
Abstract
Robust solutions combining computational chemistry and data-driven approaches are in high demand in various areas of materials science. For instance, such methods can use machine learning models trained on a limited dataset to make structure-to-property predictions over large search spaces. This paper examines the impact of data selection mechanisms on thermodynamic property assessments for chemically modified lead halide perovskite γ-CsPbI3 and non-perovskite δ-CsPbI3. For disordered states of these phases, complete composition/configuration spaces are built by adding Cd or Zn substitutions of Pb and Br substitutions of I and comprise 2,946,709 and 2,995,462 inequivalent spatial arrangements of substituents, respectively. Using the properties of 1162 entries of the built spaces evaluated by means of density functional theory, we implement independent procedures for training graph neural networks (GNNs). In each of them, a training dataset is constructed depending on the defect contents and presence of low- and high-symmetry structures. The results show that symmetries of training structures can significantly influence quality of the subsequent GNNs’ predictions and can result in twofold increase in errors due to the preferential selection of high-symmetry structures.
Similar content being viewed by others
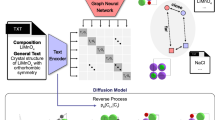


Introduction
Data-driven solutions in materials science have become powerful tools for many applications, such as predicting potentially new materials1,2,3, targeted property and composition modification4,5,6,7, exploring vast composition/configuration spaces (CCSs)8,9,10,11, studying processes12,13,14, etc. The undeniable advantage of such approaches is the ability to speed up predictions compared to direct application of density functional theory (DFT) calculations to entire search spaces. In direct structure-to-property predictions, any machine learning model can be considered as a surrogate model approximating computationally expensive solvers. For large chemical spaces, this definitely may be extremely helpful due to the enormous number possible atomic arrangements in three-dimensional space and the complexity of the energy surface under consideration15,16. Within more complex heuristics, such solutions can be easily extended to molecular dynamics applications through approximation of interatomic potentials17. Whatever the method used, there is always data behind the scenes. In turn, data sampling for training purposes may become a non-straightforward task for a separate research in some cases, e.g., for disordered systems18,19,20,21,22.
Classical machine learning algorithms can provide useful and reliable results even being trained on small or noisy datasets prepared in tabular form23,24,25,26. For such models, the successful application of active learning for materials discovery should be also noted27,28. For such solutions, specific geometrical/topological descriptors of the search space entries specially developed for a certain domain29,30,31 or degenerated automatically32,33,34 are required. On the one hand, this peculiarity requires expert knowledge in the field of certain class of functional materials, and on the other hand, reduces transferability, universality, and extendibility of the model developed for a certain task. Considering CCSs of disordered crystals, one should also keep in mind a limited descriptive power of many automatically generated and even hand-crafted descriptor sets in such spaces11,35. In contrast to the descriptor-based approaches, neural networks have the ability to use the structure representation directly, searching for the most essential features under the hood of a specific problem solution, such as predicting various properties of materials36,37. By handling atoms as nodes/vertices, chemical structures can be naturally represented as a graph and hence be processed by graph neural networks (GNNs)38,39. Thus, based on the above comparison of the classical machine learning approaches and GNN-based solutions, it can be assumed that the latter mentioned group has much more bright perspectives in materials discoveries, where descriptor-based approaches may be limited.
Among the families of materials for energy conversion and storage applications, reliable tools for thermodynamic property assessments are of practical importance for chemically modified (doped or alloyed) perovskites. Solar cells utilizing lead halide perovskites have already achieved a power conversion efficiency exceeding 25%40,41,42. Among the lead halide perovskites, CsPbI3 appears to be the most promising due to its low band gap of 1.7 eV allowing for efficient absorption of light. The γ-phase of CsPbI3 retains favorable optoelectronic characteristics, such as direct bandgap and high charge-carrier mobility, making it a potential alternative to the highly symmetrical cubic perovskite phase43. However, any large-scale applications of CsPbI3 face difficulties caused by its polymorphic transitions44,45,46 into the undesirable δ-CsPbI3 phase possessing no useful properties. γ- and δ-CsPbI3 powders are black- and yellow-colored. Thus, we use corresponding designations bellow.
One of many methods for stabilization of the γ-phase is increasing tolerance factor by means of partial Pb2+ ion substitution at B-site46,47,48,49,50,51,52,53. It is well-known that Cd and Zn cations leads to significant improvement of electro- and photoluminescence of CsPbBr3 and CsPbCl3 nanoparticles54,55,56,57,58,59,60. Additionally, substitution of Pb2+ at B-site by Cd2+/Zn2+ within the CsPbI3 structure can be seen as either isoelectronic doping at lower concentrations, or alloying at higher concentrations. Therefore, these dopants do not introduce extra charge carriers in the systems. For CsPbI3, we recently studied the effects of such chemical modifications61 from computational perspectives. This work showed that CCS complexity drastically increases when substituent content increases and can hardly be studied by means of DFT. From domain specifics point of view, partial substitution of I− by Br− also contributes to γ-phase stabilization with only a small increase in the band gap62,63.
Doping and chemical modifications not only seems promising for the γ-phase stability enhancement, but definitely lead to a dramatic increase in the complexity of the corresponding CCS from computational/predictive perspectives. The aim of the current contribution is to shed light on answering the question «How should one subsample entire range of disorder realization within particular crystal structures to train the most precise machine learning models?” On this track, simultaneous substitution of Pb2+ at B-site by Cd2+/Zn2+ and I− by Br− represent perfect ability for the development and verifications of efficient domain-inspired data sampling strategies for the subsequent training GNN models.
Methodology
Complete composition/configuration space
To address the aforementioned issue, a set of γ- and δ-CsPbI3 structures with partial substitutions of Pb and I positions by Cd/Zn and Br, respectively, was used. The pristine crystal structures were taken from the experimental data64. For the complexity reasons, we set limits of Pb and I site substitutions for both phases studied to 25% and 33.3%, espectively. To do so, the conventional crystal structures of γ- and δ-CsPbI3 were enlarged up to the 1 × 2 × 1 supercells followed by substitution of up to 2 Pb atoms and up to 8 I atoms by Cd and Br atoms, respectively. Technically, the described procedure was performed by writing corresponding partial occupancies of Wyckoff site 4b (4с for δ-phase) filled with a mix of Pb, Cd and Wyckoff sites 4c, 8d (4c, 4c, 4c for δ-phase) filled with a mix of I, Br to CIF file of γ- or δ-phase for the Supercell program65. In this way, two complete CCSs of unique symmetrically inequivalent structures were obtained with Cd content range from 0 to 25% with step 12.5% and Br content range from 0 to 33.3% with step 4.167%. Within the built CCSs, each entry has a weight, reflecting the number of symmetrically equivalent structures, obtained as a result of the full CCSs generation in a purely combinatorial way.
DFT calculations
For the structures chosen in this study for the training, validation, or test purposes, target properties used in GNN fitting were obtained using the DFT calculations. Total energies of each structure of a certain dataset and all neat constituents in their respective ground states were calculated after relaxation using Vienna Ab initio Simulation Package (VASP) with Perdew-Burke-Ernzerhof exchange-correlation functional and projector-augmented wave pseudopotentials66,67. The energy cutoff of 600 eV was employed. The calculations were performed at the Г-point of reciprocal space to eliminate the interactions between defects and due to relatively large supercell size. Stopping tolerances for energy convergence within the self-consistency loop and maximum forces acting on atoms after the relaxation process were set to 10− 5 eV and 0.01 eV/Å, respectively. Structure formation energy was chosen as a target property for the GNN models.
GNN models
To stay consistent with the previously obtained results, we used the Allegro model68, provided state-of-the-art results on the benchmarks. Allegro was compared61 with another rather pioneer model – SchNet69 – and showed the best performance obtained for doped CsPbI3. Allegro represents a many-body potential using iterated tensor products of learned equivariant edge representations without atom-centered message passing in contrast to other popular in the field of machine learning potentials models, where message passing process between neighboring nodes implemented in. For example, NequIP model70, can significantly change node embeddings and reduce information about the actual arrangement of dopants. Instead, the Allegro model operates only on graph edges, and node embeddings remain unchanged.
Firstly, the Allegro model was pretrained on a subset of the AFLOWLIB database, representing a storage of DFT-derived properties of experimentally observed and hypothetical crystals71. This database is managed by the AFLOW software that uses VASP for relaxations and thermodynamic properties evaluations. For model pretraining, all structures containing at least two species out of Cs, Pb, Zn, I, Br were selected from AFLOWLIB and split into train and validation parts in ratio of 90:10. The obtained distributions of relaxed and formation energies for the structures within the built dataset are given in Figures S1-2 of the Supplementary Information. This step helps the model to catch information about interatomic interactions in the structures similar to the title compounds.
Results and discussion
Hints of additional data sampling criteria
In the aforementioned work61, we comprehensively analyzed impact of the number of substitutions in the training and validation structures on the quality of the GGN model predictions. Each of the introduced training/validation groups comprised the structures with the same numbers of defects. The higher the defect content, the higher the number of structure realizations in the CCS. To account for this, we considered four random subsets of the structures with 2 and 3 substituted sites within each of the introduced groups. For this reason, models trained and validated on the datasets identical in terms of defect contents – training/validation groups – might provide different predictions only due to the randomness of the structures constituted them. For instance, the models validated on the maximum available number of the structures with 2 substituted sites and, consequently, trained only on the structures with other defect contents – training/validation group 9 of ref.61 – demonstrated widely scattered root-mean-square errors (RMSEs) as shown in Fig. 1. This observation can be definitely interpreted as an inconsistence of the model predictions with respect to the random subsampling rather than chemical compositions of training/validation samples.
By design, the complete CCS included all possible symmetrically inequivalent structure realizations. Despite the same chemical composition, some CCS entries can thus possess different symmetries caused by the differences in defect arrangements. Thus, the composition differences by themselves cannot be the only reason for this behavior. Therefore, one can consider space groups of the built CCS entries (prior to any relaxation of them) as the next most obvious and easily accessible metainformation in addition to the composition. The test scores in the abovementioned work were checked for the group of training/validation splits providing the highest variation of them. Such a comparison demonstrated that the most pronounce differences in the model scores might be associated to the differences in the data splits concerning presence or absence of the structures of Pc (7) and Pm (8) space groups. This fact prompted us to study the symmetry effect in more detail.

For the 9th train/validation group in the work61, distributions of Cd- and Zn-doped CsPbI3 crystal structures by their space groups determined prior to DFT relaxations. The corresponding test RMSE scored of the models trained on the 9th group are provided in meV/atom.
CCS statistics
The complete CCSs obtained in the current study included 2,946,709 and 2,995,462 inequivalent structures for the γ- and δ-CsPbI3 phases, respectively. For each entry of the built CCSs, the corresponding space symmetry was additionally determined prior to any DFT evaluations using the pymatgen library72. Furthermore, defect contents in each entry, i.e. a set of Cd and Br atoms quantity at each Wyckoff site of 1 × 2 × 1-supercell were collected as metainformations. Expectedly, the introduced substitutions lead to symmetry reduction and to a structure diversity in terms of space groups as shown in Figs. 2 and 3. The readers are also referred to Figure S3 of the Supplementary Information section for more details on the corresponding group subgroup relations obtained within the built CCSs.
Regarding the structure weights it should be added that there was no strong correlation obtained between the space group of a structure and its weight. Nevertheless, the entries with the highest available space group, namely Pnma (62), had the lowest weights of 1, and vice versa all P1 (1) entries possessed weights of 16. Thus, any random subsample of the full CCSs is unlikely to provide high symmetry structures for statistical reasoning – low numbers of high symmetry structures and low weight each of them. The graphical representation of weight dependencies via symmetries over the entire CCSs is provided in Figure S4.

Heatmap of the number of complete CCS entries of the black γ-CsPbI3 via Pb and I substitution levels and symmetry space groups. The empty fields correspond to the lack of certain combination. The color map corresponds to the natural logarithm of the number of CCS entries (the values provided as notations).

Heatmap of the number of complete CCS entries of the yellow δ-CsPbI3 via Pb and I substitution levels and symmetry space groups.
The obtained space groups within the built CCSs turned out to be severely unbalanced. In the case of black γ-CsPbI3, space symmetry groups greater than 7 (Pc) corresponded to 176 structures (less than 0.006% of the built CCS). On the other hand, 99.6% of the structures possessed space group P1. In the case of yellow δ-CsPbI3, corresponding values equaled to 0.014% and 96.4%, respectively.
To explore possible effect of space symmetries in training and validation subsets on the resulting performance of the GNN model, we added space group numbers of the entries of the CCSs built in this study and subsequently used this metainformation for the required balancing of the training/validation and test subsets.
Training, validation, and test subsets
Based on the space groups statistics, we set a threshold for space group number to split the entire CCSs into high- and low-symmetry structures. To select the former mentioned group, 176 γ- and 405 δ-phase structures with space group number greater than 7 (Pc) were considered. To select low-symmetry entries, all γ-phase structures of the full CCS were ordered in ascending order by space group number and in descending order by weight. For each unique composition i in the high-symmetry structure set, first ni structures were collected from the ordered list obtained, where ni equals to the number of high-symmetry γ-phase structures with a certain defect content. If there were no structure with required defect composition among those possessing space groups lower than the introduced threshold, structures with a higher number were used. In the same way δ-phase structures were collected. This allowed preserving similar chemical compositions among the introduced groups and directly track symmetry impact on the model performance. In such way, we follow monothetic analysis, i.e. method of designing experiments involving testing factors one at a time instead of multiple ones simultaneously.
Thus, two structure sets, referred to hereafter as predominantly high-symmetry (PHS) and predominantly low-symmetry (PLS) datasets, were created. Both of them comprised 176 γ- and 405 δ-phase structures possessing the similar defect compositions not to add excessive uncertainty, but different space group distributions. Both datasets were further doubled by substitution all Cd atoms with Zn to the size of 1162 structures each and subdivided into train and validation parts with 80/20 ratio using γ-/δ-phase stratification.
For the test purposes, 100 random γ- and 100 random δ-phase structures with P1 space group and the highest weights were taken from the complete CCS. It is worth noting here that defect compositions of the selected structures was different compared to that of train/validation structures allowing to check generalization ability of the models additionally. The obtained dataset was doubled by substitution all Cd atoms with Zn and denoted as PLS test dataset (a total of 400 structures).
To create PHS test dataset, γ-phase structures of the full CCS with space group number less than or equal to 7 (Pc) were ordered in descending order by space group number and weight. After that first ni structures with defect composition i were picked, where ni equals to amount of γ-phase structures with defect composition i in PLS test dataset with Cd dopant only. In the same way δ-phase structures were picked. Thus, the dataset comprising 100 γ- and 100 δ-phase structures was made. Its structures have the same defect compositions as in the case of the PLS test, but high space group number. One cannot take structures with space group number more than 7 (Pc) to add them to the PHS test, because these structures were included in the PHS train/validation dataset.
Furthermore, 45 (5*9) additional train/validation datasets intermediate in a sense of space group contents were created by random mixing PLS and PHS train/validation datasets in a ratio of 10:90, 20:80, etc. Besides, 4 supplementary train/validation datasets were sampled using similar procedure as in the case of PLS dataset. In each case, the subsets were considered with different random seeds to track how the changes in train/validation dataset affect quality of the corresponding models and to obtain statistically significant results. It is worth noting that each Cd-substituted structure in each one of 49 abovementioned datasets had a pair structure with Zn as substituent. Detail information about the datasets obtained is given in Table 1 and Figures S5 – S10.
The obtained distributions of DFT-derived formation energies within the built datasets are shown in Fig. 4. It can be noted that the introduced PLS and PHS datasets remained similar ion terms of the target properties, which may be associated with the similar sets of chemical compositions of the structures included. Strictly speaking, this observation has particular importance for additional checking in any other studies, since the obtained distribution of the target properties can directly impact GNN performance by themselves. It is also important to emphasize that since the PHS structures were included in our sets without randomness in their selection, while the PLS ones were chosen quasi-randomly, one can conclude that the high-symmetry structures are a reliable choice for assessing thermodynamic properties in complete CCS. In other words, one would expect that structures selected from a substantially larger set would obtain broader property distributions than that in the very limited set of high symmetry variants. However, this is not observed in any of the sets considered.

Distributions of DFT-derived formation energies for PHS and PLS structures within the introduced training/validation (left) and test (right) datasets.
In turn, for the purpose of this study, this fact allows one to conclude, that comparison of the models trained on different subsets is not influenced by any differences in target energy distributions. Thus, we were able to study symmetries impact independently on any other data peculiarities – chemical compositions and target energy ranges. Additionally, it is worth noting that formation energy distributions of training and validation data cover the entire range for that of the test dataset. Thus, we consider an interpolation task in terms of mode of GNN-based predictions.
Test scores of model predictions
For the models using random initialization of trainable parameters, the test scores of model predictions turned out to be almost identical, regardless of whether pretraining step was carried out. However, greater stability of the pretrained model could be expected, which means that model predictions were less dependent on some random factors. Thus, the pretrained Allegro model was fine-tuned on the introduced PLS and PHS datasets, 45 introduced mixtures of high- and low-symmetry structures and 4 additional PLS-like datasets resulting in a total of 51 fine-tuned model states.
All models were run on two test datasets. Then, RMSEs of predicted formation energies compared to those calculated using VASP were obtained for each model and for each test dataset. In turn, the RMSE values of the models, trained on the mixed train/validation datasets, were averaged over 5 values for each mixing ratio, and corresponding standard deviations were additionally calculated. The results obtained are given in Fig. 5 and Table 2.

Test RMSEs in formation energy assessments obtained for the PLS (left) and PHS (right) test structures using the models trained on various proportions of the PLS and PHS training/validation structures. Black lines denote RMSE values averaged over 5 independent models for each mixing ratio, blue translucent areas correspond to one standard deviation.
From Fig. 5 and Table 2, it can be seen that purposeful sampling of high symmetry structures for training GNNs increase resulting errors of assessments and thus reduces quality of model predictions. Also, it should be noted, that the models, trained on the datasets, containing more than 50% high symmetry structures, demonstrate lower formation energy RMSE on PHS test dataset. On the contrary, the other models provide more precise results on the PLS test.
As is known, the larger the size of the training sample and the higher its diversity in terms of feature space, the higher the generalizing ability and accuracy of the machine learning model. The higher performance of models trained on low symmetry train/validation dataset can be explained by the fact that a low symmetry structure has more variety of unique environment of each atom. In other words, there are more quantity of unique bi-, tri- and so on interatomic interactions in the case of a crystal structure with low symmetry. As the space group number increases, these unique local interactions degenerate due to the appearance of new symmetry elements and a decrease in the number of unique subgraphs in the crystal structure. This observation in itself clearly shows the more quantity of training samples and their diversity are available, the better generalization ability and accuracy of a model can be obtained. Thus, in addition to defect content and chemical composition, the space group of structures in training data can be used as an additional criterion for sampling of training datasets. Moreover, such metainformation obtained for the structures before structural relaxation is physically based and can be collected without expensive computations since it does not require a DFT-based evaluation of the CCS entries.
Inference results over the entire CCS
To study impact of symmetry distributions in train/validation data on the model predictions over the built CCSs for both phases, we used Allegro models in inference mode. Keeping in mind monotonic increase of the test RMSEs (see Fig. 5) by increase of the fraction of high-symmetry structures (PHS fraction) in training data, we considered limiting cases of the Allegro models fine-tuned on the PLS and PHS datasets. Distributions of the predicted formation energy over the complete CCS are shown in Fig. 6.

Violin plots of GNN-based assessments of formation energies over the complete CCSs and accounting for weights of their entries as predicted by the models trained and validated on the PLS and PHS datasets.
By comparison of the model predictions obtained for Zn- and Cd-substituted structures, one can conclude that the obtained distributions are similar for the models fine-tuned on the PLS dataset from both shape and ranges perspectives. Secondly, the predictions of the models fine-tuned on the PHS dataset have sharper distributions despite the fact that for both substituents the predicted ranges of formation energies remain similar. In the case of Cd-substituted systems, the models fine-tuned on the PHS dataset result in a number of peaks in the obtained distributions, i.e. predominant levels in formation energy predictions, which might be caused by the limited distinguishability of the composition-structure-property relationships. Thirdly, usage of high-symmetry structures in the training dataset does not affect δ-phase predictions. At the same time the model fine-tuned on the PHS dataset predict higher formation energies for the Cd-substituted structures of the γ-phase.
The differences between the models’ inference discussed above directly affects the calculated parameters, such as the energy difference between the chemically modified phases, that can be important from a practical point of view. For the models trained on the PHS and PLS structures, Fig. 7 shows relative behavior of energy difference between the γ and δ phases via substituent contents. As can be seen, the PHS group tends to underestimate the energy difference between phases for both metals substituting Pb. Moreover, the difference between the PHS and PLS models is more related to metallic substituent content rather than the Br/I ratios for the compositions studied.

For the models trained and validated on the PLS and PHS datasets, the formation energy difference between the Cd- (left) and Zn-doped (right) black (γ) and yellow (δ) phases obtained using minimum energy configurations over the complete CCSs.
Conclusions
In the present work, we comprehensively studied the impact of space symmetries of training structures on the resulting performance of the GNN-based surrogate models for structure-to-property predictions. To do so, it was suggested to build complete composition/configuration spaces of two competing CsPbI3 phases with B-site substitutions by Cd or Pb and I substitutions by Br. During data selection for training and validation of a set of independents realizations of the Allegro model, we tried to avoid any composition differences between the training datasets and made comparison of the models trained on exactly the same amounts of DFT-derived datapoints (structures).
It was shown that in the case of similar distributions of target properties and compositions, a higher fraction of high-symmetry structures resulted in a lower quality of model predictions. As the resulting statistics of the built CCS entries shows, selection of predominantly low-symmetry structures accompanied with composition constrains can be carried out in a purely random manner. Nevertheless, possessing the same composition and similar target property distributions predominantly low symmetry structures can provide better performance compared to that of high-symmetry structures in similar applications. Keeping in mind nearly the same composition and target property distributions, this observation might be caused by more diverse subgraphs of the structure graphs in the case of low symmetries. As shown, accounting for different symmetries on the training data can also significantly affect the inference mode of the trained GNN models.
Obviously, the results of the approach developed in this study and applied to the particular family of functional materials can hardly be extrapolated to any other group of crystal structures without appropriate additional check of the conclusions made. However, the developed workflow clearly shows the following points:
-
(1)
entries of vast CCSs can be severely unbalanced with respect to the space symmetries reduced compared to the original structure due to the introduced point defects and determined prior to their DFT evaluation,
-
(2)
despite the significantly lower numbers of the high-symmetry structures in the built CCS their thermodynamic properties ranges remain comparable with those of random subsets of much more numerous low-symmetry entries of the built CCS in all the cases considered,
-
(3)
when such parameters as compositions and target property ranges coincide, symmetry of crystal structures used for training models can significantly affect their resulting performance and at least can be considered as an additional easily achievable criterion for constructing training datasets in a stratified manner.
As any other deep learning models (artificial neural networks), GNNs are sensitive to quality and quantity of data available for training. By design, GNNs implicitly generate inner features of input molecular or crystal graphs and aggregates them by a number of trainable layers to produce a final prediction. This, in turn, hinders interpretation of GNN prediction pipeline, getting scientific insights on certain data importance, and searching for enhanced data selection routines73,74. Keep in mind the challenge of rationalized selection of training samples from the entire CCS to reaching a trade-off between the size of the training dataset and the quality of the model predictions, the insights of this study may additionally support development of tools efficient in terms of data amounts required for training and validation purposes.
Data availability
The Supplementary Information are available free of charge. Details on the pretraining dataset, additional statistics of the built composition/configuration spaces including group-subgroup relations analysis, and visual representation of the introduced predominantly high and low symmetry datasets. The data obtained and analyzed within this work as well as the Python scripts for processing and visualization of the results are available free of charge on GitHub: https://github.com/AIRI-Institute/doped_CsPbI3_energetics2.
Abbreviations
- CCS:
-
Composition/configuration space
- GNN:
-
Graph neural network
- DFT:
-
Density functional theory
- PLS:
-
Predominantly low symmetry
- PHS:
-
Predominantly high symmetry
- RMSE:
-
Root-mean-squared error
- VASP:
-
Vienna Ab initio Simulation Package
References
Kang, S., Kim, M. & Min, K. Discovery of superionic Solid-State electrolyte for Li-Ion batteries via machine learning. J. Phys. Chem. C Am. Chem. Soc. 127 (39), 19335–19343 (2023).
Lan, J. et al. Comprehensive and accurate prediction of band gap for Lead-Free double perovskites through Self-Modified machine learning strategy. J. Phys. Chem. C Am. Chem. Soc. 127 (48), 23412–23419 (2023).
Sun, Y. et al. Interpretable machine learning to discover perovskites with high spontaneous polarization. J. Phys. Chem. C Am. Chem. Soc. 127 (49), 23897–23905 (2023).
Xu, P. et al. Machine learning combined with weighted voting regression and proactive searching progress to discover ABO3-δ perovskites with high oxide ionic conductivity. J. Phys. Chem. C Am. Chem. Soc. 127 (34), 17096–17108 (2023).
Lyngby, P. & Thygesen, K. S. Data-driven discovery of 2D materials by deep generative models. NPJ Comput. Mater. 8 (1), 232 (2022).
Rosen, A. S. et al. High-throughput predictions of metal–organic framework electronic properties: theoretical challenges, graph neural networks, and data exploration. NPJ Comput. Mater. 8 (1), 112 (2022).
Rumiantsev, E. et al. Doping position Estimation for FeRh-based alloys. Sci. Rep. 14 (1), 20612 (2024).
Eremin, R. A. et al. Li(Ni,Co,Al)O2 cathode delithiation: A combination of topological analysis, density functional theory, neutron diffraction, and machine learning techniques. J. Phys. Chem. C Am. Chem. Soc. 121 (51), 28293–28305 (2017).
Chen, D. et al. A machine learning framework for predicting physical properties in configuration space of gate alloys. Mater. Today Commun. 37, 107526 (2023).
Huang, P. et al. Unveiling the complex structure-property correlation of defects in 2D materials based on high throughput datasets. NPJ 2D Mater. Appl. 7 (1), 6 (2023).
Eremin, R. A. et al. Hybrid DFT/data-driven approach for searching for new quasicrystal approximants in Sc-X (X = Rh, Pd, Ir, Pt) systems. Cryst. Growth Des. 22 (7), 4570–4581 (2022).
Zhuang, Z., Fox, B. L. & Barnard, A. S. Simultaneous prediction and optimization of charge transfer properties of graphene and graphene oxide nanoflakes from multitarget machine learning. J. Phys. Chem. C Am. Chem. Soc. 127 (45), 22364–22377 (2023).
Ouyang, X. et al. Quantum-Accurate modeling of ferroelectric phase transition in perovskites from Message-Passing neural networks. J. Phys. Chem. C Am. Chem. Soc. 127 (42), 20890–20902 (2023).
Stark, W. G. et al. Machine learning interatomic potentials for reactive hydrogen dynamics at metal surfaces based on iterative refinement of reaction probabilities. J. Phys. Chem. C Am. Chem. Soc. 127 (50), 24168–24182 (2023).
Brunin, G. et al. Transparent conducting materials discovery using high-throughput computing. NPJ Comput. Mater. 5 (1), 63 (2019).
Oganov, A. R. et al. Structure prediction drives materials discovery. Nat. Rev. Mater. 4 (5), 331–348 ( 2019).
Tsypin, A. et al. Gradual optimization learning for conformational energy minimization. In The Twelfth International Conference on Learning Representations. (2024).
Xu, L. et al. Data efficient and stability indicated sampling for developing reactive machine learning potential to achieve ultralong simulation in Lithium-Metal batteries. J. Phys. Chem. C Am. Chem. Soc. 127 (50), 24106–24117 (2023).
Qi, J. et al. Robust training of machine learning interatomic potentials with dimensionality reduction and stratified sampling. NPJ Comput. Mater. 10 (1), 43 (2024).
de Montes, D. et al. Training data selection for accuracy and transferability of interatomic potentials. NPJ Comput. Mater. 8 (1), 189 (2022).
Karabin, M. & Perez, D. An entropy-maximization approach to automated training set generation for interatomic potentials. J. Chem. Phys. 153 (9), 094110 (2020).
Allen, C. & Bartók, A. P. Optimal data generation for machine learned interatomic potentials. Mach. Learn. Sci. Technol. IOP Publishing. 3 (4), 045031 (2022).
Zhang, Y. & Ling, C. A strategy to apply machine learning to small datasets in materials science. NPJ Comput. Mater. 4 (1), 25 (2018).
Xu, P. et al. Small data machine learning in materials science. NPJ Comput. Mater. 9 (1), 42 (2023).
Shimakawa, H., Kumada, A. & Sato, M. Extrapolative prediction of small-data molecular property using quantum mechanics-assisted machine learning. NPJ Comput. Mater. 10 (1), 11 (2024).
Gupta, V. et al. Cross-property deep transfer learning framework for enhanced predictive analytics on small materials data. Nat. Commun. 12 (1), 6595 (2021).
Wang, A. et al. Benchmarking active learning strategies for materials optimization and discovery. Oxf. Open. Mater. Sci. 2 (1), 006 (2022).
Yuan, X. et al. Active learning to overcome exponential-wall problem for effective structure prediction of chemical-disordered materials. NPJ Comput. Mater. 9 (1), 12 (2023).
Hoock, B., Rigamonti, S. & Draxl, C. Advancing descriptor search in materials science: feature engineering and selection strategies. New. J. Phys. IOP Publishing. 24 (11), 113049 (2022).
Seko, A., Togo, A. & Tanaka, I. Descriptors for Machine Learning of Materials Data. Nanoinformatics, 3–23 (Springer, 2018).
Ghiringhelli, L. M. et al. Learning physical descriptors for materials science by compressed sensing. New. J. Phys. IOP Publishing. 19 (2), 023017 (2017).
Himanen, L. et al. DScribe: library of descriptors for machine learning in materials science. Comput. Phys. Commun. 247, 106949 (2020).
Gallegos, M. et al. An unsupervised machine learning approach for the automatic construction of local chemical descriptors. J. Chem. Inf. Model. Am. Chem. Soc. 64 (8), 3059–3079 (2024).
Liu, Y. et al. An automatic descriptors recognizer customized for materials science literature. J. Power Sources. 545, 231946 (2022).
Eremin, R. A. et al. Ionic transport in doped solid electrolytes by means of DFT modeling and ML approaches: A case study of Ti-Doped KFeO2. J. Phys. Chem. C Am. Chem. Soc. 123 (49), 29533–29542 (2019).
Fung, V. et al. Benchmarking graph neural networks for materials chemistry. NPJ Comput. Mater. 7 (1), 84 (2021).
Reiser, P. et al. Graph neural networks for materials science and chemistry. Commun. Mater. 3 (1), 93 (2022).
Duval, A. et al. A Hitchhiker’s Guide to Geometric GNNs for 3D Atomic Systems. (2023).
Korovin, A. N. et al. Boosting heterogeneous catalyst discovery by structurally constrained deep learning models. Mater. Today Chem. 30, 101541 (2023).
Wang, Y. K. et al. All-Inorganic quantum-dot leds based on a phase-stabilized α-CsPbI3 perovskite. Angew. Chem. Int. Ed. 60 (29), 16164–16170 (2021).
Duan, L. et al. Phase-Pure γ-CsPbI3 for efficient inorganic perovskite solar cells. ACS Energy Lett. Am. Chem. Soc. 7 (9), 2911–2918 (2022).
Liang, J. et al. All-Inorganic perovskite solar cells. J. Am. Chem. Soc. Am. Chem. Soc. 138 (49), 15829–15832 (2016).
Zhao, B. et al. Thermodynamically stable orthorhombic γ-CsPbI3 thin films for High-Performance photovoltaics. J. Am. Chem. Soc. Am. Chem. Soc. 140 (37), 11716–11725 (2018).
Liang, J. et al. Solution synthesis and phase control of inorganic perovskites for high-performance optoelectronic devices. Nanoscale Royal Soc. Chem. 9 (33), 11841–11845 (2017).
Marronnier, A. et al. Anharmonicity and disorder in the black phases of cesium lead iodide used for stable inorganic perovskite solar cells. ACS Nano Am. Chem. Soc. 12 (4), 3477–3486 (2018).
Ustinova, M. I. et al. Partial substitution of Pb2 + in CsPbI3 as an efficient strategy to design fairly stable All-Inorganic perovskite formulations. ACS Appl. Mater. Interfaces Am. Chem. Soc. 13 (4), 5184–5194 (2021).
Travis, W. et al. On the application of the tolerance factor to inorganic and hybrid halide perovskites: a revised system. Chem. Sci. Royal Soc. Chem. 7 (7), 4548–4556 (2016).
Sa, R. et al. Revealing the influence of B-site doping on the physical properties of CsPbI3: A DFT investigation. J. Solid State Chem. 309, 122956 (2022).
Pansa-Ngat, P. et al. Stereoelectronic effect from B-Site dopants stabilizes black phase of CsPbI3. Chem. Mater. Am. Chem. Soc. 35 (1), 271–279 (2023).
Huang, X. et al. B-site doping of CsPbI3 quantum Dot to stabilize the cubic structure for high-efficiency solar cells. Chem. Eng. J. 421, 127822 (2021).
Shen, X. et al. Zn-Alloyed CsPbI3 nanocrystals for highly efficient perovskite Light-Emitting devices. Nano Lett. Am. Chem. Soc. 19 (3), 1552–1559 (2019).
Zhao, Y. et al. Novel B-site Cd2 + doped CsPbBr3 quantum Dot glass toward strong fluorescence and high stability for wLED. Opt. Mater. (Amst). 107, 110046 (2020).
Ji, S. et al. Near-Unity red Mn2 + Photoluminescence quantum yield of doped CsPbCl3 nanocrystals with cd incorporation. J. Phys. Chem. Lett. Am. Chem. Soc. 11 (6), 2142–2149 (2020).
Skurlov, I. D. et al. Improved One- and Multiple-Photon excited photoluminescence from Cd2+-Doped CsPbBr3 perovskite NCs. Nanomaterials 12 (1), 151 (2022).
Guo, J. et al. Pb2 + doped CsCdBr3 perovskite nanorods for pure-blue light-emitting diodes. Chem. Eng. J. 427, 131010 (2022).
Imran, M. et al. Alloy CsCdxPb1–xBr3 perovskite nanocrystals: the role of surface passivation in preserving composition and blue emission. Chem. Mater. Am. Chem. Soc. 32 (24), 10641–10652 (2020).
Cai, T. et al. Synthesis of All-Inorganic Cd-Doped CsPbCl3 perovskite nanocrystals with Dual-Wavelength emission. J. Phys. Chem. Lett. Am. Chem. Soc. 9 (24), 7079–7084 (2018).
Naresh, V. & Lee, N. Zn(II)-Doped cesium lead halide perovskite nanocrystals with high quantum yield and wide color tunability for color-Conversion Light-Emitting displays. ACS Appl. Nano Mater. Am. Chem. Soc. 3 (8), 7621–7632 (2020).
Thapa, S. et al. Zn-Alloyed All-Inorganic halide Perovskite-Based white Light-Emitting diodes with superior color quality. Sci. Rep. 9 (1), 18636 (2019).
Zeng, Y. T. et al. Bright CsPbBr3 perovskite nanocrystals with improved stability by In-Situ Zn-Doping. Nanomaterials 12 (5), 759 (2022).
Eremin, R. A. et al. Graph neural networks for predicting structural stability of Cd- and Zn-doped γ-CsPbI3. Comput. Mater. Sci. 232, 112672 (2024).
Sutton, R. J. et al. Bandgap-tunable cesium lead halide perovskites with high thermal stability for efficient solar cells. Adv. Energy Mater. 6 (8), 1502458. (2016).
Yang, Z. et al. Impact of the halide cage on the electronic properties of fully inorganic cesium lead halide perovskites. ACS Energy Lett. Am. Chem. Soc. 2 (7), 1621–1627 (2017).
Sutton, R. J. et al. Cubic or orthorhombic?? Revealing the crystal structure of metastable Black-Phase CsPbI3 by theory and experiment. ACS Energy Lett. Am. Chem. Soc. 3 (8), 1787–1794 (2018).
Okhotnikov, K., Charpentier, T. & Cadars, S. Supercell program: a combinatorial structure-generation approach for the local-level modeling of atomic substitutions and partial occupancies in crystals. J. Cheminform. 8 (1), 17 (2016).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for Ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Am. Phys. Soc. 54 (16), 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. Am. Phys. Soc. 77 (18), 3865–3868 (1996).
Musaelian, A. et al. Learning local equivariant representations for large-scale atomistic dynamics. Nat. Commun. 14 (1), 579 (2023).
Schütt, K. T. et al. SchNet – A deep learning architecture for molecules and materials. J. Chem. Phys. 148 (24), 241722 (2018).
Batzner, S. et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 13 (1), 2453 (2022).
Curtarolo, S. et al. An automatic framework for high-throughput materials discovery. Comput. Mater. Sci. AFLOW, 218–226 (2012).
Ong, S. P. et al. Python materials genomics (pymatgen): A robust, open-source python library for materials analysis. Comput. Mater. Sci. 68, 314–319 (2013).
Holm, E. A. In defense of the black box. Sci. Am. Assoc. Adv. Sci. 364 (6435), 26–27. (2019).
Oviedo, F. et al. Interpretable and explainable machine learning for materials science and chemistry. Acc. Mater. Res. Am. Chem. Soc. 3 (6), 597–607 (2022).
Acknowledgements
The authors thank the hardware and software facilities of the “Christofari” heterogeneous platform.
Author information
Authors and Affiliations
Contributions
Aliaksei V. Krautsou: Methodology, Software, Investigation, Data curation, Writing – original draft, review & editing, Visualization. Innokentiy S. Humonen: Methodology, Software, Writing – review & editing. Vladimir D. Lazarev: Data curation. Roman A. Eremin: Conceptualization, Methodology, Software, Investigation, Writing – original draft, review & editing, Visualization. Semen A. Budennyy: Resources, Writing – review & editing, Supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Krautsou, A.V., Humonen, I.S., Lazarev, V.D. et al. Impact of crystal structure symmetry in training datasets on GNN-based energy assessments for chemically disordered CsPbI3. Sci Rep 15, 8856 (2025). https://doi.org/10.1038/s41598-025-92669-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-92669-3
