
Abstract
In this theoretical analysis, the pressure-dependent structural, electronic, mechanical, and optoelectronic properties of LiMCl3 (M = Mg, Be) have been calculated using density functional theory within the framework of the GGA PBE and hybrid HSE06 functional. At ambient pressure, the calculated lattice parameters of LiMCl3 match well with previously reported values, validating the accuracy of this study. Geometry optimization reveals that under increasing hydrostatic pressure, both the lattice parameters and the unit cell volume decrease. Additionally, the band structure exhibits notable phenomena over the pressure range from 0 to 100 GPa. For the LiMgCl3 compound, the bandgap decreases from an indirect bandgap of 4 eV to a direct bandgap of 2.563 eV. Similarly, LiBeCl3 shows an indirect bandgap that decreases from 2.388 eV to 0.096 eV over the pressure range from 0 to 100 GPa. The optical properties of LiMCl3, including absorption coefficient, reflectivity, refractive index, dielectric function, and conductivity, have been calculated throughout the study under varying pressure conditions. The analysis reveals that the optical properties of LiMCl3 (M = Be, Mg) enhance with increasing hydrostatic pressure, thereby rendering these materials more suitable for optoelectronic applications. To assess the stability of these compounds, elastic constants were analyzed, indicating that LiMCl3 exhibits ductile and anisotropic characteristics under different pressure conditions. These investigated materials are suitable for use in optoelectronic devices due to their favorable physical properties under different pressure circumstances.
Similar content being viewed by others
Introduction
Perovskites are demonstrating exceptional performance due to their numerous optoelectronic properties1,2,3,4,5,6,7,8,9, making them suitable for various optoelectronic applications, including solar cells10,11,12,13,14,15,16,17,18, photodetectors19, light-emitting diodes (LEDs)19, lasers20, memristors21, and artificial synapse devices22. Their properties can vary widely, ranging from metallic to semiconducting to conductive23, depending on the composition of the different materials used24,25. Perovskites are often referred to as the ‘department store of physical properties’ due to their photocatalytic26, dielectric27, ferroelectric28, pyroelectric29, piezoelectric30, magnetic31, superconductivity32, and ionic conductivity33. Perovskites often have the formula “ABX3”, in which “X” is an anion, usually oxygen or a halogen, and “A” and “B” are cations. Making sure that neither the A-site nor the B-site cations contain lead results in the formation of lead-free perovskites. Lead (Pb²⁺) is instead replaced with alternative cations including Sn²⁺, Ge²⁺, Mg²⁺, etc. Lead-free metal halide perovskites are increasingly used in various devices due to their non-toxicity, supporting the green technology revolution34,35,36. These materials also possess outstanding photoelectronic and thermoelectric properties, including a high dielectric constant, a low exciton binding energy, and a high absorption coefficient. These properties allow for efficient electron-hole pair photogeneration, which makes them suitable for using as absorber materials in solar cells as well as thermoelectric applications1,2,3,4,5,37,38,39,40. Over the past years, lead-free perovskite-based solar cells have demonstrated greater than 20% efficiency both experimentally and in theoretical studies41,42. Under light exposure, halide perovskites exhibit effective flexoelectric coefficients that are too much higher than typical dielectric materials43. Flexoelectricity involves generating energy through non-uniform deformations of dielectric materials, regardless of their symmetry. Additionally, the use of piezoelectric materials in a wide range of electronic devices is expanding the piezoelectric device market globally on a daily basis. Thus, it is imperative to employ more lead-free perovskites rather than lead perovskites in order to address environmental and health issues. Lead-free perovskites are also increasingly finding applications in energy storage systems including Li-ion and photo rechargeable batteries. Employing lightweight materials for energy conversion and storage renders them exceptionally well-suited for integration into electric vehicles and portable electronic devices44.
Numerous techniques including applying pressure45,46 and adding dopants47,48 can alter the physical properties of materials, rendering them suitable for various optoelectronic devices36,49. To increase photocurrent density, Sr is introduced into CsPbBr3 perovskite, as reported by Arindam et al.50. Linh et al. reported that (Bi0.5M0.5)TiO3’s direct band gap increased when larger ionic radius alkali metals (Li, Na, and K) were substituted51. S Kurra et al. reported enhanced photocatalytic activities of Na0.5Bi0.5TiO3 perovskite when it is doped with metal ions (Ag+, Cu2+, and Sn2+). Moreover, in a prior investigation, it was illustrated that the application of hydrostatic pressure leads to a reduction in the bandgap, shifting from the ultraviolet to the visible spectrum. This manipulation also facilitates a transition from an indirect to a direct bandgap, thereby augmenting the material’s viability for optoelectronic applications52. M. Aslam et al. also reported a decrease in bandgap and enhanced optoelectronic properties of CdZrO3 perovskite under various hydrostatic pressure conditions in their study53. Various alkaline earth metal perovskites are increasingly being considered for photovoltaic applications due to their suitable bandgaps and higher absorption coefficients54,55. Recent investigations using first-principles DFT calculations have highlighted the potential of alkaline metal-based halide perovskites, such as LiMgCl3 and LiBeCl3, for applications in energy storage devices, scintillating materials, and various modern technologies56. In a previous study, ‘Be’ and ‘Mg’ have a significant impact on the structural, elastic, and optoelectronic characteristics of ternary LiMCl3 (R = Mg and Be) perovskites. The bandgap energy of these materials indicates their insulating nature, which is a limit to employ these materials in optoelectronic devices that need comparatively narrow bandgaps for easier transition of electrons. As pressure has been widely studied for its ability to modify the physical properties of various materials, there remains a lack of theoretical or experimental investigations into the effects of pressure on the cubic phase of LiMgCl3 and LiBeCl3. Therefore, we applied hydrostatic pressure to observe the changes in their physical properties and evaluate their potential viability for optoelectronic applications.
The Generalized Gradient Approximation (GGA) and the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional have been employed within the CASTEP for this calculation. Mechanical properties are analyzed to assess the stability of the investigated materials. Band structure, total density of states (TDOS), and partial density of states (PDOS) have been calculated across a pressure range of 0 to 100 GPa. Additionally, optical properties such as reflectivity, refractive index, absorption coefficient, and dielectric functions have been evaluated to explore their potential applications in optoelectronic devices. The selection of this pressure range may have been motivated by the goal of achieving better optoelectronic properties compared to ambient pressure, specifically to enhance absorption in both the visible and UV photon energy ranges, with a primary focus on the UV region. This is especially relevant for multilayer solar cells, such as tandem solar cells, where enhanced absorption in the UV region can improve the performance of solar cell devices. Additionally, reducing the bandgap facilitates a smoother transition of electrons from the valence band to the conduction band, which benefits optoelectronic devices. Given these considerations, our study demonstrates that these outcomes are achievable within a pressure range up to 100 GPa, and therefore, further investigation beyond this pressure condition was not pursued.
Computational details
In this study, First-principles calculations based on Density Functional Theory (DFT)57, are implemented through the Cambridge Serial Total Energy Package (CASTEP) code58,59. The interactions between ions and electrons are described using ultrasoft pseudopotentials in conjunction with the GGA-PBE exchange-correlation functional60,61. For geometry optimization, the Broyden–Fletcher–Goldfarb–Shanno (BFGS) technique has been employed62, while electronic structure calculations are conducted using density mixing. Geometry optimization involves relaxing the unit cell structures while computational physical parameters are determined under conditions of zero applied pressure and a temperature of 0 K. The plane wave cut-off energy has been set as 550 eV where For Brillouin zone sampling, the Monkhorst-Pack method62 is employed with a 5 × 5 × 5 k-point mesh. This ensures sufficient convergence of both the overall energy and the geometrical configuration. The convergence criteria are defined by the following parameters: a maximum ionic Hellmann-Feynman force of 0.01 eV/Å, a maximum displacement of 5 × 10⁻⁴ Å, a maximum stress of 0.02 GPa, and a total energy difference within 5 × 10⁻⁶ eV per atom. For visualizing the optimized structures, VESTA software has been used in this study63. The “finite-strain” approach included in the CASTEP code is used to obtain the elastic constants64. The ELATE program65 is then utilized to generate three-dimensional mechanical anisotropic contour plots for various key mechanical parameters, such as Young’s modulus (E), shear modulus (G), and Poisson’s ratio (ν).
Results and discussion
Structural properties
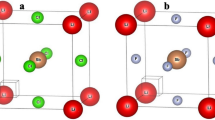
The cubic perovskite-type structure of lead-free halide LiMCl3 (M = Mg, Be) crystallizes with the space group Pm3̅m (#221). Fig. 1 illustrates that each unit cell of cubic LiMCl3 (M = Mg, Be) perovskites contains five atoms: Li occupies the corner position with the Wyckoff position 1a (0,0,0), Mg (Be) located at the body-centered position 1b (0.5, 0.5, 0.5), and Cl atoms are positioned at the face-centered positions with the Wyckoff position 3c (0, 0.5, 0.5). This study analyzes the pressure-induced structural properties of LiMCl3 compounds through geometry optimization under various hydrostatic pressures. At ambient pressure, the lattice parameters for LiMgCl3 and LiBeCl3 compounds have been calculated as 4.96 Å and 4.56 Å, respectively. These values closely align with previously reported data57, validating the accuracy of this study using the GGA-PBE approximation. The Murnaghan state equation has been employed to determine these lattice parameters, ensuring that the total energy of the investigated structures is minimized during the process. As the pressure increases from 0 GPa to 100 GPa with a 20 GPa step, the lattice parameters and unit cell volume of both compounds change. Detailed lattice parameters and cell volume of LiMCl3 compounds under varying pressures are listed in Table 1. Figs. 2(a) and (b) illustrate the decrease in lattice parameters and unit cell volume of both investigated compounds with increasing hydrostatic pressure, indicating a reduction in interatomic distances, respectively. This pressure-induced reduction in lattice parameters and unit cell volume has also been observed in previous studies55,66,67. The bond lengths of Li-Li, Li-Cl, Li-Mg (Be), and Mg (Be)-Cl decrease with increasing hydrostatic pressure, as shown in Figs. 2(c) and (d). Under increasing hydrostatic pressures, the repulsive forces between atoms become stronger, leading to an increase in the hardness of LiMCl3 (M = Mg, Be) perovskites. Under higher hydrostatic pressure, the interatomic spacing decreases, reducing the bond length and subsequently narrowing the energy gap between the valence band maximum (VBM) and conduction band minimum (CBM)68,69. The conduction band, which is more sensitive to changes in bond length than the valence band, shifts to lower energies at a faster rate, resulting in a noticeable bandgap decrease in LiMCl3 compounds. Due to the reduction in bond length, the repulsive interactions between atoms strengthen, increasing the resistance to compression under higher pressure conditions. Therefore, the relationship between bond length and hardness is inversely proportional; longer bonds result in weaker atomic attraction, while shorter bonds indicate stronger interactions. As this study shows the decreasing nature of bond length, which in turn leads to an increase in hardness, supported by the hardness values of this work. Similar effects have also been observed in previous studies70. Moreover, with the decrease in bond length, Poisson’s ratio of LiMCl3 increases, which also aligns with previously reported literature71,72. For experimental analysis, understanding the stability of the investigated LiMCl3 compounds is essential. As LiMCl3 are ABX3 type perovskite, we calculated both the Goldschmidt tolerance factor (t) and the octahedral factor (\(\:\mu\:\)) by the following formulas where X denotes the ionic radius of Cl, RB denotes the ionic radius of Mg and Be, and RA denotes the ionic radius of Li.
Stability in a cubic structure requires the tolerance factor to fall within the range of 0.81 to 1.00, while the octahedral factor should be between 0.37 and 0.859. Our calculations confirm that both materials meet these criteria, ensuring their structural stability under ambient pressure. According to earlier research, the tolerance factor (t) can also be computed using the following alternate techniques, which use the bond lengths of Li-Cl, M (Mg, Be)-Cl.
According to earlier research, the structure is stable if the t value falls between 0.93 and 1.0245; Table 2 shows that our compounds under various hydrostatic pressures fall within this range.

Crystal structure of cubic (a) LiMgCl3 and (b) LiBeCl3.

Pressure-induced variation of (a) lattice parameters, (b) unit cell volume of LiMCl3, and variation on bond length of (c) LiMgCl3 and (d) LiBeCl3.
Phonon dispersion
The interaction between phonons’ momentum and energy in a crystal lattice is known as phonon dispersion. Quantized lattice vibration is known as phonons and their dispersion offers vital insights into the thermal behavior and vibrational characteristics associated with numerous materials73. Using a phonon dispersion curve, one can observe several bands that correspond to different frequencies of the crystal structure. In this study, phonon calculation has been employed to observe the stability of the investigated compounds under different pressure circumstances. It is evident that both materials with ambient pressure and higher hydrostatic pressure conditions vibrate in the positive mode, as can be seen from the computed phonon spectra in Fig. 3. The crystal lattice’s phonon dispersion curve displays positive frequencies that correspond to significant and physically acceptable vibrational modes that can be employed in optoelectronic devices.

Phonon dispersion of LiMCl3 (M = Mg, Be) compounds for 0 GPa and 100 GPa pressure conditions.
Electronic properties
The energy bands are a representation of the different energy levels that electrons can potentially occupy. Understanding the electronic band structure and Density of states (DOS) of a material is crucial for assessing the electronic behavior of a material. Band structure offers insights into the diverse energy levels and electronic states that consist in a material, while DOS elucidates the distribution of these energy states relative to energy. The electronic band structures of LiMCl3 along the high-symmetry points “X-R- M- G- R” in the first Brillion zone at various hydrostatic pressure ranges, from 0 to 100 GPa pressure with 20 GPa interval, are shown in Figs. 4 and 5. The energy range of the band structures spans from − 5 eV to + 8 eV, with the Fermi energy level positioned at 0 eV, marked by a black dotted line. The portion of the energy band situated above the Fermi level is known as the conduction band, while the segment below the Fermi level is termed the valence band. In this investigation, the GGA-PBE & HSE06 approximation has been considered to calculate the bandgap of the investigated compounds (Table 3). While some studies express concerns about the accuracy of GGA functional for bandgap calculations, it’s noteworthy that GGA functional don’t consistently underestimate the bandgap, as evidenced by several studies74,75. Under non-pressurized conditions, both LiMgCl3 and LiBeCl3 compounds exhibit CBM at G symmetry points and VBM at R symmetry points, resulting in an indirect bandgap of 4 eV for LiMgCl3 and 2.3 eV for LiBeCl3. The calculated bandgaps of these compounds closely align with other theoretical calculations, validating the accuracy of the GGA-PBE functional utilized in this study56. However, no experimental studies have been conducted on these materials to date. When hydrostatic pressure is applied, the CBM of LiMgCl3 compounds shifts towards lower energy levels along the G symmetry point. This shifting of energy band results in a decrease in bandgaps, providing significant insights into the inverse connection between bandgap and pressure, as supported by Fig. 4. This decrease in band gap can be attributed to a myriad of factors influenced by pressure, including alterations in bond angles, atomic configuration, and interatomic distance. Moreover, increased pressure also amplifies the potential between electrons and ions, leading to a reduction in lattice parameters and contribute to bandgap reduction. The transition from an indirect to a direct bandgap can be attributed to several factors, primarily the reduction in atomic distances within the crystal under higher pressure, which enhances orbital overlap between adjacent atoms. This increased overlap alters the electronic band structure, leading to changes in the energy levels of the conduction band minimum (CBM) and valence band maximum (VBM). In the LiMCl3 compounds, the CBM and VBM are initially located at different k-points, indicating a momentum mismatch. At higher hydrostatic pressure LiBeCl3 also shows the indirect bandgap with reduced bandgap energy. A significant reduction of energy gap is observed in this compound as depicted in Fig. 5, where its band gap experiences a notable decrease from 2.4 eV at 0 GPa to only 0.096 eV under 100 GPa pressure. This leads to speculation that over 100 GPa pressure may drive the compound toward a metallic state. This process reduces the distance between atoms in the LiBeCl3 compound resulting in a narrow band gap. By making it simpler to transfer electrons from VB to the CB, this shift in the band gap may increase the effectiveness of optoelectronic devices as reported by previous studies76,77,78. However, in indirect bandgap materials such as LiBeCl3, electron transitions require both photon absorption for energy transfer and phonon interaction to provide the necessary momentum change. LiMgCl3 also demonstrates a reduction in band gap under pressure, with a notable decrease from 4 eV to 2.563 eV at 100 GPa pressure (Table 3). However, based on the HSE06 calculation, the bandgap of LiMgCl₃ decreases from indirect 4.12 eV to direct 2.61 eV, while that of LiBeCl₃ decreases from indirect 3.71 eV to indirect 1.32 eV. Indirect bandgap materials are important in many devices, but direct bandgap materials are typically chosen for optoelectronic applications because of their effective light emission and absorption capabilities. Si-based devices also remain essential in infrared photodetectors, as highlighted by previous studies79,80. When exposed to a propagating far-field light source, silicon (Si), a commonly used indirect bandgap material, requires phonon assistance to excite carriers during photoexcitation which results in a decreased excitation efficiency. In order to overcome this constraint, scientists frequently use plasmonic resonance to increase the light field’s intensity80. Additionally, indirect bandgap semiconductors have demonstrated exceptional performance in thermoelectric applications, where energy conversion efficiency is maximized by an ideal bandgap that is usually a particular multiple of the operating temperature81.

Band structures of LiMgCl3 under different hydrostatic pressure with GGA-PBE exchange-correlation functional.

Band structures of LiBeCl3 under different hydrostatic pressures with GGA-PBE exchange-correlation functional.
To delve into the electronic band structure in-depth, total density of states (TDOS) and partial density of states (PDOS) is used to elucidate the contribution of different states to the formation of conduction and valance bands. The TDOS is the sum of all the states of each atom, and PDOS is the sum of all the contributions of each sub-state to the TDOS. Fig. 6 illustrates the TDOS of LiMCl3 compounds across an energy range from − 8 eV to + 10 eV under varying hydrostatic pressures. As pressure increases, notable variations in the TDOS become apparent which can be further elucidated by analyzing the PDOS as depicted in Figs. 7 and 8. For both LiMgCl3 and LiBeCl3 compounds, the TDOS below the fermi level known as valance band mostly originated from the Cl-3p state with a tiny contribution from the Mg-2p (Be-2p) state. TDOS above the Fermi level, known as the conduction band, is predominantly dominated by the Li-2s state, which plays a crucial role in bandgap reduction in high-pressure circumstances, comparable to the influence of Mg-2p (Be-2p) and Mg-2s (Be-2s) states. The hybridization of Li-2s and Mg-2s (Be-2s) also contributes to the reduction in bandgap. It’s noteworthy that the non-zero value of DOS at 0 eV underscores the metallic nature of LiMgCl3 at high pressure (100 GPa). In contrast, for LiBeCl3, a more pronounced metallic behavior has been observed at higher pressures (40–100 GPa) circumstances.

The total density of states (TDOS) of (a) LiMgCl3 and (b) LiBeCl3 compounds under hydrostatic pressures (0-100 GPa).

The partial density of states (PDOS) of LiMgCl3 under pressures (0-100 GPa).

The partial density of states (PDOS) of LiBeCl3 under pressures (0-100 GPa).
Optical properties
The response of a material to electromagnetic radiation is crucial for assessing its potential in various industrial applications. This response is characterized by several energy/frequency-dependent parameters such as dielectric constant, absorption spectra, refractive index, reflectivity, and conductivity. To gain a comprehensive understanding, this section investigates the optical properties of LiMgCl3 and LiBeCl3 at different hydrostatic pressures (0-100 GPa) across an energy range of 0 eV to 30 eV.
The dielectric function ε(ω), which signifies a material’s linear response to an external electromagnetic field, offers valuable insights into its optical properties. The relationship between optical properties and the dielectric function, arising from light’s impact as it travels through materials, is mathematically expressed as ε(ω) = ε1(ω) + ε2(ω), with ε1(ω) and ε2(ω) representing the real and imaginary part of the dielectric function, respectively82.
In this study, the dielectric function ε(ω) of LiMCl3 (M = Mg, Be) is illustrated in Fig. 9. The real component of the dielectric function (RDF) serves as an efficient parameter, offering crucial information about the charge carrier recombination rate and thereby the overall effectiveness of optoelectronic devices83. Moreover, it is evident from the RDF that the spectral patterns of the two materials are consistent with each other.

Pressure-induced variation of Real part of DF: (a) LiMgCl3, (b) LiBeCl3 and Imaginary part of DF: (c) LiMgCl3, (d) LiBeCl3.
At ambient pressure, the static dielectric constant ε1(0) of LiMgCl3 is measured to be 2.82, which increases to 4.35 at 100 GPa pressure, while LiBeCl3 shows ε1(0) of 3.58 at ambient pressure, increasing to 5.82 under 100 GPa pressure as shown in Fig. 9(a) and (b). Materials with a substantial band gap typically exhibit a low static dielectric constant. The ε1(0) value increases with induced pressure for both compounds, correlating with a decrease in band gap under pressures. This decrease in band gap ensures a greater number of electrons can transfer from valence bands (VBs) to conduction bands (CBs), thereby reducing the recombination rate52. Additionally, both LiMgCl3 and LiBeCl3 exhibit negative ε1(ω) values within an energy range from 9.2 eV to 18 eV and 10 eV to 18 eV, respectively, suggesting drude-like characteristic of these compounds, confirming their increasing metallic nature under high-pressure circumstances84.
The imaginary component of the dielectric function (IDF) plays a crucial role in elucidating the absorption behavior of materials85. It is directly influenced by the band structure and DOS of a material. As depicted in Figs. 9(c) and (d), the peak of ε2(ω) for LiMCl3 (M = Mg, Be) compounds increases as pressure rises. This behavior is in agreement with the absorption spectra depicted in Figs. 10(a) and (b). Additionally, these peaks shift towards higher energies with increased pressure, attributed to the widening of the valence band and the downward shift of the lower valence band86. ε2(ω) tends to decrease from its peak value with increasing photon energy, eventually reaching zero. The distinctive pattern of higher ε2(ω) values at lower frequencies and smaller ε2(ω) values in the higher energy range highlights the potential applicability of LiMCl3 (X = Mg, Be) in microelectronics and integrated circuits. This superiority is notably amplified under pressure, indicating the material’s enhanced performance characteristics in such applications75.

Pressure-induced variation of Absorption coefficient: (a) LiMgCl3, (b) LiBeCl3 and Conductivity: (c) LiMgCl3, (d) LiBeCl3.
Studying the absorption coefficient (α) is pivotal in understanding how a material interacts with specific photon energies, influencing the attenuation of light87. This investigation provides valuable insights into a material’s compatibility and potential for enhanced performance in various device applications. The absorption coefficient variations of LiMgCl3 and LiBeCl3 across different hydrostatic pressures are depicted in Figs. 10(a) and (b). In both materials, absorption does not begin at 0 eV due to the presence of optical bandgap. At ambient pressure, absorption starts at about 2.391 eV and 3.978 eV of photon energy for LiBeCl3 and LiMgCl3 respectively, signifying their specific optical bandgap energies. For both compounds, a major absorption peak has been observed within a broad region of photon energy. By applying pressure, the absorption spectrum for both compounds becomes narrower, heightened, and broadened, falling within the ultraviolet region. This indicates the potentiality of LiMgCl3 and LiBeCl3 compounds in fabricating devices for sterilizing surgical equipment88. These two materials demonstrate higher absorption in the UV region, with their absorption rate increasing as hydrostatic pressure rises, despite a decrease in their bandgap under incremental pressure. This behavior underscores the motivation to apply higher hydrostatic pressure to these materials. These materials are suitable for use in tandem solar cells, where each layer is designed to absorb different parts of the solar spectrum. These materials can be used as the top layer to absorb high-energy UV photons, while other layers are optimized for absorbing visible and infrared light. This configuration helps reduce thermal losses, resulting in more efficient utilization of these materials. Under high pressure, LiMgCl3 exhibits a direct bandgap, while LiBeCl3 shows an indirect bandgap under varying pressure conditions. As a result, LiMgCl3 is likely to be more efficient in these conditions, as electron transitions in direct bandgap materials do not require phonon assistance, eliminating the momentum mismatch between the valence and conduction bands.
The optical conductivity (σ) stands as a fundamental parameter that characterizes a material’s electromagnetic response89. Figs. 10(c) and (d) depict the real part of conductivity under various hydrostatic pressures (0–100 GPa). It is observed for both materials that σ does not initiate at 0 eV. This may be attributed to the specific optical bandgap of these materials under different hydrostatic pressures. However, beyond their optical bandgap, both materials smoothly reach their peaks under different hydrostatic pressures. The figures clearly show that the optical conductivity enhances with increasing pressure, correlating with the heightened absorption coefficient under pressures. This enhancement in conductivity is attributed to electron transitions from the valence to the conduction band triggered by light absorption53. Intriguingly, the peaks in σ become sharper under hydrostatic pressure. For LiMgCl3, the conductivity peak shifts from 9.18 eV at zero pressure to 9.83 eV at 100 GPa, while for LiBeCl3, the peak shifts from 8.9 eV to 10.15 eV under similar pressure conditions. These shifts align with the pressure-induced changes in the band structure, characterized by a narrowing band gap with increasing pressure90.
The reflectivity spectra (R) of LiMgCl3 and LiBeCl3 under varying pressures are depicted in Figs. 11(a) and (b). R is a key optical parameter, indicating a material’s ability to reflect light energy, and is inversely related to its light absorption91. Both materials show similar trends in their reflectivity spectra. At lower photon energies, the reflectivity remains relatively low and experiences minimal changes with increasing pressure. However, within the energy range of 10 eV to 20 eV, a noticeable variation in reflectivity has appeared under pressure. This observation implies that LiMgCl3 and LiBeCl3 can be effectively controlled using external pressure, making them suitable coating materials within this energy range92.

Pressure-induced variation of Reflectivity: (a) LiMgCl3, (b) LiBeCl3 and Refractive index: (c) LiMgCl3, (d) LiBeCl3.
The refractive index, a dimensionless parameter indicating electromagnetic wave speed in a material, is illustrated in Fig. 11(c) and (d) across the energy of 0 eV to 100 eV and pressures from 0 to 100 GPa. Analysis of the figures reveals that, at ambient pressure, normal dispersion is observed between 0 eV – 7.5 eV and 16.3 eV – 30 eV, while abnormal dispersion is noted in the 7.5 eV-16.3 eV photon energy range. Additionally, LiBeCl3 exhibits a consistently higher maximum refractive index compared to LiMgCl3 at 0 GPa pressure. This difference signifies a stronger electronic polarization effect in LiBeCl3, leading to a greater delay in light propagation. When applying pressure, both materials display an increase in refractive index, reaching a maximum of around 10 eV due to the growing number of electronic oscillators per unit volume93. However, at a photon energy of 11.35 eV, the refractive index falls below unity for both materials, regardless of pressure, due to the group velocity of incident radiation surpassing the speed of light94. This indicates that the investigated materials transform into a superluminal medium when subjected to high-energy photons.
Mechanical properties
Elastic constants (Cij) are crucial parameters for understanding the mechanical properties of a material, as they provide valuable insights into the material’s stability and stiffness95,96. By analyzing the Cij values, one can determine a crystal’s ability to resist external forces, thereby gaining practical information about its potential applications. Thermal expansion, Gruneisen factor, specific heat, and Debye temperature are among the thermodynamic properties closely intertwined with elastic constants. These parameters also offer valuable insights into elastic properties such as thermo-elastic stress, internal strain, load deflection, sound velocity, and fracture toughness66. To calculate Cij, the finite strain theory has been used66. There are three independent elastic constants for a cubic phase of a compound including, longitudinal compression (C11), shear modulus (C44), and transverse expansion (C12). Under varying pressures, a material’s lattice parameters undergo alterations, accentuating the necessity to calculate elastic constants across various pressure conditions. This study also delves into the determination of pressure-induced elastic constants for LiMCl3, aiming to discern changes in elastic constants and mechanical properties. Table 4 presents the elastic constant values under distinct hydrostatic pressures. The stability of the cubic structure is evaluated using the Born stability criteria97,98,99: (C11 + 2C12) > 0, C44 > 0, and (C11 – C12) > 0. However, the Cij values of both investigated compounds satisfy these criteria, thereby confirming the stability of these compounds under varying hydrostatic pressures.
The mechanical robustness of different optoelectronic devices under thermal cycles is greatly influenced by the stiffness of the material, which should be carefully chosen to prevent mechanical delamination. Based on Young’s and bulk modulus values, the stiffness of the studied LiMCl3 compounds increases with rising hydrostatic pressure. Higher stiffness reduces deformation, which can improve the structural integrity of the device by reducing cracks and delamination, two main failure modes in optoelectronic devices100.
Cauchy pressure is used to determine the ductility or brittleness of a material based on its positive or negative values101. A positive value of Cauchy pressure indicates that the material is ductile, while a negative value signifies that the material is brittle. The investigated compounds exhibit positive Cauchy pressure values under different hydrostatic pressures, confirming their ductile behavior. As the pressure increases, the positive Cauchy pressure values become even more positive, highlighting the increased ductility of the LiMCl3 (M = Mg, Be) compounds.
The bulk modulus and shear modulus are calculated using the Voigt102, Reuss103, and Hill approximations104, with the Hill approximation often considered the final value due to its closer alignment with experimental results, as supported by several studies. The bulk modulus, Young’s modulus, and shear modulus using the Voigt, Reuss approximations can be determined from the following relationships105,106:
Mechanical properties, such as bulk modulus, Young’s modulus, Poisson’s ratio, and Pugh’s ratio, determined in this study are summarized in Table 5. It’s clear from the data that as pressure escalates, the values of bulk modulus (B), Poisson’s ratio (ν), and shear modulus (G) also increase. This trend signifies a rise in the stiffness and hardness of LiMCl3 (M = Mg, Be) compounds under increasing pressures.
Poisson’s and Pugh’s ratios are calculated as well in this study to provide precise insights into a material’s brittleness or ductility. According to Table 5, the threshold value for Poisson’s ratio is 0.26; above this value, a material is considered ductile, while below it, the material is considered brittle. The investigated compounds in this study exhibit ductile behavior under varying hydrostatic pressures. Moreover, Poisson’s ratio provides information on whether a material’s bonding force is central or non-central107. A central force is directed toward a fixed point and its magnitude depends on the distance of the particle from that point. In contrast, a non-central force does not simply depend on the distance between the centers of two interacting bodies108. Bonding in solids is considered central when the Poisson’s ratio is between 0.25 and 0.50. This study’s findings confirm that the LiMCl3 (M = Mg, Be) compounds exhibit central force characteristics. Similarly, to Poisson’s ratio, Pugh’s ratio (B/G) serves as a critical indicator for discerning the ductility and brittleness of a material. The materials above the threshold value of 1.26 are considered ductile, while those below are brittle. Figs. 12(a) and (b) visually presented the variations in Poisson’s ratio and Pugh’s ratio respectively under different hydrostatic pressures. Notably, at ambient pressure, the materials exhibit ductile behavior, with ductility further increasing as pressure rises which is also supported by the analysis of Poisson’s ratio and Cauchy pressure, proofing the accuracy of this study. This observation aligns with the analyses of Poisson’s ratio and Cauchy pressure, validating the accuracy and robustness of this study.

(a) Pugh’s ratio and (b) Poisson ratio of LiMgCl3 and LiBeCl3 under pressures.
The machinability index (\(\:{\mu\:}_{M}\)) stands as a crucial mechanical property, defining the ease or difficulty with which a material can be machined using a cutting tool. It closely associates with both the bulk modulus and the C44 elastic constant, as delineated by the following equation:
Machinability holds paramount importance for materials that are earmarked for industrial applications. It governs various factors, including the selection of cutting tools, the optimal cutting speed, and the geometry of the cutting tool, etc. Enhanced machinability results in heightened lubricating properties, diminished friction values, and higher plastic strain. Pressure-induced variations in machinability have been investigated in this study and are presented in Table 5. A minor variation in the machinability index has been observed under increasing hydrostatic pressure for the LiBeCl3 compound. Conversely, higher \(\:{\mu\:}_{M}\) values are observed for LiMgCl3 under high-pressure circumstances, suggesting exceptional lubricating properties of this compound.
Vickers hardness is indeed a crucial mechanical property, reflecting a material’s resistance to deformation. Understanding a material’s elastic and plastic behaviors significantly depends on knowing its hardness value. There’s a close relationship between hardness and Poisson’s ratio, which is established by the following relation109:
Table 5 suggests that the hardness of LiMCl3 (M = Mg, Be) compounds typically increases with rising pressure. Notably, LiMgCl3 shows a significant increase under high-pressure conditions. In contrast, the hardness LiBeCl3 demonstrates a comparatively milder increasing trend, suggesting lower hardness compared to LiMgCl3.

3D anisotropic representation of (a) Young’s modulus, (b) shear modulus, and (c) Poisson’s ratio of LiMgCl3 and LiBeCl3 under 0 GPa and 100 GPa pressures.
The anisotropy factor of a crystal is crucial as it strongly correlates with the likelihood of inducing microcracks in materials. The performance of a compound in a particular technological application depends significantly on its level of anisotropy110. As the investigated compounds possess cubic symmetry, A1, A2, and A3 are equal. The anisotropy of a material can be evaluated using its elastic constants through the following relation111,112:
When the anisotropic factor value is 1, a material is considered isotropic, while deviation from this unity value indicates the degree of anisotropy present in the material. Anisotropic factor values of LiMCl3 under hydrostatic pressures are presented in Table 5. It is evident that the degree of anisotropy increases with pressure for LiMgCl3, whereas the opposite trend has been noticed in LiBeCl3 where the value of the anisotropy factor tends to approach towards unity as pressure rises. To provide a clearer visualization, Fig. 13 presents the 3D contour plots, illustrating the directional dependence of the anisotropy factor on Young’s modulus, shear modulus, and Poisson’s ratio for LiMCl3 (M = Mg, Be) compounds under pressures of 0 GPa and 100 GPa. Typically, an isotropic material displays a spherical shape in these plots. However, any deviations from this spherical shape indicate the degree of anisotropy of a material. For LiMgCl3, a significant deviation from the spherical shape is observed at 100 GPa, indicating a higher degree of anisotropy. In contrast, LiBeCl3 shows a more spherical shape at 100 GPa, implying that this compound becomes more isotropic under high-pressure circumstances.
To further elucidate the anisotropy nature of LiMCl3, the universal anisotropy factor (\(\:{A}^{U}\)) has been used. It can also be obtained through the following relation113:
The value of \(\:{A}^{U}\) for isotropic compounds is zero whereas any deviation from this value indicates the degree of anisotropy. Pressure-induced \(\:{A}^{U}\) for different hydrostatic pressure has been calculated and listed in Table 5. It is evident that LiMgCl3 exhibits increased anisotropy under rising hydrostatic pressures, whereas LiBeCl3 becomes more isotropic under higher pressure conditions. However, these observations are also supported by Fig. 12.
Conclusion
In summary, the physical properties of the investigated compounds LiMgCl3 and LiBeCl3 change under varying hydrostatic pressure, ranging from 0 GPa to 100 GPa. The lattice parameters and unit cell volume decrease with increasing pressure, likely due to the reduction in bond length between constituent atoms. The electronic properties, including band structure, TDOS, and PDOS, are also examined under different pressure conditions. LiMgCl3 exhibits a transition from an ultrawide bandgap to a narrow bandgap, while LiBeCl3 transitions from a semiconductor to a metallic state under 100 GPa pressure. This reduction in energy bandgap enhances the efficiency of these materials in optoelectronic applications. Improved dielectric function, higher reflectivity, and refractivity are observed under high-pressure conditions, with absorption peaks shifting upwards in the UV region, indicating their potential use in optoelectronic devices. Additionally, the elastic constants of both materials suggest ductile behavior, which increases with applied pressure. The anisotropy factor shows an increasing trend for LiMgCl3 with rising pressure, while it decreases for LiBeCl3, indicating different anisotropic behaviors under pressure. This study highlights the potential application of alkali earth metal-based perovskites, particularly Li-based compounds, in future optoelectronic device applications.
Data availability
The data used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Pandit, N. et al. Study of chalcogenide-based metal perovskites BaZrX3 (X = S and Se): DFT insight into fundamental properties for sustainable energy generation using AMPS-1D. J. Comput. Electron. 23, 1014–1028 (2024).
Boutramine, A. et al. A theoretical investigation of the Ba2CePtO6 double perovskite for optoelectronic and thermoelectric applications. Opt. Quantum Electron. 56, 395 (2024).
Khuili, M. et al. First-principles study of halide double perovskite cs 2 SnX 6 (X = cl, br, I) for solar cell applications. Mod. Phys. Lett. B. 38, 2350223 (2024).
Abbas, Z. et al. DFT insights for structural, opto-electronic, thermodynamic and transport characteristics of Tl2TeX6 (X = at, br, cl, I) double perovskites for low-cost solar cell applications. Inorg. Chem. Commun. 163, 112293 (2024).
Boutramine, A. et al. First-principles investigations of structural, thermodynamic, optoelectronic and thermoelectric properties of Rb2CuMF6 (M = As3+, Bi3+) eco-friendly halide double perovskites: materials for green energy applications. J. Inorg. Organomet. Polym. Mater. 34, 4374–4391 (2024).
Rahman, M. F. et al. Exploring the impact of strain on the electronic and optical properties of inorganic novel cubic perovskite Sr3PI3. Phys. Scr. 98, 115105 (2023).
Rahman, M. F. et al. The optical and electronic properties of inorganic halide perovskite Sr3NCl3 under applied biaxial strain. J. Mater. Sci. 58, 13100–13117 (2023).
Reza, M. S. et al. Boosting efficiency above 28% using effective charge transport layer with Sr3SbI3 based novel inorganic perovskite. RSC Adv. 13, 31330–31345 (2023).
Rahman, M. F. et al. Unraveling the strain-induced and spin–orbit coupling effect of novel inorganic halide perovskites of Ca3AsI3 using DFT. AIP Adv. 13, (2023).
Sabbah, H. Numerical simulation of 30% efficient lead-free perovskite CsSnGeI3-Based solar cells. Materials (Basel) 15, 3229 (2022).
Cao, X. et al. Stability improvement of tin-based halide perovskite by precursor-solution regulation with dual-functional reagents. Adv. Funct. Mater. 31 (2021).
Diau, E. W. G., Jokar, E. & Rameez, M. Strategies to improve performance and stability for tin-based perovskite solar cells. ACS Energy Lett. 4, 1930–1937. https://doi.org/10.1021/acsenergylett.9b01179 (2019).
Masannan, V. et al. Experimental Investigation on the Drilling Characteristics of Kenaf/PLA-Based Laminates. Eng. Proc. 61, 9 (2024).
Bouazizi, S., Tlili, W., Bouich, A., Soucase, B. M. & Omri, A. Design and efficiency enhancement of FTO/PC60BM/CsSn0.5Ge0.5I3/Spiro-OMeTAD/Au perovskite solar cell utilizing SCAPS-1D Simulator. Mater. Res. Express 9, 0–12 (2022).
Hossain, M. K. et al. Deep insights into the coupled optoelectronic and photovoltaic analysis of lead-free CsSnI3 perovskite-based solar cell using DFT calculations and SCAPS-1D simulations. ACS Omega 8, 22466–22485 (2023).
Hossain, M. K. et al. Photovoltaic performance investigation of Cs3Bi2I9-based perovskite solar cells with various charge transport channels using DFT and SCAPS-1D frameworks. Energy Fuels 37, 7380–7400 (2023).
Hossain, M. K. K. et al. Harnessing the potential of CsPbBr3-based perovskite solar cells using efficient charge transport materials and global optimization. RSC Adv. 13, 21044–21062 (2023).
Hossain, M. K. et al. Achieving above 24% efficiency with non-toxic CsSnI3 perovskite solar cells by harnessing the potential of the absorber and charge transport layers. RSC Adv. 13, 23514–23537 (2023).
Wang, H. & Kim, D. H. Perovskite-based photodetectors: materials and devices. Chem. Soc. Rev. 46, 5204–5236 (2017).
Liu, Y. et al. Surface-emitting perovskite random lasers for speckle-free imaging. ACS Nano. 13, 10653–10661 (2019).
Park, Y. & Lee, J. S. Metal halide perovskite-based memristors for emerging memory applications. J. Phys. Chem. Lett. 13, 5638–5647 (2022).
Li, B. et al. Metal halide perovskites for resistive switching memory devices and artificial synapses. J. Mater. Chem. C. 7, 7476–7493 (2019).
Hossain, M. K. et al. A review of applications, prospects, and challenges of proton-conducting zirconates in electrochemical hydrogen devices. Nanomaterials 12, 3581 (2022).
Wang, X. et al. Metal halide semiconductors beyond lead-based perovskites for promising optoelectronic applications. J. Phys. Chem. Lett. 12, 10532–10550 (2021).
Bouhmaidi, S. et al. A DFT study of electronic, optical and thermoelectric properties of Ge-halide perovskites CsGeX3 (X = F, cl and br). Comput. Condens. Matter. 31, e00663 (2022).
Yang, Y., Sun, Y. & Jiang, Y. Structure and photocatalytic property of perovskite and perovskite-related compounds. Mater. Chem. Phys. 96, 234–239 (2006).
Su, R. et al. Dielectric screening in perovskite photovoltaics. Nat. Commun. 12, 2479 (2021).
Gao, W., Zhu, Y., Wang, Y., Yuan, G. & Liu J.-M. A review of flexible perovskite oxide ferroelectric films and their application. J. Mater. 6, 1–16 (2020).
Guo, W. et al. Broadband Photoresponses from Ultraviolet to Near-Infrared (II) Region through light‐induced pyroelectric effects in a hybrid perovskite. Angew Chemie. 134, e202213477 (2022).
Takenaka, T., Nagata, H., Hiruma, Y., Yoshii, Y. & Matumoto, K. Lead-free piezoelectric ceramics based on perovskite structures. J. Electroceram. 19, 259–265 (2007).
Yoshii, K. Magnetic properties of perovskite GdCrO3. J. Solid State Chem. 159, 204–208 (2001).
Rubel, M. H. K. et al. First-principles calculations to investigate physical properties of single-cubic (Ba0.82K0.18)(Bi0.53Pb0.47)O3 novel perovskite superconductor. Mater. Today Commun. 33, 104302 (2022).
Hayashi, H. et al. Structural consideration on the ionic conductivity of perovskite-type oxides. Solid State Ionics. 122, 1–15 (1999).
Rehman, Z. et al. A DFT study of structural, electronic, mechanical, phonon, thermodynamic, and H2 storage properties of lead-free perovskite hydride MgXH3(X = cr, Fe, Mn). J. Phys. Chem. Solids. 186, 111801 (2024).
Mera, A. & Rehman, M. A. First-principles investigation for the hydrogen storage properties of AeSiH3 (ae = Li, K, na, mg) perovskite-type hydrides. Int. J. Hydrogen Energy. 50, 1435–1447 (2024).
Rehman, M. A. et al. Pressure-induced modulation of structural, electronic, and optical properties of LiCaF3 fluoro perovskite for optoelectronic applications. Solid State Commun. 380, 115447 (2024).
Hossain, M. K. et al. An extensive study on charge transport layers to design and optimization of high-efficiency lead-free Cs2PtI6-based double-perovskite solar cells: a numerical simulation approach. Results Phys. 61, 107751 (2024).
Hossain, M. K. et al. Exploring the optoelectronic and photovoltaic characteristics of lead-free Cs2TiBr6 double perovskite solar cells: a DFT and SCAPS‐1D investigations. Adv. Electron. Mater. 11, 2400348 (2025).
Hossain, M. K. et al. Design and simulation of CsPb.625Zn.375IBr2-based perovskite solar cells with different charge transport layers for efficiency enhancement. Sci. Rep. 14, 30142 (2024).
Hossain, M. K. et al. Enhancing efficiency and performance of Cs2TiI6-based perovskite solar cells through extensive optimization: a numerical approach. Inorg. Chem. Commun. 168, 112964 (2024).
Vasudevan, B. et al. Experimental study, modeling, and parametric optimization on abrasive waterjet drilling of YSZ-coated Inconel 718 superalloy. J. Mater. Res. Technol. 29, 2024, 4675 (2024).
Tan, S. et al. Sustainable thermal regulation improves stability and efficiency in all-perovskite tandem solar cells. Nat. Commun. 15, 4136 (2024).
Shu, L. et al. Photoflexoelectric effect in halide perovskites. Nat. Mater. 19, 605–609 (2020).
Zhang, L., Miao, J., Li, J. & Li, Q. Halide perovskite materials for energy storage applications. Adv. Funct. Mater. 30, 1–20 (2020).
Batool, A. et al. The pressure-induced indirect to direct bandgap transition and thermoelectric response in SrTiO3: an ab-initio study. J. Phys. Chem. Solids. 123, 70–75 (2018).
Hou, X. Y., Tan, J., Hu, C. E., Chen, X. R. & Geng, H. Y. Thermoelectric properties of strontium oxide under pressure: first-principles study. Phys. Lett. A. 390, 127083 (2021).
Gillani, S. S. A. et al. Effect of magnesium doping on band gap and optical properties of SrZrO3 perovskite: a first-principles study. Optik (Stuttg). 191, 132–138 (2019).
Gillani, S. S. A. et al. Structural stability of SrZrO3 perovskite and improvement in electronic and optical properties by ca and Ba doping for optoelectronic applications: a DFT approach. Philos. Mag. 99, 3133–3145 (2019).
Rehman, M. A., Rehman, J. & Tahir, M. B. Density functional theory study of structural, electronic, optical, mechanical, and thermodynamic properties of halide double perovskites Cs2AgBiX6 (X = cl, br, I) for photovoltaic applications. J. Phys. Chem. Solids. 181, 111443 (2023).
Mondal, A., Lata, A. & Gupta, S. Effect of Sr2 + doping on optical, thermal and photocatalytic behaviour of CsPbBr3. Mater. Lett. 309, 131314 (2022).
Linh, N. H., Tuan, N. H., Dung, D. D., Bao, P. Q. & Cong, B. T. Alkali metal-substituted bismuth-based perovskite compounds: a DFT study. J. Sci. Adv. Mater. Devices. 4, 492–498 (2019).
Shuvo, I. K., Saiduzzaman, M., Asif, T. I., Ali Haq, M. & Hossain, K. M. Band gap shifting of halide perovskite CsCaBr3 from ultra-violet to visible region under pressure for photovoltaic applications. Mater. Sci. Eng. B. 278, 115645 (2022).
Aslam, M. et al. Physical characteristics of CdZrO3 perovskite at different pressure for optoelectronic application. J. Mater. Res. Technol. 9, 9965–9971 (2020).
Dimesso, L., Wussler, M., Mayer, T., Mankel, E. & Jaegermann, W. Inorganic alkali lead iodide semiconducting APbI3 (A = Li, na, K, Cs) and NH4PbI3 films prepared from solution: structure, morphology, and electronic structure. AIMS Mater. Sci. 3, 737–755 (2016).
Tang, Z. et al. Modulations of various alkali metal cations on organometal halide perovskites and their influence on photovoltaic performance. Nano Energy 45, 184–192 (2018).
Rahman, N. et al. Appealing perspectives of the structural, electronic, elastic and optical properties of LiRCl3 (R = be and mg) halide perovskites: a DFT study. RSC Adv. 13, 18934–18945 (2023).
Chamorro, E., Pérez, P., Duque, M., De Proft, F. & Geerlings, P. Dual descriptors within the framework of spin-polarized density functional theory. J. Chem. Phys. 129 (2008).
Clark, S. J. et al. First principles methods using CASTEP. Z. für Krist Cryst. Mater. 220, 567–570 (2005).
Rahman, M. S. et al. Insights from computational analysis on novel lead-free FrGeCl3 perovskite solar cell using DFT and SCAPS-1D. Inorg. Chem. Commun. 171, 113578 (2025).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Datta, A. K. et al. Pressure induced tunable physical properties of cubic KHgF3 fluoro-perovskite: a first principle study. Inorg. Chem. Commun. 170, 113424 (2024).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-Zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011).
Fan, C. Z. et al. Potential superhard osmium dinitride with fluorite and pyrite structure: first-principles calculations. Phys. Rev. B 74, 125118 (2006).
Gaillac, R., Pullumbi, P. & Coudert, F. X. ELATE: an open-source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter 28, 275201 (2016).
Islam, M. A., Rahaman, M. Z. & Sen, S. K. A comparative study of hydrostatic pressure treated environmentally friendly perovskites CsXBr3 (X = Ge/Sn) for optoelectronic applications. AIP Adv. 11, 75109 (2021).
Hossain, M. S. et al. Pressure induced semiconductor to metal phase transition in cubic CsSnBr3perovskite. AIP Adv. 11, (2021).
Hossain, M. A. et al. Insights into the pressure-dependent physical properties of cubic Ca3MF3 (M = as and sb): first-principles calculations. Heliyon 10, e38898 (2024).
Miglio, A., Heinrich, C. P., Tremel, W., Hautier, G. & Zeier, W. G. Local bonding influence on the band edge and band gap formation in quaternary chalcopyrites. Adv. Sci. 4 (2017).
Li, D. et al. The mechanical properties, structural stability and thermal conductivities of Y, sc doped AuIn2 by first – principles calculations. Metals (Basel) 12, 2121 (2022).
Islam, J. & Hossain, A. K. M. A. Semiconducting to metallic transition with outstanding optoelectronic properties of CsSnCl3 perovskite under pressure. Sci. Rep. 10, 14391 (2020).
Hosen, A., Islam, M. R. & Park, J. Pressure-induced band gap shifting from ultra-violet to visible spectrum of non-toxic RbCaBr3 cubic perovskite for enhancing optoelectronic applications. J. Inorg. Organomet. Polym. Mater. 34, 2378–2387 (2024).
Ghosh, A. et al. Inorganic novel cubic halide perovskite Sr3AsI3: strain-activated electronic and optical properties. Heliyon 9, e19271 (2023).
Haq, M. A., Saiduzzaman, M., Asif, T. I., Shuvo, I. K. & Hossain, K. M. Ultra-violet to visible band gap engineering of cubic halide KCaCl 3 perovskite under pressure for optoelectronic applications: insights from DFT. RSC Adv. 11, 36367–36378 (2021).
Alam, M. S. et al. Tuning band gap and enhancing optical functions of AGeF3 (A = K, rb) under pressure for improved optoelectronic applications. Sci. Rep. 12, 8663 (2022).
Gong, J. et al. Pressure-induced indirect‐direct bandgap transition of CsPbBr 3 single crystal and its effect on photoluminescence quantum yield. Adv. Sci. 9, 2201554 (2022).
Wang, T. et al. Indirect to direct bandgap transition in methylammonium lead halide perovskite. Energy Environ. Sci. 10, 509–515 (2017).
Jellil, Z. & Ez-Zahraouy, H. Pressure-induced band gap engineering and enhanced optoelectronic properties of non-toxic Ca-based perovskite CsCaCl3: insights from density functional theory. Comput. Condens. Matter 38, e00879 (2024).
Zhuge, F. et al. Nanostructured materials and architectures for Advanced Infrared Photodetection. Adv. Mater. Technol. 2, 1700005 (2017).
Yatsui, T. et al. Enhanced photo-sensitivity in a Si photodetector using a near-field assisted excitation. Commun. Phys. 2, 62 (2019).
Sofo, J. O. & Mahan, G. D. Optimum band gap of a thermoelectric material. Phys. Rev. B. 49, 4565–4570 (1994).
Asiri, A. M. et al. Analysis of XGaO3 (X = ba and cs) cubic based perovskite materials for photocatalytic water splitting applications: a DFT study. Heliyon 9, e14112 (2023).
Liu, X. et al. A high dielectric constant non-fullerene acceptor for efficient bulk-heterojunction organic solar cells. J. Mater. Chem. A. 6, 395–403 (2018).
Yaseen, M. et al. Phase transition and thermoelectric properties of cubic KNbO3 under pressure: DFT approach. J. Mater. Res. Technol. 11, 2106–2113 (2021).
Hayatullah et al. Physical properties of CsSnM 3 (M = cl, br, I): a first principle study. Acta Phys. Pol. A. 124, 102–107 (2013).
Ali, M. A. et al. Insight into pressure tunable structural, electronic and optical properties of via DFT calculations. Eur. Phys. J. Plus 135, 309 (2020).
Islam, M. A., Islam, J., Islam, M. N., Sen, S. K. & Hossain, A. K. M. A. enhanced ductility and optoelectronic properties of environment-friendly CsGeCl3 under pressure. AIP Adv. 11, 45014 (2021).
Noor, N. A., Mahmood, Q., Rashid, M., Ul Haq, B. & Laref, A. The pressure-induced mechanical and optoelectronic behavior of cubic perovskite PbSnO3 via ab-initio investigations. Ceram. Int. 44, 13750–13756 (2018).
Sakr, G. B., Yahia, I. S., Fadel, M., Fouad, S. S. & Romčević, N. Optical spectroscopy, optical conductivity, dielectric properties and new methods for determining the gap states of CuSe thin films. J. Alloys Compd. 507, 557–562 (2010).
Rashid, M. A. First-principles calculations to explore the metallic behavior of semiconducting lead-free halide perovskites RbSnX 3 (X cl, br) under pressure. Eur. Phys. J. Plus 123 (2022).
Mitro, S. K., Saiduzzaman, M., Asif, T. I. & Hossain, K. M. Band gap engineering to stimulate the optoelectronic performance of lead-free halide perovskites RbGeX3 (X = cl, br) under pressure. J. Mater. Sci. Mater. Electron. 33, 13860–13875 (2022).
Rahman, M. A., Rahaman, M. Z. & Sarker, M. A. R. First principles investigation of structural, elastic, electronic and optical properties of HgGeB2 (BP, as) chalcopyrite semiconductors. Comput. Condens. Matter. 9, 19–26 (2016).
Ullah, M. et al. Pressure induced variations in the optoelectronic response of ASnX3 (A = K, rb; X = cl, br, I) perovskites: a first principles study. Mater. Sci. Semicond. Process. 150, 106977 (2022).
Hossain, M. K. et al. Numerical analysis in DFT and SCAPS-1D on the influence of different charge transport layers of CsPbBr3 perovskite solar cells. Energy Fuels. 37, 6078–6098 (2023).
Roknuzzaman, M., Ostrikov, K., Wang, H., Du, A. & Tesfamichael, T. Towards lead-free perovskite photovoltaics and optoelectronics by ab-initio simulations. Sci. Rep. 7, 14025 (2017).
Vaitheeswaran, G., Kanchana, V., Svane, A. & Delin, A. Elastic properties of MgCNi3—a superconducting perovskite. J. Phys. Condens. Matter. 19, 326214 (2007).
Mouhat, F. & Coudert, F. X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B. 90, 224104 (2014).
Born, M. On the stability of crystal lattices. I. Math. Proc. Camb. Philos. Soc. 36, 160–172 (1940).
Waller, I. Dynamical theory of crystal lattices by M. Born and K. Huang. Acta Crystallogr. 9, 837–838 (1956).
Tu, Q., Kim, D., Shyikh, M. & Kanatzidis, M. G. Mechanics-coupled stability of metal-halide perovskites. Matter 4, 2765–2809 (2021).
Pettifor, D. G. Theoretical predictions of structure and related properties of intermetallics. Mater. Sci. Technol. 8, 345–349 (1992).
Voigt, W. & Voigt, W. Wechselbeziehungen Zwischen Zwei Tensortripeln.(Elastizität Und Innere Reibung) (Springer, 1966).
Reuß, A. Berechnung Der fließgrenze Von Mischkristallen Auf Grund Der plastizitätsbedingung für einkristalle. ZAMM-Journal Appl. Math. Mech. für Angew Math. und Mech. 9, 49–58 (1929).
Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. Sect. A 65, 349–354 (1952).
Stalin, P. M. J. et al. Innovative cinque rib-roughened stimulators on performance improvement in triangular channel solar air heater. Int. J. Low-Carbon Technol. 19, 227–235 (2024).
Gao Huang, Z. Mechanical and thermodynamics properties of Al 2 ca and mg 2 ca under various pressure: a first principle study. Mater. Express 11, 1571–1578 (2021).
Al-Qaisi, S. et al. Opto‐electronic and thermophysical characteristics of < scp > A 2 TlAgF 6 (A = rb, Cs) for green technology applications. J. Comput. Chem. 45, 1576–1586 (2024).
Belhachi, S. et al. DFT Analysis of Ba2NbRhO6: a promising double perovskite for sustainable energy applications. J. Inorg. Organomet. Polym. Mater. https://doi.org/10.1007/s10904-024-03336-5 (2024).
Masood, M. K. et al. Theoretical investigation of XSnH3 (X: rb, Cs, and Fr) perovskite hydrides for hydrogen storage application. Int. J. Hydrogen Energy. 63, 1248–1257 (2024).
Rony, J. K. et al. TlBX3 (B = Ge, Sn; X = cl, br, I): promising non-toxic metal halide perovskites for scalable and affordable optoelectronics. J. Mater. Res. Technol. 29, 897–909 (2024).
Rahaman, M. Z., Rahman, M. A. & Sarker, M. A. R. Prediction of a new transition metal oxide MgRhO3 with SrTiO3-type structure: Stability, structure and physical characteristics. Chin. J. Phys. 55, 1489–1494 (2017).
Zener, C. M. & Siegel, S. Elasticity and anelasticity of metals. J. Phys. Chem. 53, 1468 (1949).
Ranganathan, S. I. & Ostoja-Starzewski, M. Universal elastic anisotropy index. Phys. Rev. Lett. 101, 055504 (2008).
Acknowledgements
This work was funded by the Researchers Supporting Project Number (RSPD2025R667), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
A.K. Datta, and M.K. Hossain: Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Data curation, Writing – original draft, Writing – review & editing, Supervision, Project administration; M. S. Rahman, P. Paramasivam, Adel El-marghany, and V. K. Mishra: Validation, Formal analysis, Writing – review & editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Datta, A., Hossain, M., Rahman, M. et al. DFT insights into bandgap engineering of lead-free LiMCl3 (M = Mg, Be) halide perovskites for optoelectronic device applications. Sci Rep 15, 6944 (2025). https://doi.org/10.1038/s41598-025-90621-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-90621-z
