estimated glomerular filtration rate and proteinuria and long-term outcomes after ischemic stroke: A longitudinal observational cohort study. Stroke 54 (5), 1268–1277 (2023).
Nlandu, Y. et al. Reverse epidemiology of elevated blood pressure among chronic Hemodialysis black patients with stroke: a historical cohort study. BMC Nephrol. 18 (1), 277 (2017).
Briet, M. & Burns, K. D. Chronic kidney disease and vascular remodelling: molecular mechanisms and clinical implications. Clin. Sci. 123 (7), 399–416 (2012).
Tuttle, K. R. et al. Clinical characteristics of and risk factors for chronic kidney disease among adults and children: an analysis of the CURE-CKD registry. JAMA Netw. Open. 2 (12), 1918169 (2019).
Riella, L. V. & Cravedi, P. Kidney immunology: embracing the complexity to advance the field. Clin. Kidney J. 15 (2), 366–367 (2021).
Mikolasevic, I., Žutelija, M., Mavrinac, V. & Orlic, L. Dyslipidemia in patients with chronic kidney disease: etiology and management. Int. J. Nephrol. Renovasc Dis. 10, 35–45 (2017).
Covic, A. & Voroneanu, L. Chronic kidney disease and stroke: more observations but no trials. Nephrol. Dial Transpl. 33 (3), 367–370 (2018).
Masson, P. et al. Chronic kidney disease and the risk of stroke: a systematic review and meta-analysis. Nephrol. Dial Transpl. 30 (7), 1162–1169 (2015).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43 (7), e47 (2015).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16 (5), 284–287 (2012).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat. Genet. 25 (1), 25–29 (2000).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30 (2000).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12 (5), 453–457 (2015).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 14 (11), 1083–1086 (2017).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13 (11), 2498–2504 (2003).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14 (1), 7 (2013).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 102 (43), 15545–15550 (2005).
KDIGO. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 3 (1), 1–150 (2013).
Powers, W. J. et al. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke. Stroke 50 (12), 440–441 (2019).
Foley, R. N. et al. Long interdialytic interval and mortality among patients receiving Hemodialysis. N Engl. J. Med. 365 (12), 1099–1107 (2011).
Hemmelgarn, B. R. et al. Relation between kidney function, proteinuria, and adverse outcomes. JAMA 303 (5), 423–429 (2010).
Aono, T. et al. Changes in the risk of stroke in dialysis patients: A retrospective analysis over the last 40 years. Toxins (Basel). 13 (5), 350 (2021).
Pawluk, H. et al. Selected mediators of inflammation in patients with acute ischemic stroke. Int. J. Mol. Sci. 23 (18), 10614 (2022).
Sarnak, M. J. et al. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American heart association councils. Hypertension 42 (5), 1050–1065 (2003).
Qin, C. et al. Signaling pathways involved in ischemic stroke: molecular mechanisms and therapeutic interventions. Signal. Transduct. Target. Ther. 7 (1), 215 (2022).
Zhong, Y. et al. The bidirectional role of the JAK2/STAT3 signaling pathway and related mechanisms in cerebral ischemia-reperfusion injury. Exp. Neurol. 341, 113690 (2021).
Lee, H. et al. Advances in uremic toxin detection and monitoring in the management of chronic kidney disease progression to end-stage renal disease. Analyst 149 (10), 2784–2795 (2024).
Frąk, W. et al. Role of uremic toxins, oxidative stress, and renal fibrosis in chronic kidney disease. Antioxid. (Basel). 13 (6), 687 (2024).
Huang, G., Li, H. & Zhang, H. Abnormal expression of mitochondrial ribosomal proteins and their encoding genes with cell apoptosis and diseases. Int. J. Mol. Sci. 21 (22), 8879 (2020).
Richman, T. R. et al. Mitochondrial ribosomes in cancer. Semin. Cancer Biol. 47, 67–81 (2017).
Gopisetty, G. & Thangarajan, R. Mammalian mitochondrial ribosomal small subunit (MRPS) genes: A putative role in human disease. Gene 589 (1), 27–35 (2016).
Jiang, J. et al. MRPS9-mediated regulation of the PI3K/Akt/mTOR pathway inhibits neuron apoptosis and protects ischemic stroke. J. Mol. Neurosci. 74 (1), 23 (2024).
Pang, Y. et al. Cuproptosis-related lncRNA-based prediction of the prognosis and immunotherapy response in papillary renal cell carcinoma. Int. J. Mol. Sci. 24 (2), 1464 (2023).
Tao, J. et al. Identification of key biomarkers in ischemic stroke: single-cell sequencing and weighted co-expression network analysis. Aging (Albany NY). 15 (13), 6346–6360 (2023).
Cheong, A., Lingutla, R. & Mager, J. Expression analysis of mammalian mitochondrial ribosomal protein genes. Gene Expr Patterns. 38, 119147 (2020).
Itoh, Y. et al. Mechanism of mitoribosomal small subunit biogenesis and preinitiation. Nature 606 (7914), 603–608 (2022).
Zhao, M., Liu, A., Mo, L., Wan, G. & Lu, F. The value of RPS15 and MRPS27 in ischemic stroke. Med. (Baltim). 102 (33), 34706 (2023).
Cañadas-Garre, M. et al. Genetic variants affecting mitochondrial function provide further insights for kidney disease. BMC Genom. 25 (1), 576 (2024).
Hashad, D., Elgohry, I. & Dwedar, F. Nuclear respiratory factor-1 (NRF-1) gene expression in chronic kidney disease patients undergoing Hemodialysis. Clin. Lab. 62 (11), 2149–2154 (2016).
Sharoyko, V. V. et al. Loss of TFB1M results in mitochondrial dysfunction that leads to impaired insulin secretion and diabetes. Hum. Mol. Genet. 23 (21), 5733–5749 (2014).
Johar, K. et al. Neuron-specific specificity protein 4 bigenomically regulates the transcription of cytochrome C oxidase subunit genes. J. Neurochem. 127 (4), 496–508 (2013).
Heldin, C. H. & Moustakas, A. Role of Smads in TGFβ signaling. Cell. Tissue Res. 347 (1), 21–36 (2012).
Chen, L. et al. Central role of dysregulation of TGF-β/Smad in CKD progression and potential targets of its treatment. Biomed. Pharmacother. 101, 670–681 (2018).
Akhter, M. S. et al. P53-induced reduction of lipid peroxidation supports brain microvascular endothelium integrity. J. Pharmacol. Sci. 141 (1), 83–85 (2019).
Peng, J. et al. Hyperglycemia, p53, and mitochondrial pathway of apoptosis are involved in the susceptibility of diabetic models to ischemic acute kidney injury. Kidney Int. 87 (1), 137–150 (2015).
Sofroniew, M. V. Astrocyte reactivity: subtypes, States, and functions in CNS innate immunity. Trends Immunol. 41 (9), 758–770 (2020).
Zhen, X. et al. Nicotinamide supplementation attenuates renal interstitial fibrosis via boosting the activity of sirtuins. Kidney Dis. (Basel). 7 (3), 186–199 (2021).
López-Lluch, G. et al. Mitochondrial biogenesis and healthy aging: therapeutic potential of metabolic modulators. Exp. Gerontol. 183, 112327 (2023).
Lee, T. H. et al. Immunosenescence, gut dysbiosis, and chronic kidney disease: interplay and implications for clinical management. Biomed. J. 47 (2), 100638 (2024).
Jayaraj, R. L. et al. Neuroinflammation: friend and foe for ischemic stroke. J. Neuroinflammation. 16 (1), 142 (2019).
Cai, Z., Hussain, M. D. & Yan, L. J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 124 (5), 307–321 (2014).
Humphreys, B. D. Mechanisms of renal fibrosis. Annu. Rev Physiol. 80, 309–326 (2018).
Jang, H. R. & Rabb, H. Immune cells in experimental acute kidney injury. Nat. Rev. Nephrol. 11 (2), 88–101 (2015).
Jickling, G. C. et al. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J. Cereb. Blood Flow. Metab. 35 (6), 888–901 (2015).
Selvaraj, U. M. & Stowe, A. M. Long-term T cell responses in the brain after an ischemic stroke. Discov Med. 24 (134), 323–333 (2017).
Wytrykowska, A., Prosba-Mackiewicz, M., Nyka, W. M. & IL-1β TNF-α, and IL-6 levels in gingival fluid and serum of patients with ischemic stroke. J. Oral Sci. 58 (4), 509–513 (2016).
Monsour, M. et al. Contemplating IL-6, a double-edged sword cytokine: which side to use for stroke pathology? CNS Neurosci. Ther. 29 (2), 493–497 (2023).
Kadatane, S. P. et al. The role of inflammation in CKD. Cells 12 (12), 1581 (2023).
Canaud, B. et al. The Janus-faced nature of complement in hemodialysis: interplay between complement, inflammation, and bioincompatibility. Front. Nephrol. 4, 1455321 (2024).
Zhang, L. et al. Extracellular vesicles from hypoxia-preconditioned microglia promote angiogenesis and repress apoptosis in stroke mice via the TGF-β/Smad2/3 pathway. Cell. Death Dis. 12 (11), 1068 (2021).
Meng, X. M., Nikolic-Paterson, D. J. & Lan, H. Y. TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol. 12 (6), 325–338 (2016).
Xu, S., Li, X. & Wang, Y. Regulation of the p53–mediated ferroptosis signaling pathway in cerebral ischemia stroke (Review). Exp. Ther. Med. 25 (3), 113 (2023).
Liu, L. et al. Downregulation of fatty acid oxidation led by Hilpda increases G2/M arrest/delay-induced kidney fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 1869 (5), 166701 (2023).
Mu, J. et al. Multicenter validation of LncRNA and target mRNA diagnostic and prognostic biomarkers of acute ischemic stroke. J. Am. Heart Assoc. 13 (14), 034764 (2024).
Cai, S. C. et al. Oleanolic acid inhibits neuronal pyroptosis in ischaemic stroke by inhibiting miR-186-5p expression. Exp. Neurobiol. 30 (6), 401–414 (2021).
Gao, X. et al. Extracellular vesicle-mediated transfer of miR-21-5p alleviates early brain injury via the PTEN/Akt pathway after subarachnoid hemorrhage. Cell. Death Dis. 11 (5), 363 (2020).
Sun, Y. et al. Baicalin attenuates lipopolysaccharide-induced renal tubular epithelial cell injury by inhibiting the TXNIP/NLRP3 signalling pathway via increasing miR-223-3p expression. J. Biol. Regul. Homeost. Agents. 34 (1), 69–82 (2020).
Rossetti, G. et al. A common genetic variant of a mitochondrial RNA processing enzyme predisposes to insulin resistance. Sci. Adv. 7 (39), 7514 (2021).
Acknowledgements
We thank all medical staff of the Department of Organ Transplantation at The Affiliated Guangdong Second Provincial General Hospital of Jinan University, for their assistance with this research.
Funding
This study was supported by the Natural Science Foundation of Guangdong Province, China (No. 2020A1515011249) and the Guangzhou Science and Technology Program Project, China (No. 201804010421).
Author information
Authors and Affiliations
Contributions
W and SS conceived the study. W, L, and S designed the experiments, and W performed data analysis and interpretation. L, W, S, and M conducted bioinformatics and statistical analyses. W and L drafted the manuscript, and Z revised it. Z, Z, and Z supervised the research. All authors have read, edited, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study involving human participants was reviewed and approved by the Ethics Committee of The Affiliated Guangdong Second Provincial General Hospital of Jinan University (Approval No:2024-KY-KZ-211-01). The research was conducted in accordance with local legislation and institutional requirements. All the patients have been informed and signed informed consent before the experiments.
Consent for publication
All authors have read and approved the final manuscript. We confirm that we have obtained consent from all individuals whose datas are included in the manuscript, and all authors agree with the publication of this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, S., Li, Z., Wang, X. et al. Bioinformatics analysis of comorbid mechanisms between ischemic stroke and end stage renal disease. Sci Rep 15, 17060 (2025). https://doi.org/10.1038/s41598-025-01049-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-01049-4
