
Abstract
Magnetic Tunnel Junction-based molecular spintronics devices (MTJMSDs) hold great potential for integrating paramagnetic molecules with ferromagnetic electrodes, creating a diverse array of metamaterials with novel magnetic behaviors. Understanding interactions, especially between molecules and electrode materials, is essential to advancing this field. In this study, we used Monte Carlo simulation (MCS) to examine the influence of Dzyaloshinskii-Moriya interaction (DMI) on the MTJMSDs. Our simulations reveal that the presence of DMI interaction significantly lowered the magnetization of the ferromagnetic (FM) electrode. This DMI effect on the FM electrode provides a potential mechanism to explain the experimental observations of losing magnetic contrast on one FM electrode of the MTJMSD. A cross-junction-shaped MTJMSD, where several thousands of paramagnetic Octametallic Molecular Complexes are covalently bonded between two FM electrodes along the junction edges, exhibited loss of magnetic contrast on one ferromagnet in MFM imaging. DMI’s impact on FM electrode properties resembles the experimental observation on MTJMSD. Our MCS showed that the strong DMI induced alternating magnetic bands aligned in opposite directions on a ferromagnetic electrode. Molecule bridges transported the effect of the DMI-induced magnetic phases onto the FM electrode connected to the other end of the molecule. For the specific range of DMI, the direction of magnetization of the FM electrode present on the other end of the molecular channel could switch based on the nature of the DMI-induced magnetic phase present in the junction area. This study underscores the importance of antisymmetric interactions, like DMI, in influencing the magnetic properties of MTJMSD systems. In future MSD experimental studies, DMI on FM electrodes can be achieved using suitable molecule-FM interfaces or multilayer FM electrodes harnessing spin-orbit coupling. MTJMSD test bed provides excellent opportunities for creating unprecedented strong molecule-FM electrode coupling and using multilayer electrodes.
Similar content being viewed by others

Introduction
Molecular spintronics devices (MSDs) are promising candidates for next-generation, energy-efficient memory applications spanning both classical and quantum computing systems1,2,3,4,5,6,7,8. This promise stems from the unique properties of molecules, which serve as fundamental units in MSDs, possessing distinct quantum behaviors9,10,11 and highly variable resistive states12. Molecules also exhibit low hyperfine splitting and pronounced Zeeman effects, enabling precise control over atomic-scale interactions1,13,14. This growing interest in MSDs as components for modern quantum state-dependent computers has further propelled research into integrating molecular elements with ferromagnetic materials4,5,6,7,8. Ferromagnetic electrodes are thermally stable spin sources and detectors and are already part of commercially successful spin valve technology. Most importantly, ferromagnetic thin films keep exhibiting novel phenomena depending on the properties of the material they are interfacing15,16,17,18,19,20.
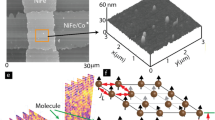
A critical step in developing successful MSDs21,22 is establishing reliable molecular spin channels between ferromagnetic electrodes23. Various methods have been explored for this purpose, including creating molecular junctions with a conducting atomic force microscope (CAFM) probe24,25, sandwiching molecules between two metal films26, and constructing electronic devices with multilayer edges27. However, these conventional methods of forming molecular conduction bridges between two electrodes have substantial limitations. Molecular junctions often struggle to produce stable and diverse configurations and are generally unsuitable for scalable mass production25,28,29,30. To overcome these challenges, a magnetic tunnel junction-based molecular spintronics device (MTJMSD) approach has been developed[31]. MTJMSDs feature ferromagnetic electrodes with exposed side edges separated by a tunneling barrier, typically AlOx (Fig. 1a). In this configuration (Fig. 1b), magnetic molecules are covalently bonded to both the top and bottom ferromagnetic electrodes across the tunnel barrier. As a unique advantage of this approach, any molecule of suitable length functionalized with thiol-like end groups can be chemically bonded with two arbitrarily chosen ferromagnetic electrodes. MTJMSD involves covalent-like bonding between the molecule and ferromagnetic electrodes and is much stronger and reproducible than the present at metal-insulator interfaces in typical MTJ. Hence, MTJMSD architecture allows molecules to influence the overall magnetic properties of the MTJMSD system depending on their interactions with each electrode31,32. Unique attributes of MTJMSD have led to unprecedented observations of several orders of resistance change with the magnetic field33, room temperature current suppression34, and spin photovoltaic effect35.
An intriguing observation was a stable loss of magnetic contrast at room temperature on cross-junction MTJMSD-shaped MTJMSD36. Notably, a partial disappearance of magnetic contrast in one electrode of cross-junction MTJs was observed following the attachment of Octametallic Molecular Complexes (OMCs), as shown by magnetic force microscopy (MFM) imaging (Fig. 1d)36.In the same study36, we also reported molecule-induced non-magnetic phases over larger areas of the MTJMSD electrodes, suggesting that molecular bonding significantly altered the magnetic ordering of ferromagnetic electrodes36. However, we could not gain mechanistic insight into underlying physics, causing the disappearance of magnetic contrast on one ferromagnetic electrode of MTJMSD. To date, we have investigated the effect of (i) type of molecular coupling and strength31, (ii) magnetic electrode anisotropy37, (iii) paramagnetic molecule spin state38, (iv) magnetic electrode materials24, (v) MTJMSD shape and electrode dimensions39,40. Although previous MCS studies provided valuable insights, their applicability was limited. Concurrently, our attempts at density functional theory (DFT) modeling through collaboration faced challenges due to the prohibitive complexity of MTJMSD. Consequently, we redirected our efforts to expand MCS studies in the alternative direction.
To further investigate the disappearance of magnetic contrast on cross-junction-shaped MTJMSD electrodes36, we aimed to examine additional magnetic interactions that could explain the experimentally observed phenomenon. Here, we turn our focus to the Dzyaloshinskii-Moriya interaction (DMI), an antisymmetric exchange interaction typically associated with interfaces between antiferromagnetic and ferromagnetic materials41,42,43. Our earlier experimental and MCS studies demonstrated that paramagnetic molecules on cylindrical-shaped MTJs induced antiferromagnetic coupling, resulting in a room-temperature stable antiparallel magnetic orientation between two electrodes40. In contrast, a cross-junction-shaped MTJMSD consisted of two long ferromagnetic electrodes intersecting at the junction. We surmise that the antiferromagnetic coupling between electrodes at junctions impacts the ferromagnetic behavior of the electrodes away from the junction. The interplay between this antiferromagnetic interaction at the junction and the ferromagnetic nature of the extended electrodes may induce DMI, which we conjecture as a potential contributor to the observed magnetic contrast loss.
DMI is known to promote the formation of chiral magnetic spin textures, such as chiral domain walls and skyrmions, which are crucial for next-generation, high-capacity data storage devices 44,45,46,47,48,49. In this study, we explored the role of DMI within one of the electrodes of MTJMSDs and its influence on the magnetic behavior of MTJMSDs, particularly at the molecule-electrode interfaces in cross-junction configurations where antiferromagnetic coupling is prevalent. Using MCS, we investigated how varying DMI strengths impact the magnetic phases of one electrode and its interaction with the molecular channel. This study offers insights into the potential mechanisms underlying magnetic contrast loss in MTJMSDs.
Methodology
This Monte Carlo simulation (MCS) study was inspired by experimental observations of unique magnetic contrasts on the electrodes of the magnetic tunnel junction-based molecular spintronics device (MTJMSD), as shown in Fig. 1d36. In the experiment, the cross-junction-shaped MTJ was fabricated with exposed edges, consisting of a bottom electrode stack of Ta(~ 2 nm)/Co(~ 5 nm)/NiFe(~ 5 nm) and a top electrode stack NiFe (~ 10 nm). An AlOx(~ 2 nm) insulating layer was sandwiched between the electrodes, as illustrated in the schematic (Fig. 1a). Paramagnetic Octametallic Molecular Complexes (OMCs) were covalently bonded to both the top and bottom electrodes, establishing the MTJMSD structure (Fig. 1b). The OMCs feature cyanide-bridged Ni and Fe ions with the chemical formula: [(pzTp) FeIII(CN)3]4[NiII(L)]4[O3SCF3]4, where (pzTp) = tetra(pyrazol-1-yl)borate; L = 1-S(acetyl)tris(pyrazolyl)decane]50. These OMCs contain long carbon chains terminated with thiol groups, facilitating covalent bonding with NiFe electrodes51. Figure 1c shows the topology of an MTJMSD junction. The full molecule attachment procedure is detailed elsewhere52, and the complete experimental methods are published in a separate work36.
MCS24,31,53,54 was employed to explore the influence of DMI on the magnetic properties of cross-junction-shaped MTJMSD.In our MCS study, the configuration of the MTJMSD rising model was set as 11 × 50 × 50 (width height length). The molecular plane was represented by a 5 × 5 atomic square and positioned at the cross junction between two 5 monolayers thick and 50 atoms long ferromagnetic metal electrodes as illustrated in Fig. 2b,e,h. Upon running the simulation, the equilibrium state was achieved based on the following Hamiltonian equation 31,54:
where \(\:{\varvec{S}}_{\varvec{i}}\) and \(\:{\varvec{S}}_{\varvec{i}+1}\:\)represented the neighboring spin vectors within the electrode or at the interface between the electrode and molecule. \(\:{J}_{l}\), \(\:{J}_{R}\), \(\:{J}_{ml}\) and \(\:{J}_{mR}\) denote the Heisenberg exchange coupling strengths of the left, right, molecule-left, and molecule-right electrodes, respectively. \(\:\varvec{D}\) represents the Dzyaloshinskii -Moriya vector for the left electrode. All of these parameters are unitless quantities.
For the simulation model setup, the \(\:{J}_{l}\) and \(\:{J}_{R}\) were fixed at 1 to ensure both electrodes were ferromagnetic. The molecule-metal electrode coupling strengths \(\:{J}_{ml}\) ane \(\:{J}_{mR}\) were chosen to be 1 and − 1, respectively, based on our prior findings that the organic molecule induces opposite coupling effects with the two electrodes40. Additionally, the atomic interaction between two ferromagnetic electrodes \(\:{J}_{lR}\)was set to 0 to avoid interference between them and solely study the molecule’s effect on individual ferromagnetic electrodes. The study on the impact of \(\:{J}_{lR}\) in such MTJMSDs system was published elsewhere53.

Schematics of Magnetic tunnel junction (MTJ) (a) Bare (b) With molecule. (c) Atomic force microscopy image of molecule treated MTJ junction (d) Magnetic force microscopy image of the same junction showing partial loss of identity of one electrode.
The simulation thermal energy was KBT = 0.1. This energy corresponded to a 50–90 °C temperature range since several practical ferromagnetic materials have Curie Temperature between 500 °C to 900 °C range55. During the simulation, the left electrode was subjected to varying DMI values between 0 and 1 along the x-direction. The temporal evolution, 3D lattice structure and spatial correlation for each FM electrode and whole MTJMSD system were investigated. The spins of MTJMSD fluctuated widely, and each Monte Carlo simulation ran for 500 million iterations to reach a stable state. The system’s equilibrium energy state was determined through Metropolis algorithms and the Markov process56. At equilibrium, each ferromagnetic electrode could achieve a maximum moment of 1250 for homogeneous spin alignment on each. Similarly, 16 molecules were present along the perimeter, allowing 16 moments when the spins were aligned in the same direction.
In our MCS study, the paramagnetic complex molecule was treated as a single atomic analog unit, following the approach used in prior MCS studies to interpret experimental results40,57. The molecule was represented as an atomic analog of the complex molecules employed in the experiment (Fig. 1b), allowing the computation of intramolecular symmetric exchange interactions (Jmol) and antisymmetric Dzyaloshinskii-Moriya interactions (Dmol), as expressed in Eq. (1). Dmol was set to zero to focus on the role of DMI at the FM electrode. These interactions captured the essential spin dynamics within the electrode-molecule junction. While determining the exact molecular spin state in experimental MTJMSDs was complex due to measurement constraints and challenges in probing exposed edges, our simulation provided a valuable framework for understanding spin interactions in these systems. This approach was consistent with earlier theoretical frameworks based on a simplified atomic analog of the complex molecular models40,57, which helped our group explain several experimental observations on MTJMSDs. It is noteworthy that other groups have also relied on representing the prohibitively complex molecular bridges between the metal electrodes with quantum dots58. Representing molecule with well-known generic quantum dots enabled the use of quantum dot-related single-electron transistor theory58 to interpret experimental observations. A quantum dot-based spintronics device showing Kondo level splitting59 was applied to explain the Kondo level splitting with C60 molecular spintronics devices60.
Furthermore, simulating complex molecules connected to two ferromagnetic electrodes at room temperature is prohibitively complex. It is noteworthy that DFT like alternative approaches have been challenged by numerous approximations while dealing with complex molecules involving transition metal atoms61. While our MCS approach also has several limitations, it provides a practical way to investigate a wide range of well-established magnetic interactions to analyze experimental observations. In this study, we specifically investigated the role of DMI in the MTJMSD system by varying the DMI of the left electrode to analyze its influence on the system’s magnetic properties and spin configuration. The magnetic properties of MTJMSD were investigated for eight different magnitudes of the Dzyaloshinskii-Moriya interaction (DMI) parameter. The DMI parameter is denoted as D. We varied the D values for the left electrode along the x-direction ( D = 0, 0.05, 0.1, 0.2, 0.4, 0.6, 0.8 ) within the range of 0 to 1.
Results and discussion
We first studied the temporal evolution of MTJMSD as a function of time. Figure 2a,d,g represent the temporal evolution of magnetic moments. In these graphs, red data points indicate the left ferromagnetic electrode, blue data points represent the right ferromagnetic electrode, and black data points indicate the whole MTJMSD as a function of iteration counts for D = 0, 0.1 and 1, respectively. Additionally, Fig. 2b,e, and Fig. 2h display the stabilized states of magnetic moments at the end of simulations in the form of 3D lattice plots of cross-type MTJMSD for the respective D values, while Fig. 2c,f,i depict the corresponding zoomed in pictures showing the spin alignment of molecules. In the color scale of 3-D lattice plots, blue and red colors represent two oppositely aligned spins in reference to the molecule.

Monte Carlo simulations result investigating the effect of DMI on the MTJMSD devices. Temporal progression of magnetic moments of the left electrode, right electrode, and MTJMSD at the magnitude of DMI (D) value (a) 0 (d) 0.1 (g) 1. Equilibrium states of a simulated spatial 3-D lattice model of the MTJMSD measured at KT = 0 0.1 for DMI values (b) 0, (e) 0.1 (h) 1. Zoomed-in a snapshot of the equilibrium state of the molecule between single layers of right and left electrode for DMI values (c) 0, (f) 0.1, (i) 1. Spin direction along the x-axis for the magnitude of DMI 0 and along the y-axis for both DMI values 0.1 and 0.
The total magnetic moments of the MTJMSD system plotted in Fig. 2a,d, and Fig. 2g are the sum of contributions from individual electrodes. For D = 0 (Fig. 2a), the magnitude of magnetic moments of the left (red data points) and right ferromagnetic electrode (blue data points) overlaps and were nearly equal to the total possible moment of 1250. However, the total magnetic moments of MTJMSD (black data points) stabilize to a lower value since the moments on the left and right ferromagnets are opposite, as depicted in 3-D lattice plots of Fig. 2b,c. In the case of D = 0.1 (Fig. 2d), the moment of the individual left electrode (red data points) decreases as the DMI, antisymmetric exchange interaction, begins to dominate, causing spins within the left electrode to align in two opposite directions (Fig. 2e,f) thereby reducing the net moment of the left electrode. Consequently, the whole MTJMSD’s moment (black data points) begins to follow that of right electrode (blue data points), commencing left electrode to be in a multidomain state (Fig. 2e). As the D value approaches 1 (Fig. 2g), the moments of left electrode tend towards zero (red data points) since the magnetic moments within the entire electrode align uniformly with two oppositely aligned moments in the form of red and blue stripes (Fig. 2h,i). So, the right electrode and the entire MTJMSD’s moments overlap, each being 1250 (blue and black data points).
We observed that the presence of DMI interaction on the FM electrode produced alternating bands with the magnetization direction of adjacent bands aligning in the opposite direction (Fig. 2b,e,h). We quantified the number of bands on the ferromagnetic electrode and plotted them as a function of D (Fig. 3). We observed that the number of bands in the form of multiple domain states increases with an increase in DMI. The maximum number of bands,14, is achieved for the strongest DMI, i.e., D = 1. This trend of increasing band numbers on the left electrode is quite interesting as it reveals the clear interplay between DMI and symmetric exchange coupling for a particular electrode.
In the absence of DMI, the left and right electrodes are coupled ferromagnetically and anti-ferromagnetically with respect to the molecule, as the \(\:{J}_{ml}\:and\:\)\(\:{J}_{mR}\) values are 1 and − 1, respectively (Fig. 2b,c). Therefore, the electrodes are in a single-domain state. For higher DMI values, long-range antiferromagnetic coupling with the molecule remains as such for the right electrode. However, due to the DMI effect on the left electrode, only the tiny stripes of this electrode, which is closer to the molecule plane, get ferromagnetically coupled with each molecule, yielding the multiple bands of oppositely aligned magnetic moments within the electrode itself (Fig. 2e,h).
DMI is a non-symmetric exchange interaction in which energy is minimized when the spins are perpendicular to each other instead of ferromagnetic and antiferromagnetic states, as preferred by symmetric exchange interaction44. DMI field is anisotropic along the direction perpendicular to the spin plane. Due to this reason, magnetic moments of MTJMSD are settled down along the y or z direction at the end of the simulation as the DMI is applied along the x direction (Fig. 2h,i). However, in the absence of DMI, MTJMSD’s magnetization normally settles in any distinct (x, y, or z) states34,36. The strong competition between symmetric and antisymmetric exchange interaction put the spins into the whirling state, resulting in the spin texture. In the presence of DMI, the left electrode of MTJMSD consists of oppositely magnetized bands that might represent the extreme ends of spin textures as they appear when spins turn from one direction to another.

Total band numbers on the left electrode as a function of magnitude of DMI (D).
We observed the switching phenomenon on the right FM electrode as a function of molecule interaction with the DMI-created specific orientation. Figure 4a,b shows an equilibrium state of moments settled along the y direction in a three-dimensional lattice model at the end of simulations conducted for D = 0.4, observed with two distinct, independent runs. In Fig. 4a, the molecule’s moments align along the negative y direction (red color), while in Fig. 4b, they align along the positive y direction (blue color). Notably, in both cases, the molecule’s moments have opposite alignment with the right ferromagnet, displaying antiferromagnetic coupling. Additionally, DMI-induced magnetic bands within the left electrode influenced the molecules’ magnetic orientation. DMI impact on molecules’ spin is evident in the switching of red and blue colored bands between Fig. 4a,b. Interestingly, the DMI impact of the left electrode traveled to the right electrode via the molecular bridges (Fig. 4a,b). One can hypothesize the computation strategy on the molecule-entangled system of MTJMSD (Fig. 4a,b) if DMI created a suitable size band that can be moved in and out of the junction area using external energy. Notably, the width of the DMI-induced bands must exceed the junction area to enable practical switching. If DMI is strong, the bandwidth becomes narrower than the junction area, causing molecules to interact with multiple bands simultaneously, resulting in a rather indifferent ferromagnetic ordering on the right electrode (Fig. 2h,i).
We have investigated the correlation between the molecule’s spin and distinct regions of the two ferromagnetic electrodes. To quantify the molecule-correlated magnetic phases, we utilized autocorrelation methods, as detailed in the previous publications31. This involved optimizing and studying the spatial correlation (SC) factor between the molecule and different layers of the ferromagnetic electrode. Mathematically, the SC was calculated as the dot product between the spin vectors of the molecule and atomic spin vectors of the different layers of the ferromagnet, as expressed in the equation31,62:
where \(\:{\varvec{S}}_{\varvec{m}}\) and \(\:{\varvec{S}}_{\varvec{F}\varvec{M}}\:\)are the molecule’s spin vectors and atomic spin vectors of the ferromagnetic electrodes, respectively. The 2D spin correlation contour plots in Fig. 4c,d correspond to 3D lattice plots of two distinct runs presented in Fig. 4a,b, respectively. To shrink the FM electrodes from 3D to 2D we averaged the left electrode and molecule along the y direction and the right electrode along the Z direction. Right FM’s correlated data was displayed after rotating them by 90 degree to display in the same plane. The color bars in these plots represent the magnitude of SC, ranging from − 1 to 1. Here, a value of 1 indicates the parallel alignment of the spins of the molecule with the ferromagnetic electrode, while − 1 denotes the antiparallel coupling.

3-dimensional spatial lattice model at the end of simulation for DMI magnitude (D) = 0.4 for the (a) first and (b) third run. The spins are along the y direction. (c) and (d) are the 2-dimensional spatial correlation (SC) contour plots for the respective runs.
In Fig. 4c,d, it is evident that segments of molecules adjacent to the right electrode are strongly negatively correlated (SC factor ~-0.80) with the right electrode. Conversely, the molecule shows a strong positive correlation (SC factor ~ 0.75) with the adjacent narrow bands of the left ferromagnet. This SC difference also highlights the molecule establishing antiferromagnetic coupling31 between the two FM electrodes. Moreover, in Fig. 4d, it is observed that the 40th to 50th atomic layers of the right electrode exhibit a weak negative correlation (SC factor ~-0.50) with the segment of the molecule. Multiple magnetic phases are apparent within the bands of the left electrodes, with SC factors spanning from ~ 0.60 to 0.80 (Fig. 4d) despite all stripes being in the same magnetic phases with SC factor of ~ 0.75 (Fig. 4c).
We also investigated the temporal evolution of energies and magnetic moments as a function of iteration counts for different DMI. Figure 5a shows that the left FM electrode’s energy (UL) is stabilized at different points for distinct DMI. UL stabilized at around − 3.1 for D = 0 and 0.05; however, UL decreased with increasing D (Fig. 5a) and stabilized at the lowest value of − 4.4 for D = 1 (brown data points). Decreasing UL indicates that DMI becomes highly dominant over symmetric exchange interaction. However, right electrode energies (UR) settle at − 3.1 (Fig. 5b) for all D values since no DMI field is applied on this electrode. This corresponds to our 3D lattice plots that always put the right electrode into the single domain state and is independent of DMI (Fig. 1). The total energy (U) of the MTJMSD system (Fig. 5c) represents the stabilization of the left FM, right FM, and molecules. U is rather constant until 0.2; however, a more stable state is realized with an increase in DMI magnitude. Stabilized energy is diminished as the D value increases, with maximum energy settling around − 6.25 for D = 0 and approaching − 7.6 for D = 1, as shown in Fig. 5c.However, molecule-left electrode interaction energy (UmL) shown in Fig. 5d escalates with rise in D value. The same trend appears in molecule-right FM electrode interface energy (UmR) variation with DMI (Fig. 5e).
It is observed that UmL (Fig. 5d) and UmR (Fig. 5e) oscillate around − 15 (black data points) for D = 0 and − 12 (blue data points) for D = 1. Although UL and UR variation with different DMI are different, but the molecule-electrode interface energies UmL (Fig. 5d) and UmR (Fig. 5e) exhibited similar profile. The noise in UmL (Fig. 5d) and UmR (Fig. 5e) data indicate the wide range of coupling between different molecules/FM electrode interfaces. To delve into the variation pattern in the molecule’s moment, we quantified and plotted the molecule moment (Msm) as a function of iteration count (inset of Fig. 5f) for all DMI values. We extracted the average of Msm values from 200 M to 500 M iterations; we ignored the first 200 million to avoid including a non-equilibrium state at any MTJMSD’s component. The dependence of Msm with D, presented in Fig. 5d, shows the diminishing trend of Msm with increasing magnitude DMI. It is observed that Msm remains almost invariant around 15 for 0.05 DMI, which is very close to the maximum attainable moment of 16 in our Monte Carlo simulation. Beyond this critical value D, Msm starts to decrease and achieves the minimum value of around 8.5 for D = 1. Such an inverse relationship of DMI with magnetic moment is contributed by the increase in band numbers on the left electrode, as shown in Fig. 3. As the band number increases on the left electrode, their width narrows, and the molecule’s sublattices interact with two oppositely aligned bands. When molecules simultaneously interact with left FM’s band with multiple spin directions, corresponding molecules spins become inhomogeneous. All the molecules are perfectly aligned in the specific direction only if the two FM electrodes have uniform magnetic phases around molecular interfaces31,62. The error bar in the measurement represents the fluctuations in molecule moment as the simulation evolves over time. The length of error bars also increased for higher values of DMI, which means fluctuations in Msm increased with escalating DMI (Fig. 5d,e,f inset) as it couples with different stripes of the left electrode.

Time evolution of left ferromagnetic electrode, right ferromagnetic electrode and MTJMSD for discrete DMI magnitudes, plotted energy (U) as a function of iteration counts: (a) Left Electrode (b) Right electrode (c) MTJMSD (d) Molecule-left electrode (e) Molecule -right electrode. (f) Dependence of spin magnetic moment (Msm) of the molecule as a function DMI magnitude (D). The inset of Fig. (f) shows the variation of Msm as a function of iteration count at D = 0, 0.4, and 1.
Conclusion
Monte Carlo simulations have been effectively utilized to investigate Dzyaloshinskii-Moriya interaction (DMI) variation on cross-junction-shaped magnetic tunnel junction-based molecular spintronics devices. In the simulations, we varied the DMI values applied to the left electrode from 0 to 1. It is observed that the entire left electrode exhibited alternate bands of two oppositely coupled magnetic moments. Furthermore, a short-range ferromagnetic coupling between the molecule and the nearby band of the left electrode was examined, while long-range antiferromagnetic coupling was maintained with the right electrode. In the presence of maximum DMI, net magnetic moments are canceled, resulting in zero for the left electrode, supporting earlier experimental findings. Similarly, we analyzed the temporal evolution of energy for both electrodes, the entire MTJMSD system, the molecule-left ferromagnet, the molecule-right ferromagnet, and the magnetic moment of the molecule. We observed that the impact of DMI on the molecule’s moment is significant, as it is decreased with increasing DMI. Molecules’ magnetic moment reached a value of 8.5 for the highest DMI magnitude of 1, which is ~ 50% of the maximum attainable value of 16 obtained for D < 0.2. Additionally, the interaction energy between electrode and molecule also varied for different DMI values, indicating the influence of DMI on molecule-FM electrode interfaces of the MTJMSD devices.
We surmise that dissimilar molecule/FM electrode couplings are responsible for producing a DMI-like effect in our experimental studies, leading to the disappearance of magnetic contrast (Fig. 1). We hypothesize that DMI has subsequently created multiple bands of alternating magnetic phases that locally canceled the magnetic moments along the FM electrodes. Future work is necessary to conduct high spatial resolution MFM to verify the presence of alternating phases.
Future work is needed to realize switchability on MTJMSD with FM electrodes showing optimum DMI. The fabrication of successful DMI-based switchable MTJMSD will also require the encapsulation of MTJMSD for reliable long-term operation. The interdependent encapsulation process and the design of FM electrode configurations for achieving DMI in MTJMSD are complex multivariable processes that need significant planning and effort and are part of our future work.
Data availability
The data presented in this paper are available upon reasonable request by contacting the corresponding author at: ptyagi@udc.edu.
References
Sanvito, S. The rise of spinterface science. Nat. Phys. 6, 562–564 (2010).
Rocha, A. R. et al. Towards molecular spintronics. Nat. Mater. 4, 335–339 (2005).
Ghirri, A., Candini, A. & Affronte, M. Molecular spins in the context of quantum technologies. Magnetochemistry 3, 12 (2017).
Coronado, E. & Epsetin, A. J. Molecular spintronics and quantum computing. J. Mater. Chem. 19 (2009).
Chowrira, B. et al. Quantum advantage in a molecular spintronic engine that harvests thermal fluctuation energy. Adv. Mater. 34, 2206688 (2022).
Leuenberger, M. N. & Loss, D. Quantum computing in molecular magnets. Nature 410, 789–793 (2001).
Lehmann, J., Gaita-Arino, A., Coronado, E. & Loss, D. Quantum computing with molecular spin systems. J. Mater. Chem. 19, 1672–1677 (2009).
Leuenberger, M. N., Meier, F. & Loss, D. Quantum spin Dynamics in Molecular Magnets (Springer, 2003).
Bogani, L. & Wernsdorfer, W. Molecular spintronics using single-molecule magnets. Nat. Mater. 7, 179–186 (2008).
Liang, W., Shores, M. P., Bockrath, M., Long, J. R. & Park, H. Kondo resonance in a single-molecule transistor. Nature 417, 725–729 (2002).
Heersche, H. et al. Electron transport through single mn 12 molecular magnets. Phys. Rev. Lett. 96, 206801 (2006).
Chen, J., Reed, M., Rawlett, A. & Tour, J. Large on-off ratios and negative differential resistance in a molecular electronic device. Science 286, 1550–1552 (1999).
Sanvito, S. Molecular spintronics. Chem. Soc. Rev. 40, 3336–3355 (2011).
Sanvito, S. & Rocha, A. R. Molecular-spintronics: the art of driving spin through molecules. J. Comput. Theor. Nanosci. 3, 624–642 (2006).
Taylor, C. S. et al. Sputter gas damage in nanolayered Pt/Co/Ir-based synthetic antiferromagnets for top-pinned magnetic tunnel junctions. ACS Appl. Nano Mater. 6, 131–139. https://doi.org/10.1021/acsanm.2c03917 (2023).
Yuasa, S., Nagahama, T., Fukushima, A., Suzuki, Y. & Ando, K. Giant room-temperature magnetoresistance in single-crystal Fe/MgO/Fe magnetic tunnel junctions. Nat. Mater. 3, 868–871. https://doi.org/10.1038/nmat1257 (2004).
Urdampilleta, M., Klayatskaya, S., Ruben, M. & Wernsdorfer, W. Magnetic interaction between a radical spin and a single-molecule magnet in a molecular spin-valve. ACS Nano. 9, 4458–4464. https://doi.org/10.1021/acsnano.5b01056 (2015).
Tiusan, C. et al. Spin filtering effects in monocristalline Fe/MgO/Fe magnetic tunnel junctions. Mater. Sci. Eng. B-Solid State Mater. Adv. Technol. 126, 112–119. https://doi.org/10.1016/j.mseb.2005.09.054 (2006).
Faure-Vincent, J. et al. Interlayer magnetic coupling interactions of two ferromagnetic layers by spin polarized tunneling. Phys. Rev. Lett. 89, 107206 (2002).
Bollero, A., Baltz, V., Buda-Prejbeanu, L., Rodmacq, B. & Dieny, B. Origin and control of exchange-bias-like phenomenon in coupled ferromagnetic [Pt/Co]/NiFe bilayers. Phys. Rev. B. 84, 094423 (2011).
Barraud, C. et al. Phthalocyanine based molecular spintronic devices. Dalton Trans. 45, 16694–16699 (2016).
Katcko, K. et al. Encoding information on the excited state of a molecular spin chain. Adv. Funct. Mater. 31, 2009467 (2021).
Benini, M. et al. The Ferromagnetic Glass State: collapse of the standard ferromagnetic domain structure. (2024).
Mutunga, E., D’Angelo, C., Grizzle, A., Lamberti, V. & Tyagi, P. Dramatic effect of electrode type on tunnel junction based molecular spintronic devices. Org. Electron. 106, 106526. https://doi.org/10.1016/j.orgel.2022.106526 (2022).
Prokopuk, N. & Son, K. A. Alligator clips to molecular dimensions. J. Phys.: Condens. Matter. 20, 374116 (2008).
Metzger, R. M. Unimolecular electrical rectifiers. Chem. Rev. 103, 3803–3834 (2003).
Ashwell, G. J., Wierzchowiec, P., Bartlett, C. J. & Buckle, P. D. Molecular electronics: connection across nano-sized electrode gaps. Chem. Commun., 1254–1256 (2007).
Tyagi, P., Friebe, E. & Baker, C. Advantages of prefabricated tunnel junction-based molecular spintronics devices. Nano 10, 1530002 (2015).
Walker, A. V. Toward a new world of molecular devices: making metallic contacts to molecules. J. Vacuum Sci. Technol. A 31 (2013).
LI, J. C., ZHOU, W. U. J. Z., GONG, X. & C. & Latest studies on metal-molecule-metal junctions. Acta Phys. Chim. Sin. 29, 1123–1144 (2013).
Savadkoohi, M., Dahal, B. R., Grizzle, A., D’Angelo, C. & Tyagi, P. Interaction between magnetic molecules and two ferromagnetic electrodes of a magnetic tunnel junction (MTJ). J. Magn. Magn. Mater. 529, 167902 (2021).
Kronik, L. & Morikawa, Y. Understanding the metal–molecule interface from first principles. Mol.–Metal Interface. 51–89 (2013).
Tyagi, P. & Friebe, E. Large resistance change on magnetic tunnel junction based molecular spintronics devices. J. Mag. Mag. Mater. 453, 186–192. https://doi.org/10.1016/j.jmmm.2018.01.024 (2018).
Tyagi, P., Riso, C. & Friebe, E. Magnetic tunnel Junction Based Molecular Spintronics devices Exhibiting current suppression at room temperature. Org. Electron. 64, 188–194 (2019).
Tyagi, P. & Riso, C. Molecular spintronics devices exhibiting properties of a solar cell. Nanotechnology 30, 495401 (2019).
Tyagi, P. & Riso, C. Magnetic force microscopy revealing long range molecule impact on magnetic tunnel junction based molecular spintronics devices. Org. Electron. 75, 105421. https://doi.org/10.1016/j.orgel.2019.105421 (2019).
Dahal, B. R. et al. Easy axis anisotropy creating high contrast magnetic zones on magnetic tunnel junctions based molecular spintronics devices (MTJMSD). Sci. Rep. 12, 1–14 (2022).
Grizzle, A., D’Angelo, C., Martínez-Lillo, J. & Tyagi, P. Spin state of a single-molecule magnet (SMM) creating long-range ordering on ferromagnetic layers of a magnetic tunnel junction—a Monte Carlo study. RSC Adv. 11, 32275–32285. https://doi.org/10.1039/D1RA05473B (2021).
Savadkoohi, M., D’Angelo, C., Grizzle, A., Dahal, B. & Tyagi, P. Impact of ferromagnetic electrode length and thickness on magnetic tunnel junction-based molecular spintronic devices (MTJMSD). Org. Electron. 102, 106429. https://doi.org/10.1016/j.orgel.2022.106429 (2022).
Tyagi, P. & Baker, C. D’Angelo, C. Paramagnetic molecule induced strong antiferromagnetic exchange coupling on a magnetic tunnel junction based molecular spintronics device. Nanotechnology 26, 305602 (2015).
Wang, Z., Zhang, B., Cao, Y. & Yan, P. Probing the Dzyaloshinskii-Moriya interaction via the propagation of spin waves in ferromagnetic thin films. Phys. Rev. Appl. 10, 054018 (2018).
Ma, X. et al. Dzyaloshinskii-Moriya interaction across an antiferromagnet-ferromagnet interface. Phys. Rev. Lett. 119, 027202 (2017).
Yun, J. et al. Anisotropic interlayer dzyaloshinskii–Moriya interaction in synthetic ferromagnetic/antiferromagnetic sandwiches. Adv. Funct. Mater. 33, 2301731 (2023).
Dzyaloshinsky, I. A thermodynamic theory of weak ferromagnetism of antiferromagnetics. J. Phys. Chem. Solids. 4, 241–255 (1958).
Moriya, T. New mechanism of anisotropic superexchange interaction. Phys. Rev. Lett. 4, 228 (1960).
Wei, W. S., He, Z. D., Qu, Z. & Du, H. F. Dzyaloshinsky–Moriya interaction (DMI)-induced magnetic skyrmion materials. Rare Met. 40, 3076–3090 (2021).
Sankhi, B. R. et al. Engineering Pt/Co/AlO x heterostructures to enhance the dzyaloshinskii–Moriya interaction. J. Phys. Condens. Matter. 35, 145802 (2023).
Parkin, S. S., Hayashi, M. & Thomas, L. Magnetic domain-wall racetrack memory. Science 320, 190–194 (2008).
Fert, A., Reyren, N. & Cros, V. Magnetic skyrmions: advances in physics and potential applications. Nat. Rev. Mater. 2, 1–15 (2017).
Li, D. et al. An S = 6 cyanide-bridged octanuclear FeIII4NiII4 complex that exhibits slow relaxation of the magnetization. J. Am. Chem. Soc. 128, 4214–4215 (2006).
Li, D., Parkin, S., Clérac, R. & Holmes, S. M. Ancillary ligand functionalization of cyanide-bridged S = 6 FeIII4NiII4 complexes for molecule-based electronics. Inorg. Chem. 45, 7569–7571 (2006).
Tyagi, P., Li, D., Holmes, S. M. & Hinds, B. J. Molecular electrodes at the exposed edge of metal/insulator/metal trilayer structures. J. Am. Chem. Soc. 129, 4929–4938 (2007).
Brown, H., Grizzle, A., D’Angelo, C., Dahal, B. R. & Tyagi, P. Impact of direct exchange coupling via the insulator on the magnetic tunnel junction based molecular spintronics devices with competing molecule induced inter-electrode coupling. AIP Adv. 11 (2021).
Grizzle, A., D’Angelo, C. & Tyagi, P. Monte Carlo simulation to study the effect of molecular spin state on the spatio-temporal evolution of equilibrium magnetic properties of magnetic tunnel junction based molecular spintronics devices. AIP Adv. 11 (2021).
Coey, J. M. Magnetism and Magnetic Materials (Cambridge University Press, 2010).
Newman, M. E. & Barkema, G. T. Monte Carlo Methods in Statistical Physics (Clarendon, 1999).
Dahal, B. R. et al. Easy axis anisotropy creating high contrast magnetic zones on magnetic tunnel junctions based molecular spintronics devices (MTJMSD). Sci. Rep. 12, 5721 (2022).
Selzer, Y. & Allara, D. L. Single-molecule electrical junctions. Annu. Rev. Phys. Chem. 57, 593–623 (2006).
Martinek, J. et al. Kondo effect in the presence of itinerant-electron ferromagnetism studied format? with the numerical renormalization group method. Phys. Rev. Lett. 91, 247202 (2003).
Pasupathy, A. N. et al. The Kondo effect in the presence of ferromagnetism. Science 306, 86–89 (2004).
Park, K. Exchange coupling and contribution of induced orbital angular momentum of low-spin Fe3 + ions to magnetic anisotropy in cyanide-bridged Fe2M2 molecular magnets: spin-polarized density-functional calculations. Phys. Rev. B. 74, 224440 (2006).
Dahal, B. R., Grizzle, A., D’Angelo, C., Lamberti, V. & Tyagi, P. Competing easy-axis anisotropies impacting magnetic tunnel junction-based molecular spintronics devices (MTJMSDs). Int. J. Mol. Sci. 23, 14476 (2022).
Acknowledgements
We acknowledge our funding sources: the National Science Foundation CREST Award, Grant number HRD- 1914751, NSF-MRI grant 1920097, Department of Energy/ National Nuclear Security Agency (DE-FOA-0003945) and NASA MUREP Institutional Research Opportunity Grant under Cooperative Agreement #80NSSC19M0196.
Funding
This research was funded by the National Science Foundation CREST Award, grant number HRD- 1914751, NSF-MRI grant 1920097, Department of Energy/ National Nuclear Security Agency (DE-FOA-0003945) and NASA MUREP Institutional Research Opportunity Grant under Cooperative Agreement #80NSSC19M0196.
Author information
Authors and Affiliations
Contributions
P.T. conceived the concept of MCS, supervised the project, and conducted the MTJMSD experiment. B.R.S. studied and analyzed the DMI impact on MTJMSD utilizing the Monte Carlo Simulation program developed by C.D., and prepared the manuscript, which P.T. then edited. D.T. assisted B.R.S. in conducting simulations and analyzing the data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sankhi, B.R., D’Angelo, C., Thompson, D. et al. Effect of Dzyaloshinskii Moriya interaction on magnetic tunnel junction based molecular spintronics devices (MTJMSD). Sci Rep 15, 5576 (2025). https://doi.org/10.1038/s41598-025-88741-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-88741-7
