
Abstract
Advances in cold storage and cryopreservation of amphibian sperm are critical for the genetic management and conservation of threatened species. This study represents the first investigation into the sperm of the yellow spotted mountain newt (Neurergus derjugini), focusing on both short-term and long-term storage for future reproductive efforts. We examined the effects of seven extenders on sperm motility over time at three storage temperatures (4 ± 1 °C, 9 ± 1 °C, and 20 ± 1 °C). Additionally, we assessed the impact of 16 cryoprotectants on sperm motility and morphology post-thawing. Following the identification of the most effective freezing medium, we evaluated sperm DNA fragmentation to ensure viability. Our results indicate that 10% Holtfreter’s solution is the optimal extender for short-term storage at all three temperatures, maintaining sperm motility for up to 15 days at 4 °C. For long-term storage, a combination of 10% Holtfreter’s solution and 10% DMSO was found to best preserve sperm motility, morphology, and minimize DNA fragmentation after thawing. These findings underscore the importance of specific extenders and temperature treatments in enhancing sperm functionality, thereby supporting successful assisted reproductive technologies (ART) for endangered species.
Similar content being viewed by others

Introduction
The current global amphibian extinction crisis necessitates the use of sperm storage as a valuable strategy for supporting at-risk populations and potentially reviving recently extinct species1. While recent years have seen the implementation of conservation breeding programs for threatened amphibians, these efforts can often lead to a decrease in genetic diversity2. This loss of natural genetic variation negatively impacts a species’ survival both in the wild and in captivity, leading to reduced reproductive capabilities, limited adaptability to environmental changes, and lower success rates of conservation breeding and restocking programs3,4,5. Cryopreservation of amphibian sperm offers a promising tool for maintaining and restoring amphibian populations, particularly for species that are challenging to breed in captivity or have low genetic variation6. Furthermore, sperm freezing can reduce the need for maintaining large numbers of animals in captivity, which is costly and stressful7. It also facilitates the exchange of genetic material between different captive facilities or regions, thereby enhancing genetic health and offering a relatively efficient, secure, and reliable method8,9. Moreover, cold storage and cryopreservation of sperm are practical methods for ex situ propagation10,11.
Over the past few decades, techniques for preserving amphibian sperm through freezing and thawing have significantly improved7,12,13. Research has demonstrated that sperm from various species can remain viable after cryopreservation, and this preserved sperm can be used for artificial fertilization to produce viable embryos and offspring14,15,16. Notably, these methods have been effective in the conservation of critically endangered species such as the Mississippi gopher frog (Lithobates sevosus)17 and the axolotl (Ambystoma mexicanum)13. However, the process of cryopreserving amphibian sperm is challenging due to their unique characteristics compared to sperm from other species. Amphibian sperm require specific environmental conditions, such as water and optimal ionic concentrations, for motility and fertilization5. Upon activation, they transform rapidly to swim toward the egg but have a limited lifespan to reach it18. Environmental factors and metabolic changes can affect this lifespan, making timing crucial for successful fertilization19. In externally fertilizing species such as frogs and toads, sperm activation is primarily triggered by a decrease in osmolality, rather than by direct contact with water or specific ions20. As a result, researchers must carefully optimize the freezing and thawing methods to preserve the viability and fertility of amphibian sperm7. Different species require specific cryoprotectants, freezing rates, and storage conditions to preserve sperm quality21. But this is not always the case; some protocols can be transferred between species and genera22. For example, most protocols for cryopreservation of anuran sperm have been based on protocols for freshwater fish, because fish sperm have similar osmolality properties to anuran sperm23,24. Consequently, further research is needed to refine protocols for amphibian sperm cryopreservation for conservation purposes.
Post-thaw motility is a critical factor for the success of sperm cryopreservation, directly influencing the chances of fertilizing eggs13. Factors such as cryoprotectant choice, cooling and thawing rates, and medium selection all play essential roles in preserving sperm motility25,26. Cryoprotectants act as shields to protect sperm integrity during freezing by reducing ice crystal formation that can damage cell structures27. Studies indicate that dimethyl sulfoxide (DMSO) and dimethyl formamide (DMFA) are effective cryoprotectants for certain amphibian species15,28,29. Additionally, incorporating antioxidants into cryopreservation extenders mitigates detrimental effects30,31. Enzymatic antioxidants, such as catalase and superoxide dismutase, have been shown to enhance post-thaw sperm motility and acrosome integrity by minimizing oxidative damage32. Non-enzymatic antioxidants, including vitamins C and E, have also been explored, though their efficacy varies depending on the species and specific cryopreservation conditions33. These advancements are crucial for developing effective biobanking strategies to conserve genetic material from endangered amphibians. Research on amphibian sperm cryopreservation typically focuses on two post-thaw parameters: motility and viability34,35. Recent studies also indicate that sperm cryopreservation procedures can cause DNA fragmentation, a concern for genome integrity crucial for successful embryonic development36. However, assessments of DNA integrity have been limited to anuran amphibians37,38,39, and similar studies on caudate amphibians are lacking. Understanding how to handle and store sperm properly is essential for improving assisted reproduction technologies (ART) and conservation efforts40. Cold storage techniques, such as maintaining sperm at 0–4 °C, have been developed to extend sperm lifespan for in vitro fertilization (IVF) or cryopreservation41,42. However, there is a limited understanding of caudate sperm storage, and improving cold storage techniques for sperm could offer a more reliable and cost-effective method for conservation43,44. The choice of extenders also significantly impacts sperm motility and morphology, affecting sperm preservation efforts13,44.
Among amphibians, caudate species like the yellow spotted mountain newt (Neurergus derjugini) face severe threats24. Endemic to the Zagros Mountain chain across western Iran and northeast Iraq, this species is listed as threatened (IUCN Red List, 2023) due to factors such as habitat loss, agricultural practices, illegal pet trade, and climate change45,46. While steps have been taken for captive breeding, conservation, and reintroduction, these efforts must address genetic diversity preservation for successful species reintroduction47. Cryopreservation of sperm offers a means to establish a genetic resource for future breeding programs, enabling the reintroduction of diverse genetic lineages48,49. This research is the first study focused on developing ART for N. derjugini, aiming to create protocols for short and long-term sperm storage to support conservation and breeding efforts. Specifically, this study evaluates sperm motility in seven extenders at three temperatures for short-term storage and examines 16 cryoprotectants for long-term storage, assessing motility, morphology, and DNA fragmentation to determine the best sperm-freezing environment.
Result
Sperm volume and concentration
The average sperm volume produced by the newts (n = 17) was 50.88 ± 10.93 μL, with a mean sperm concentration of 1.64 × 10⁶ ± 6.99 sperm/mL.
Short-term sperm storage
Sperm motility
Sperm motility in different types of solutions and different temperatures showed that these factors are very effective on sperm motility. In general, the highest percentage of sperm motility in all three storage temperatures was shown in 10% Holt and the lowest in 5% sucrose.
Sperm motility at 4 °C
At 4 °C, the highest sperm motility observed across all extenders was with 10% Holt extender, whereas the lowest motility was recorded with 5% sucrose. Initially, the motility was highest with 10% Holt (95.83 ± 3.76%), followed by PBS, SAR, HBSS, 1% BSA, and the lowest in Distilled Water (77.5 ± 5.24%) and 5% Sucrose (75 ± 7.07%). At six hours, the highest motility remained with 10% Holt (86.66 ± 2.58%), while Distilled Water (31.66 ± 6.83%) and 5% Sucrose (17.50 ± 5.24%) showed the lowest motility (Fig. 1A1). Over the first 24 h, 10% Holt consistently showed the highest motility (79.54 ± 3.50%), followed by SAR (71.81 ± 9.81%), HBSS (64.09 ± 11.13%), PBS (66.81 ± 8.14%), and 1% BSA (63.63 ± 6.36%). Distilled Water and 5% Sucrose showed no motility and were not evaluated beyond this period. The complete cessation of motility was observed on Day 11 for BSA, Day 12 for HBSS, Day 14 for SAR and PBS, and Day 15 for 10% Holt (Fig. 1A2). Statistical analysis showed significant differences in motility at 4 °C (P ≤ 0.001).

Variation in sperm motility for seven extenders at different temperatures. (A1) Average sperm motility over time (in hours) from 0 to 6 h at 4 °C. (A2) Average sperm motility over time (in days) from 1 to 15 days at 4 °C. (B1) Average sperm motility over time (in hours) from 0 to 6 h at 9 °C. (B2) Average sperm motility over time (in days) from 1 to 5 days at 9 °C. (C1) Average sperm motility over time (in hours) from 0 to 6 h at 20 °C. (C2) Average sperm motility over time (in days) from 1 to 4 days at 20 °C. Twelve different sperm samples (n = 12) were analyzed for each extender from 7 male newts (N = 7). Data represent mean values ± SD. The asterisks on sucrose and water in figures (A1, B1, and C1) indicate a significant p-values (P ≤ 0.001) between these two extenders and the other five extenders (HBSS, SAR, PBS, BSA, Holt) according to Mann–Whitney U tests.
Sperm motility at 9 °C
At 9 °C, the highest initial motility was observed with 10% Holt (95.00 ± 3.16%), and the lowest with Distilled Water (75.83 ± 5.84%). After six hours, 10% Holt maintained the highest motility (85.83 ± 2.04%), followed by PBS (78.33 ± 5.16%), SAR (74.16 ± 4.91%), 1% BSA (68.33 ± 5.16%), and HBSS (59.16 ± 5.84%), with Distilled Water (6.66 ± 5.16%) and 5% Sucrose (0%) showing minimal motility (Fig. 1B1). Over 24 h, 10% Holt exhibited the highest motility (62.50 ± 5.24%), followed by SAR (61.66 ± 14.02%), HBSS (49.16 ± 5.38%), PBS (45.00 ± 7.07%), and 1% BSA (25.00 ± 13.03%). The longest sperm survival in terms of motility was observed for 10% Holt and SAR, with motility lasting up to five days (Fig. 1B2). Statistical analysis revealed significant differences at this temperature (P ≤ 0.001).
Sperm motility at 20 °C
At 20 °C, the highest initial motility was with 10% Holt (95.00 ± 4.47%), followed by SAR (93.33 ± 6.05%), HBSS (90.83 ± 5.84%), PBS (90.00 ± 7.07%), 1% BSA (87.50 ± 5.24%), with Distilled Water (67.50 ± 5.24%) and 5% Sucrose (63.33 ± 6.05%) being the lowest. After 6 h, the highest motility was again with 10% Holt (72.50 ± 5.24%), and the lowest was observed in Distilled Water and 5% Sucrose (immotile) (Fig. 1C1). Over 24 h, the highest motility was recorded for HBSS (64.16 ± 7.35%), followed by 10% Holt (63.33 ± 10.32%), SAR (60.00 ± 9.48%), PBS (18.33 ± 8.16%), and 1% BSA (12.50 ± 2.73%). The lowest sperm survival was at 20 °C, with motility only observed on Day 3 for HBSS and Holt (Fig. 1C2). The data for motility at 20 °C were also significant (P ≤ 0.001).
Long-term sperm storage
Sperm motility post-thawing
Sperm samples frozen in C1 to C11 were immotile after thawing, and thus were not suitable for long-term storage. In contrast, C12 exhibited around 10% motility after 24 h, but lost motility by Day 3. C13 showed 31% ± 6.51 motility after 24 h, with motility declining to 14% ± 6.51 on Day 3 and 5% ± 3.53 by Day 7. C14, C15, and C16 showed sustained motility with C15 and C16 maintaining motility up to 21- and 30-days post-thawing, respectively (Table 1). From Day 15, motility showed flagellar movement and limited forward motion. Statistical analysis confirmed significant differences in post-thaw motility (P ≤ 0.001) (Fig. 2).

Variation in sperm motility post-thawing in three Cryoprotectants C14, C15 and C16 over one month. Twelve different sperm samples (n = 12) were analyzed from 5 male newts (N = 5). Data represent mean values ± SD, significant p values are shown by an asterisk (*p ≤ 0.05, **p ≤ 0.001).
Sperm morphology post-thawing
Morphological analysis of sperm from C14, C15, and C16 revealed that C16 had the lowest rate of abnormalities, followed by C15 and C14 (Table 1). The percentages of abnormalities for other extenders at 24 h post-thawing were: C1 (5.4 ± 1.14%), C2 (15 ± 3.87%), C3 (12.56 ± 1.26%), C4 (7.8 ± 1.52%), C5 (5.4 ± 1.14%), C6 (7.5 ± 1.30%), C7 (8.7 ± 2.57%), C8 (11.2 ± 1.76%), C9 (12.0 ± 2.57%), C10 (7.3 ± 2.00%), and C11 (7.5 ± 1.30%). The C14, C15, and C16 yielded the lowest abnormality rates, suggesting their effectiveness in preserving sperm morphology (Fig. 3A–D). Also, for C12, abnormal sperm was estimated for the first day (5.00 ± 1.58%) and for the third day (8.75 ± 1.70%). Moreover, for C13, the abnormality percentage was estimated on the first day (5.20 ± 2.38%), the third day (6.60 ± 2.88%), and the seventh day (7.40 ± 2.40%) (P ≤ 0.001).

Neurergus derjugini sperm stained by Diff-Quik. (A) Morphologically normal sperm; (B–D) Abnormalities including head and tail damage, such as head and tail cutting, breaking, and tail twisting. Images are shown at 400 × magnification. Individual images of each sperm were captured using AmScope Version x.64, 3.7.10246.20171109 (https://amscope.com/pages/software-downloads. Irvine, CA, 92602, USA). Ten different sperm samples (n = 10) were analyzed from 5 male newts (N = 5).
Sperm DNA fragmentation post-thawing
The DNA analysis results for the three cryoprotectants (C14, C15, and C16) over days 1, 3, 7, 15, 21, and 30 indicate significant differences in their effectiveness in preserving sperm DNA integrity. C16 demonstrated the highest proportion of large halos around the sperm head after one month, with a percentage of non-fragmented DNA at 60.00 ± 5%. Initially, on Day 7, C16 exhibited a high non-DNA fragmentation rate of 73.33 ± 2.88%. This rate decreased to 66.66 ± 2.88% by the second week and to 63.33 ± 2.88% by the third week. C15 showed the next highest proportion of large halos, with 45.00 ± 5%. On Day 7, the non-DNA fragmentation rate for C15 was 68.33 ± 2.88%, decreasing to 60.00 ± 5% by Day 15, and to 55.00 ± 5% by Day 21. In contrast, C14 displayed a more pronounced decrease in non-DNA fragmentation, starting from 60.00 ± 5% on Day 7, reducing to 50.00 ± 5% by Day 15, and further dropping to 36.66 ± 7.63% by Day 21.
Results indicate that C16 is the most effective in reducing DNA fragmentation over time compared to C14 and C15. Furthermore, C16 resulted in the highest percentage of morphologically normal sperm (non-fragmented DNA) at 79.28 ± 4.49%, showing the greatest similarity to the control group (Fig. 4. A-F). Statistical analysis confirmed that the differences in DNA fragmentation among the cryoprotectants and over time are significant (P ≤ 0.001).

Sperm DNA integrity in Neurergus derjugini using a DNA fragmentation test. (A, B): Sperm with DNA fragmentation showing no halo around the sperm head. (C, D): Morphologically normal sperm without DNA fragmentation displaying a medium-sized halo around the head. (E, F): Morphologically normal sperm without DNA fragmentation with a large halo around the head. Individual images of each sperm were captured using AmScope Version x.64, 3.7.10246.20171109 (https://amscope.com/pages/software-downloads, Irvine, CA, 92602, USA). Ten different sperm samples (n = 10) were analyzed for each diluent, from 5 male newts (N = 5).
Discussion
In this study, we observed and evaluated the sperm of the N. derjugini species for the first time. Our primary aim was to explore methods for preserving sperm for future fertilization studies under laboratory conditions and to address spawning asynchrony in captivity. This research also established a connection between sperm extender media, motility duration, and storage temperatures. Furthermore, we investigated long-term sperm storage using different cryoprotectants to develop optimal protocols for amphibian genetic diversity programs in captivity. Our study highlights the critical role of selecting an appropriate extender solution, as it significantly impacts both sperm motility and morphology. Notably, this research is pioneering in its use of a DNA fragmentation kit to assess sperm health following the freezing process. This approach underscores the importance of integrating advanced technologies into sperm storage strategies for breeding programs and the conservation of endangered species.
Few studies have explored extenders for diluting sperm in caudate amphibians. Investigations in this area have focused on various extenders, such as those used by Mansour et al.50, who examined sperm motility in the axolotl (A. mexicanum) and reported a motility duration of 4 h at 0 °C and 1 h at room temperature when using water or fertilization solution as extenders. Similarly, Marcec 201624 evaluated 10% Holt’s solution and 1% BSA as sperm extenders in tiger salamanders (A. tigrinum). Guy et al.11 employed 2% trehalose and 0.2% BSA for three newt species, and McGinnity et al.51 used SAR solution, which aligns with our findings showing SAR as the second most effective extender at various temperatures. Additionally, Gillis et al.40 demonstrated that 2% trehalose + 0.2% BSA maintained sperm motility better at 0 °C compared to 20 °C, reinforcing our results that favor lower temperatures for sperm storage. Other studies, like those by Arregui et al.52, who stored sperm from Fowler’s toad (Anaxyrus fowleri) at 4 °C for up to 8 days, and Browne et al.8, who observed over 50% motility for cane toad (Bufo marinus) sperm stored at 0 °C for up to 7 days, have further informed the field. Our study confirms that 10% Holt’s solution is effective for sperm extenders, particularly for extended storage at 4 °C, and that it is superior to other extenders like SAR, PBS, and HBSS at this temperature. At 9 °C, 10% Holt’s solution showed the highest daily motility, followed by SAR, HBSS, PBS, and 1% BSA, respectively. At 21 °C, 10% Holt’s solution and HBSS maintained motility for two days, whereas other extenders like sucrose and distilled water showed less than 24 h of motility. Our study builds upon recent research, such as Coxe et al.13 which compared deionized water and HBSS for sperm extenders and found that HBSS provided better motility. Chen et al.44 demonstrated that 10% Holt’s solution was effective for maintaining progressive motility. We expanded on these findings by evaluating multiple extenders at three different temperatures, confirming that 10% Holt’s solution supports long-term sperm storage at 4 °C.
Osmolality plays a significant role in influencing sperm motility in amphibians, which in turn impacts fertilization success and reproductive strategies7,20. Research has shown that the optimal osmolality for activating sperm motility in amphibians varies by species53. For example, in the common eastern froglet (Crinia signifera), optimal sperm motility was observed between 10 and 50 mOsm kg, suggesting that amphibians have adapted their sperm traits to function effectively within the osmolality ranges typical of their natural breeding environments. Conversely, high osmolality environments can negatively affect sperm motility20. A study on myobatrachid frog (Limnodynastes tasmaniensis) found that fertilization success was optimized at low osmolalities (0–7 mOsm kg), with higher concentrations leading to decreased motility and fertilization rates54. This is consistent with findings indicating that excessive osmolality can cause detrimental effects on cellular integrity and function due to osmotic stress19. In tiger salamanders (A. tigrinum), sperm motility was best maintained in media that closely matched the osmolality of their reproductive tract conditions44. Similarly, our research on N. derjugini demonstrated that sperm motility remained unaffected by both low and high osmolalities, aligning with findings by Chen et al.44. However, we found that sucrose at 5% and distilled water resulted in the lowest motility at three different temperatures, whereas BSA at 1%, with the lowest osmolality, maintained motility across various temperatures, particularly at 4 °C. This highlights the importance of temperature as a factor influencing sperm motility, especially in species with internal fertilization. Our results suggest that N. derjugini sperm exhibit resilience and adaptability in different osmolality solutions, potentially due to their ability to cope with varying environmental conditions. This adaptability may be an evolutionary trait allowing sperm to navigate diverse environments, such as the internal reproductive tracts of females and aquatic habitats like ponds, where they are often found within spermatophores4,44,55.
Research has shown that the choice and concentration of cryoprotectants significantly impact the post-thaw motility and survival of sperm. The use of specific cryoprotectants, such as DMSO and DMFA, at certain concentrations can lead to high rates of recovery of motility and fertilizing capacity in amphibian sperm5,6,29. Our study found that C14, C15, and C16 were the most effective in preserving sperm motility and morphology. C16 showed the best results in terms of motility, morphology, and DNA integrity, highlighting the importance of selecting optimal cryoprotectants for long-term sperm storage. The studies that have been used in recent years for freezing the sperm of caudate amphibians have used a combination of both penetrative and non-penetrative cryoprotectants, which have brought different results on motility and morphology sperm11,24,51. The combination of penetrative and non-penetrative cryoprotectants is crucial in achieving superior post-thaw recovery with high proportions of forward progressive motility, live cells, and intact acrosomes in various amphibian species56. Our results showed that no significant motility was observed for N. derjugini sperm using the compounds that were previously used to investigate the motility after thawing of tailed amphibian sperm (C1-C3). The mechanism of cold protection for amphibian sperm involves the use of cryoprotectants to minimize the detrimental effects of cooling, freezing, and thawing on sperm function. The use of DMSO as a cryoprotectant is effective in reducing sperm lysis and promoting post-thaw recovery of sperm motility and vitality21. Additionally, the combination of DMSO and sucrose is particularly effective in preserving sperm viability and motility in various amphibian species. However, the concentration of non-penetrative cryoprotectants such as sucrose should be carefully considered, as excessive osmolality may cause damage to cells57.
Cryoprotectants play a vital role in preserving sperm viability during freezing by preventing the formation of ice crystals, which can damage cellular structures58. Membrane-permeable cryoprotectants, such as DMSO, glycerol, and ethylene glycol, are commonly used due to their ability to easily penetrate sperm membranes. This property allows them to stabilize cellular membranes and reduce the formation of ice crystals inside the cells. At low concentrations, these permeating agents help manage the rate of dehydration and prevent osmotic stress59,60. However, at higher concentrations, they can become toxic, highlighting the need for optimal concentrations to minimize their detrimental effects on sperm61. These agents control the movement of water during the dehydration and rehydration processes, reducing the damage that may result from the use of permeating agents. By stabilizing the cellular structure and minimizing the damage from excessive dehydration, non-permeating cryoprotectants help preserve sperm motility and vitality during thawing62,63. The combination of these cryoprotectants both permeable and non-permeable is particularly effective in amphibian sperm preservation. Specifically, DMSO and sucrose, when used together, have demonstrated superior results in maintaining sperm viability, motility, and morphology. It is, however, important to carefully regulate the concentration of non-permeating cryoprotectants like sucrose, as excessive osmolality may lead to osmotic stress and subsequent damage to the cells. Therefore, the use of cryoprotectants, particularly DMSO in combination with sucrose and other chemical compounds, has proven feasible for preserving amphibian sperm. This combination is a valuable tool for the conservation and genetic management of endangered species.
DNA fragmentation, characterized by the breaking of DNA strands into smaller pieces, occurs during cellular processes such as apoptosis and necrosis64. Oxidative stress, resulting from an imbalance between reactive oxygen species (ROS) and antioxidant defenses, plays a critical role in inducing DNA damage, including strand breaks and base modifications, thereby promoting apoptosis and contributing to DNA fragmentation65,66. Sperm DNA fragmentation is strongly linked to oxidative stress, as defective spermatogenesis often results in elevated ROS levels, which compromise DNA integrity67 In the context of ART, sperm DNA fragmentation has substantial implications. Elevated fragmentation levels are associated with reduced pregnancy rates in ART procedures68,69,70. For amphibians, DNA fragmentation in thawed sperm has been shown to negatively impact reproductive outcomes71. Structural changes to sperm during cryopreservation, including compromised chromatin integrity, increase susceptibility to DNA fragmentation38,72,73. These findings underscore the importance of optimizing cryoprotectant protocols to minimize DNA damage and ensure the reproductive viability of offspring produced via ART. The evolutionary consequences of sperm DNA fragmentation in amphibians, though largely unexplored in empirical studies, have significant theoretical implications. For endangered species, the establishment of genetic resource banks and the application of ART highlight the necessity of addressing sperm DNA damage6. Evaluating and mitigating DNA fragmentation is essential to enhancing the efficacy of cryopreservation protocols, which are pivotal for maintaining genetic diversity and supporting amphibian conservation efforts. This study emphasizes the need for a comprehensive understanding of the interplay between DNA fragmentation, spermatogenesis, and preservation techniques. Such insights can inform strategies to improve cryopreservation outcomes and ART success rates. Furthermore, these advancements not only strengthen conservation strategies but also contribute to a broader understanding of amphibian reproductive biology and its evolutionary dimensions. By mitigating the impacts of DNA fragmentation, we can support the long-term sustainability of amphibian populations and ensure the success of conservation initiatives.
SDF kits like the Halosperm test use the sperm chromatin dispersion assay to assess DNA integrity by measuring halo formation, which indicates DNA fragmentation levels74. This method provides a straightforward and reliable approach to evaluating sperm DNA quality. In contrast, flow cytometry offers a more comprehensive analysis by examining multiple sperm characteristics, including motility, concentration, and morphology, but it often requires additional protocols for assessing specific DNA damage75. Both techniques are crucial for sperm evaluation, with DNA fragmentation assays providing critical insights into genetic quality and potential fertility issues, while cytometric methods excel in efficiency and broad data collection. The choice between these methods depends on clinical or research needs, available resources, and specific circumstances76,77. Our findings demonstrate that cryoprotectants yielding the highest post-thaw motility also resulted in the lowest levels of DNA fragmentation. These results underscore the importance of cryopreservation techniques that minimize genetic damage, thereby supporting the use of advanced methods in conservation efforts for endangered species. Improved sperm preservation techniques can enhance breeding programs and contribute to the recovery of threatened amphibian populations7,78. Furthermore, our study not only sheds light on the preservation of N. derjugini sperm but also lays the groundwork for future research in amphibian reproductive biology. By integrating advanced technologies and refining cryopreservation protocols, we aim to bolster the conservation of this species and deepen our understanding of amphibian reproductive mechanisms.
Conclusion
Our study demonstrated that different sperm extenders have varying effects on sperm motility, and their effectiveness in preserving motility differs across temperatures. We successfully identified several protocols for both short-term and long-term sperm storage, providing a foundation for future studies in vitro fertilization and addressing the challenge of synchronizing sperm production with oviposition in captive newts. One of the significant findings of our research is that the choice of cryoprotectant and storage conditions profoundly affects sperm DNA integrity. Our study identified a cryoprotectant that minimizes DNA damage over the long term, highlighting its potential for improving cryopreservation techniques in amphibians. This is particularly important as technologies for gamete cryopreservation can adversely affect cellular functions, and understanding the effects of the freeze–thaw process on sperm DNA integrity remains an underexplored area in amphibian reproductive biology. Moving forward, further research is needed to explore the long-term effects of cryopreservation on sperm DNA integrity in amphibians. Developing standardized methods for assessing sperm DNA fragmentation will be crucial for advancing amphibian conservation efforts and improving techniques for artificial reproduction. By refining these methods, we can enhance our ability to preserve genetic diversity and support successful breeding programs for endangered amphibian species. Overall, our study provides valuable insights into the optimization of sperm storage protocols and offers a new approach for evaluating sperm DNA health, which can be applied to future research and conservation efforts in amphibian species.
Materials and methods
Ethics approval
The permission to collect newts and all animal care protocols and all the experimental protocols used in this study were handled according to the guidelines and regulations provided by was obtained the research ethics approval committee, Faculty of Science, Razi University [IR.RAZI.REC.1402.064]. Also, the manuscript follows the recommendations in the ARRIVE guidelines. Furthermore, after the experiment, the newts were released into the wild at the same location with the cooperation of conservationists.
Animal’s management
Adult males of Neurergus derjugini (n = 17; aged 3–4 years; mean body weight: 6.71 ± 0.65 g; mean body length: 190.65 ± 9.87 mm) were collected during the breeding season (March–April 2023) from the Kavat River (34° 53′ N, 46° 31′ E), located along the Ravansar-Paveh road, approximately two kilometers from Qori-Qale Cave in Kermanshah Province, Iran. Following collection, the specimens were transported to the laboratory under controlled conditions for further study. The newts were housed in a 10 L glass tank at a temperature of 14–15 °C, with a photoperiod of 12 h light and 12 h darkness, using a 0.25 HP chiller (Hailea, HC-250) to maintain the temperature44. They were fed daily with a variety of small mealworms and bloodworms79. Water was changed weekly, and water quality parameters (temperature and pH) were monitored daily.
Sperm collection
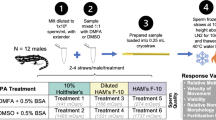
Adult male N. derjugini were identified based on cloacal prominence and collected from their natural habitat. For the experiment, the newts were randomly assigned to different test groups: 7 individuals for sperm motility and short-term storage experiments, 5 individuals for long-term storage experiments, and 5 individuals for morphology and DNA fragmentation analysis of fresh sperm. Prior to the experiments, sperm quality was assessed for each sample, ensuring homogeneity across tests, as all samples were collected simultaneously from the environment. Following an acclimatization period, sperm collection was performed according to previously described protocols15,20. The animals were held ventral side up and blotted dry, and their sides and abdomen were gently massaged from anterior to posterior towards the cloaca. The milt was collected with a micropipette and transferred to 1.5 mL centrifuge tubes. To analyze the concentration of spermatozoa, a 10 μL sample of a 1:100 diluted milt solution was placed on a hemocytometer. The spermatozoa within the four corner squares, each measuring 1 mm2, were counted. To calculate the concentration of spermatozoa per milliliter, the total count from these four squares was divided by 0.4 to adjust for the volume of the squares. This result was then multiplied by 100 to account for the dilution factor and further multiplied by 1000 to convert to a per-milliliter basis24. For anesthesia, pour 350 ppm of clove oil into 200 ml of distilled water and place the animal in the solution for 3–5 min until it is anesthesia80.
Short-term sperm storage
To determine the optimal temperature and extender for short-term sperm storage, collected sperm (an opaque and white liquid) was mixed in a 1:10 ratio in 1.5 mL microcentrifuge Eppendorf tubes with seven extenders: (1) Phosphate Buffered Saline (PBS) [Houston, Texas 77396]16; (2) Simplified Amphibian Ringer (SAR)51,79; (3) Bovine Serum Albumin [(1% BSA) SIGMA, CAS 9048-46-80]24; (4) Holtfreters Solution (10% Holt)24,44; (5) Hanks Balanced Salts Modified (HBSS; 200, SIGMA Powder H-2387)13; (6) 5% Sucrose [SIGMA, CAS 57-50-1]7; (7) Distilled Water (DW)15 (Table 2). Distilled water was used as a solvent to prepare diluents containing powdered materials (e.g., sucrose, BSA, and, …)81,82. The sperm was stored at three temperatures: 4 ± 1 °C (refrigerator temperature), 10 ± 1 °C (collection area temperature), and 20 ± 1 °C (room temperature).
To determine the best extender for sperm suspension, motility was assessed immediately after mixing with each extender (Time 0) and at intervals of 1, 2, 3, 4, 5, and 6 h for motility decay40. After that, our investigations were carried out daily, so that the sperm at each storage temperature and each extenders solution were checked for motility every 24 h. A 10 µL sample from each treatment was pipetted onto a glass slide, covered with a cover slip, and examined under a 200 × magnification using an Olympus® CX41 microscope44.
Sperm motility was assessed by two researchers in an alternating manner to reduce potential bias, with 100 sperm cells randomly selected and analyzed for each sample. Motility parameters were categorized as follows: sperm cells moving with velocity in a circular motion were classified as “progressive motile” (PM), while those exhibiting tail movement without circular motion were categorized as “non-progressive motile” (NPM). The percentage of total motility was calculated by summing the counts of progressive and non-progressive motile cells24,40. Sperm motility evaluation was conducted under a Zeiss AXIO Scope A1 microscope (Carl Zeiss, Oberkochen, Germany) at 200 × magnification, with an experienced observer recording videos of motility for detailed analysis83,84.
Long-term sperm storage
Sperm motility post-thawing
Sperm samples were mixed with one of the 16 cryoprotectant solutions (C1–C16) listed in Table 3 at a 1:1 extender-to-sperm ratio. Each treatment included 12 technical replicates, derived from 20 biological samples. After preparation, the extenders containing sperm were stored at 4 °C until use. The solution from each treatment was drawn into a 0.25 cc cryo-straw, sealed with hematocrit paste, and equilibrated at 4 °C for five minutes21. The straws were then placed in liquid nitrogen vapor, positioned 20 cm above the liquid surface, and held there for 15 min to ensure they reached a temperature of approximately − 80 °C. Following this, the straws were submerged into liquid nitrogen to achieve a storage temperature of − 196 °C using a Taylor Wharton 34HC Cryogenic Storage Dewar (USA)11,13,24.
Sperm motility and morphology were evaluated at 1, 3, 7, 15, 21, and 30 days after freezing. Each cryo-straw was thawed only once at the designated time point and assessed immediately after thawing13,85. No post-thaw storage at non-cryogenic temperatures (e.g., 4 °C) was performed. If a sample exhibited motility at a given time point, fresh sperm was collected from the newt, mixed with the same extender, and frozen again for evaluation at the next time point. This ensured that the sperm used for each time point was fresh and not subjected to multiple freeze–thaw cycles. Extenders that failed to maintain sperm motility within the first 24 or 72 h were excluded from further evaluation. Only extenders demonstrating the ability to preserve motility at earlier time points were assessed at longer intervals (e.g., Days 7, 15, 21, and 30).
Thawing was performed by gently rotating the cryo-straws in a 25 °C water bath for 10–15 s13. Immediately after thawing, a drop of sperm was placed on a slide, covered with a coverslip, and assessed for motility under a microscope at room temperature (17–18 °C) at 200 × magnification24. The remaining sperm in the straws was used to evaluate morphology.
Sperm morphology post-thawing
For morphological examination, fresh and thawed sperm samples from days 1, 3, 7, 15, 21, and 30 were spread on slides (3 repeats per method) and air-dried. Slides were stained with Diff-Quik86,87. A total of 100 sperm cells were examined for regular (normal structure) and abnormal (malformed or broken) sperm, evaluated at 400 × magnification24.
Sperm DNA fragmentation post-thawing
Sperm DNA fragmentation was assessed using the SDFA kit (Idehvarzanfarda-IVF Co, Iran). A 20 µL sample of extenders sperm suspension was mixed with agarose and loaded onto the slide, which was then refrigerated for 5 min. Solution A was applied for 7 min, followed by Solution B for 15 min. After draining excess solutions, the slides were washed with distilled water, dehydrated through ethanol series (70%, 90%, 100%), and stained with Solutions C, D, and E in sequence for 2, 3, and 2 min respectively. The slides were examined under a light microscope at 400 × magnification, and DNA fragmentation was assessed by the presence of a halo around the sperm (Fig. 4). First, DNA fragmentation was performed to examine fresh sperm before freezing, and then DNA fragmentation analysis was performed at time intervals of 1, 3, 7, 15, 21, and 30 days after thawing frozen sperm for the best extenders. Observations were made by an expert in the field. Individual pictures of each sperm were taken using AmScope Version x.64, 3.7.10246.20171109 (https://amscope.com/pages/software-downloads. Irvine, CA, 92602, USA)88.
Sperm with DNA fragmentation: Sperm with DNA fragmentation shows either no halo around the sperm head or a small halo. Small halo: The width of the halo is less than or equal to one-third of the smallest diameter of the sperm head89. Morphologically normal sperm without DNA fragmentation: These sperm exhibit a large halo or a medium halo around the sperm head. Large Halo: The halo around the sperm head is greater than or equal to one-third of the smallest diameter of the sperm head. Medium halo: The size of the halo is intermediate, between that of a large halo and a small halo89.
Statistical analysis
Data normality was tested using the Shapiro–Wilk and Kolmogorov–Smirnov test25,52. Nonparametric data (fresh sperm motility rate) were analyzed using the Kruskal–Wallis test followed by the Mann–Whitney U comparisons test and Univariate Analysis of Variance. Parametric data (sperm motility post-thawing, sperm morphology and DNA fragmentation) were analyzed using one-way ANOVA with Tukey’s post hoc test44,90. Data are presented as mean ± SD, with P ≤ 0.05 considered statistically significant. A computer program (SPSS for Windows, Version 27.0.1, SPSS Inc., IL, USA) was used for statistical analyses. Excel® version 2021 (Microsoft Corporation, Washington, USA) was used for graph design.
Data availability
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
References
Clulow, J., Trudeau, V. L. & Kouba, A. J. Amphibian declines in the twenty-first century: why we need assisted reproductive technologies. In Reproductive Sciences in Animal Conservation: Progress and Prospects (eds Holt, W. V. et al.) 275–316 (Springer New York, 2014). https://doi.org/10.1007/978-1-4939-0820-2_12.
Browne, R. K. et al. Reptile and amphibian conservation through gene banking and other reproduction technologies. Russ J Herpetol 18, 165e74 (2011).
Browne, R. K. & Figiel, C. Cryopreservation in amphibians. Cryopreserv. Aquat. species 8, 450 (2010).
Browne, R. K. et al. The sixth mass extinction and amphibian species sustainability through reproduction and advanced biotechnologies, biobanking of germplasm and somatic cells, and conservation breeding programs (RBCs). Animals 14, 3395 (2024).
Browne, R. K. et al. Sperm collection and storage for the sustainable management of amphibian biodiversity. Theriogenology 133, 187–200 (2019).
Lampert, S. S. et al. Sperm cryopreservation as a tool for amphibian conservation: Production of F2 generation offspring from cryo-produced F1 progeny. Animals 13, 53 (2022).
Anastas, Z. M. et al. The increasing role of short-term sperm storage and cryopreservation in conserving threatened amphibian species. Animals 13, 2094 (2023).
Browne, R. K., Clulow, J., Mahony, M. & Clark, A. Successful recovery of motility and fertility of cryopreserved cane toad (Bufo marinus) sperm. Cryobiology 37, 339–345 (1998).
Della Togna, G. et al. Evaluating amphibian biobanking and reproduction for captive breeding programs according to the Amphibian Conservation Action Plan objectives. Theriogenology 150, 412–431 (2020).
Figiel, C. R. Cryopreservation of sperm from the axolotl Ambystoma mexicanum: Implications for conservation. Herpetol. Conserv. Biol. 8, 748 (2013).
Guy, E. L. et al. Sperm collection and cryopreservation for threatened newt species. Cryobiology 94, 80–88 (2020).
Poo, S. & Hinkson, K. M. Amphibian conservation using assisted reproductive technologies: Cryopreserved sperm affects offspring morphology, but not behavior, in a toad. Glob. Ecol. Conserv. 21, e00809 (2020).
Coxe, N. et al. Establishment of a practical sperm cryopreservation pathway for the axolotl (Ambystoma mexicanum): A community-level approach to germplasm repository development. Animals 14, 206 (2024).
Browne, R. K., Davis, J., Clulow, J. & Pomering, M. Storage of cane toad (Bufo marinus) sperm for 6 days at 0 C with subsequent cryopreservation. Reprod. Fertil. Dev. 14, 267–273 (2002).
Shishova, N. R., Uteshev, V. K., Kaurova, S. A., Browne, R. K. & Gakhova, E. N. Cryopreservation of hormonally induced sperm for the conservation of threatened amphibians with Rana temporaria as a model research species. Theriogenology 75, 220–232 (2011).
Burger, I. J., Lampert, S. S., Kouba, C. K., Morin, D. J. & Kouba, A. J. Development of an amphibian sperm biobanking protocol for genetic management and population sustainability. Conserv. Physiol. 10, coac032 (2022).
Graham, K. M. Conserving the Mississippi Gopher Frog (Lithobates Sevosa) through the Use of Assisted Reproductive Technologies. (Mississippi State University, 2015).
Browne, R. K. et al. Sperm motility of externally fertilizing fish and amphibians. Theriogenology 83, 1–13 (2015).
Chen, D. M., Moore, M. G., Willis, E. L., Kouba, A. J. & Kouba, C. K. The impact of time and environmental factors on the mitochondrial vesicle and subsequent motility of amphibian sperm. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 268, 111191 (2022).
Byrne, P. G., Dunne, C., Munn, A. J. & Silla, A. J. Environmental osmolality influences sperm motility activation in an anuran amphibian. J. Evol. Biol. 28, 521–534 (2015).
Hobbs, R. J. et al. Cryopreservation cooling rate impacts post-thaw sperm motility and survival in Litoria booroolongensis. Animals 13, 3014 (2023).
Garde, J. J. et al. Sperm cryopreservation in three species of endangered gazelles (Gazella cuvieri, G. dama mhorr, and G. dorcas neglecta). Biol. Reprod. 69, 602–611 (2003).
Cabrita, E., Horváth, Á., Marinović, Z. & Asturiano, J. F. Technologies and strategies for ex situ conservation of aquatic organisms: the role of cryopreservation in long-term management. In Cellular and Molecular Approaches in Fish Biology 1–48 (Elsevier, 2022).
Marcec, R. M. Development of Assisted Reproductive Technologies for Endangered North American Salamanders (Mississippi State University, 2016).
Lemma, A. Effect of cryopreservation on sperm quality and fertility. Artif. Insemin. farm Anim. 12, 191–216 (2011).
Sharma, R., Kattoor, A. J., Ghulmiyyah, J. & Agarwal, A. Effect of sperm storage and selection techniques on sperm parameters. Syst. Biol. Reprod. Med. 61, 1–12 (2015).
Costanzo, J. P., Reynolds, A. M., do Amaral, M. C. F., Rosendale, A. J. & Lee, R. E. Jr. Cryoprotectants and extreme freeze tolerance in a subarctic population of the wood frog. PLoS One 10, e0117234 (2015).
Mansour, N., Lahnsteiner, F. & Patzner, R. A. Optimization of the cryopreservation of African clawed frog (Xenopus laevis) sperm. Theriogenology 72, 1221–1228 (2009).
Arregui, L., Koch, J. C. & Tiersch, T. R. Transitioning from a research protocol to a scalable applied pathway for Xenopus laevis sperm cryopreservation at a national stock center: The effect of cryoprotectants. J. Exp. Zool. Part B Mol. Dev. Evol. 342, 291–300 (2024).
Kolyada, M. N., Osipova, V. P. & Berberova, N. T. Use of cryoprotectors and antioxidants in sturgeon semen cryopreservation. Cryobiology 111, 30–39 (2023).
Chelewani, A. P., Takahashi, E., Nishimura, T. & Fujimoto, T. Optimizing the post-thaw quality of cryopreserved masu salmon (Oncorhynchus masou) sperm: Evaluating the effects of antioxidant-supplemented extender. Aquaculture 593, 741332 (2024).
Dessouki, S. M. et al. Effect of Exogenous curcumin on post-thaw sperm parameters, antioxidant status, the expression of antioxidants and antifreeze-related genes in rabbits. Egypt. J. Vet. Sci. 1–12 (2024)
Nurlatifah, A. et al. Comparison enzymatic and non-enzymatic antioxidant in sperm quality of Garut ram chilled semen to enhance rural livestock commodity. Adv. Anim. Vet. Sci 11, 1918–1926 (2023).
Shishova, N. R., Uteshev, V. K., Kaurova, S. A., Browne, R. K. & Gakhova, E. N. Mansour N, Lahnsteiner F, Patzner R. Optimization of the cryopreservation of African clawed frog (Xenopus laevis) sperm. Theriogenology 72:1221–28 (2009). Theriogenology 75, 220–232 (2011).
Langhorne, C. J. et al. 026 Cryoconservation: Successful sperm cryopreservation and develop-mental outcomes using endangered North American amphibians. Cryobiology 67, 405 (2013).
Kopeika, J., Thornhill, A. & Khalaf, Y. The effect of cryopreservation on the genome of gametes and embryos: Principles of cryobiology and critical appraisal of the evidence. Hum. Reprod. Update 21, 209–227 (2015).
Morrow, S. et al. Effects of freezing and activation on membrane quality and DNA damage in Xenopus tropicalis and Xenopus laevis spermatozoa. Reprod. Fertil. Dev. 29, 1556–1566 (2017).
Pollock, K., Gosálvez, J., López-Fernández, C. & Johnston, S. D. Amphibian sperm chromatin structure and function and its relevance to sperm preservation. J. Herpetol. 52, 486–491 (2018).
Arregui, L., Bóveda, P., Gosálvez, J. & Kouba, A. J. Effect of seasonality on hormonally induced sperm in Epidalea calamita (Amphibia, Anura, Bufonidae) and its refrigerated and cryopreservated storage. Aquaculture 529, 735677 (2020).
Gillis, A. B. et al. Short-term storage of tiger salamander (Ambystoma tigrinum) spermatozoa: The effect of collection type, temperature and time. PLoS One 16, e0245047 (2021).
Browne, R. K., Clulow, J. & Mahony, M. The short-term storage and cryopreservation of spermatozoa from hylid and myobatrachid frogs. CryoLetters 23, 129–136 (2002).
Germano, J. M., Arregui, L. & Kouba, A. J. Effects of aeration and antibiotics on short-term storage of Fowler’s toad (Bufo fowleri) sperm. Aquaculture 396, 20–24 (2013).
Chen, D. et al. Oral administration of GnRH via a cricket vehicle stimulates spermiation in tiger salamanders (Ambystoma tigrinum). bioRxiv 2008–2023 (2023).
Chen, D. M. et al. Comparing novel sperm extenders for the internally-fertilizing tiger salamander (Ambystoma tigrinum). Front. Amphib. Reptil. Sci. 1, 1320803 (2024).
Sharifi, M., Papenfuss, T., Rastegar-Pouyani, N., Anderson, S. & Kuzmin, S. Neurergus kaiseri. IUCN Red List Threat. species. Version (2009).
Niknaddaf, Z., Hemami, M.-R., Pourmanafi, S. & Ahmadi, M. An integrative climate and land cover change detection unveils extensive range contraction in mountain newts. Glob. Ecol. Conserv. 48, e02739 (2023).
Sharifi, M. & Vaissi, S. Captive breeding and trial reintroduction of the endangered Yellow-spotted Mountain Newt Neurergus microspilotus in western Iran. Endanger. Species Res. 23, 159–166 (2014).
Burger, I. J. The ART of amphibian conservation: linking in-situ and ex-situ populations of endangered species through genome banking. Conservat. Sci. Prac. https://doi.org/10.1111/csp2.525 (2021).
Clulow, J. & Clulow, S. Cryopreservation and other assisted reproductive technologies for the conservation of threatened amphibians and reptiles: Bringing the ARTs up to speed. Reproduction, Fertility and Development 28(8), 1116–1132 (2016).
Mansour, N., Lahnsteiner, F. & Patzner, R. A. Collection of gametes from live axolotl, Ambystoma mexicanum, and standardization of in vitro fertilization. Theriogenology 75, 354–361 (2011).
McGinnity, D., Reinsch, S. D., Schwartz, H., Trudeau, V. & Browne, R. K. Semen and oocyte collection, sperm cryopreservation and IVF with the threatened North American giant salamander Cryptobranchus alleganiensis. Reprod. Fertil. Dev. 34, 470–477 (2021).
Arregui, L. et al. Fertilization potential of cold-stored Fowler’s toad (Anaxyrus fowleri) spermatozoa: temporal changes in sperm motility based on temperature and osmolality. Reprod. Fertil. Dev. 34, 461–469 (2021).
Silla, A. J., Keogh, L. M. & Byrne, P. G. Sperm motility activation in the critically endangered booroolong frog: The effect of medium osmolality and phosphodiesterase inhibitors. Reprod. Fertil. Dev. 29, 2277–2283 (2017).
Edwards, D. L., Mahony, M. J. & Clulow, J. Effect of sperm concentration, medium osmolality and oocyte storage on artificial fertilisation success in a myobatrachid frog (Limnodynastes tasmaniensis). Reprod. Fertil. Dev. 16, 347–354 (2004).
Sato, T. et al. Differences of extracellular cues and Ca2+ permeable channels in the signaling pathways for inducing amphibian sperm motility. Zoolog. Sci. 38, 343–351 (2021).
Upton, R., Clulow, S., Colyvas, K., Mahony, M. & Clulow, J. Paradigm shift in frog sperm cryopreservation: reduced role for non-penetrating cryoprotectants. Reproduction 165, 583–592 (2023).
Upton, R. et al. Generation of reproductively mature offspring from the endangered green and golden bell frog Litoria aurea using cryopreserved spermatozoa. Reprod. Fertil. Dev. 33, 562–572 (2021).
Sieme, H., Oldenhof, H. & Wolkers, W. F. Mode of action of cryoprotectants for sperm preservation. Anim. Reprod. Sci. 169, 2–5 (2016).
Best, B. P. Cryoprotectant toxicity: Facts, issues, and questions. Rejuvenation Res. 18, 422–436 (2015).
Pingale, P. L. et al. Toxicity and toxicodynamics of cryoprotectant used in pharmaceutical products. In Public Health and Toxicology Issues Drug Research 493–521 (Elsevier, 2024).
Kumar, A., Prasad, J. K., Srivastava, N. & Ghosh, S. K. Strategies to minimize various stress-related freeze–thaw damages during conventional cryopreservation of mammalian spermatozoa. Biopreserv. Biobank. 17(6), 603–612 (2019).
Murray, A., Kilbride, P. & Gibson, M. I. Trehalose in cryopreservation. Applications, mechanisms and intracellular delivery opportunities. RSC Med. Chem. 15, 2980–2995 (2024).
Sudagar, M., Keivanloo, S. & Hajibeglou, A. Effect of different permeable and non-permeable cryoprotectants on the hatching rate of rainbow trout (Oncorhynchus mykiss) embryos. Aquac. Int. 26, 75–84 (2018).
Majtnerová, P. & Roušar, T. An overview of apoptosis assays detecting DNA fragmentation. Mol. Biol. Rep. 45, 1469–1478 (2018).
Ozougwu, J. C. The role of reactive oxygen species and antioxidants in oxidative stress. Int. J. Res. 1, 1–8 (2016).
Aitken, R. J. & Koppers, A. J. Apoptosis and DNA damage in human spermatozoa. Asian J. Androl. 13, 36 (2010).
Bui, A. D., Sharma, R., Henkel, R. & Agarwal, A. Reactive oxygen species impact on sperm DNA and its role in male infertility. Andrologia 50, e13012 (2018).
Benchaib, M. et al. Sperm DNA fragmentation decreases the pregnancy rate in an assisted reproductive technique. Hum. Reprod. 18, 1023–1028 (2003).
Gosálvez, J., Holt, W. V. & Johnston, S. D. Sperm DNA fragmentation and its role in wildlife conservation. In Reproductive Sciences in Animal Conservation: Progress and Prospects (eds Holt, W. V. et al.) 357–384 (Springer New York, 2014).
Newman, H., Catt, S., Vining, B., Vollenhoven, B. & Horta, F. DNA repair and response to sperm DNA damage in oocytes and embryos, and the potential consequences in ART: A systematic review. Mol. Hum. Reprod. 28, gaa071 (2022).
Pollock, K. Amphibian sperm DNA fragmentation. (2016).
Paoli, D., Pelloni, M., Lenzi, A. & Lombardo, F. Cryopreservation of sperm: Effects on chromatin and strategies to prevent them. In Genetic Damage in Human Spermatozoa (eds Baldi, E. & Muratori, M.) 149–167 (Springer, Cham, 2019).
Figueroa Villalobos, E., Lee Estévez, M., Valdebenito, I., Farías, J. G. & Romero Ormazábal, J. Potential biomarkers of DNA quality in cryopreserved fish sperm: impact on gene expression and embryonic development. Rev. Aquacult. 12(1), 382–391 (2020).
Dutta, S., Henkel, R. & Agarwal, A. Comparative analysis of tests used to assess sperm chromatin integrity and DNA fragmentation. Andrologia 53, e13718 (2021).
Dolník, M., Mudroňová, D., Pošivák, J., Lazar, G. & Mudroň, P. Flow cytometry in assessment of sperm integrity and functionality–a review. Acta Vet. Brno 88, 169–175 (2019).
Hossain, M. S. et al. Flow cytometry for the assessment of animal sperm integrity and functionality: State of the art. Asian J. Androl. 13, 406 (2011).
Aghaeepour, N. et al. Critical assessment of automated flow cytometry data analysis techniques. Nat. Methods 10, 228–238 (2013).
Comizzoli, P. & Holt, W. V. Recent progress in spermatology contributing to the knowledge and conservation of rare and endangered species. Annu. Rev. Anim. Biosci. 10, 469–490 (2022).
Rivera-Pacheco, J. et al. Ambystoma mexicanum sperm cryopreservation (Shaw & Nodder, 1798). Abanico Vet. https://doi.org/10.21929/abavet2021.12 (2021).
Sirimanapong, W. et al. A comparison of 3 anesthetic agents (ms-222, benzocaine, and clove oil) in east Asian bullfrog (Hoplobatrachus rugulosus). (2020).
Kratochvílová, I. et al. Theoretical and experimental study of the antifreeze protein AFP752, trehalose and dimethyl sulfoxide cryoprotection mechanism: Correlation with cryopreserved cell viability. RSC Adv. 7, 352–360 (2017).
Jin, B. et al. Rapid movement of water and cryoprotectants in pig expanded blastocysts via channel processes: Its relevance to their higher tolerance to cryopreservation. Biol. Reprod. 89, 81–87 (2013).
Abinawanto, A., Yimastria, S. & Pertiwi, P. Sperm analysis of Lukas fish (Puntius bramoides): Motility, viability and abnormalities. In AIP Conference Proceedings vol. 2023 (AIP Publishing, 2018).
Taheri-Khas, Z., Gharzi, A., Vaissi, S., Heshmatzad, P. & Kalhori, Z. Hormone-driven temperature optimization for elevated reproduction in goldfish (Carassius auratus) under laboratory conditions. Animals 14, 2701 (2024).
Stuart, C. C., Vaughan, J. L., Kershaw, C. M., De Graaf, S. P. & Bathgate, R. Effect of diluent type, cryoprotectant concentration, storage method and freeze/thaw rates on the post-thaw quality and fertility of cryopreserved alpaca spermatozoa. Sci. Rep. 9, 12826 (2019).
Turani, B., Aliko, V. & Shkembi, E. Characterization of Albanian water frog, Pelophylax shqipericus, sperm traits and morphology, by using phase contrast microscopy. Microsc. Res. Tech. 82, 1802–1809 (2019).
Keskin, İ et al. Detailed morphological analysis of axolotl sperm. Korean J. Vet. Res. 61, e25 (2021).
García-Vargas, D., Juárez-Rojas, L., Rojas Maya, S. & Retana-Márquez, S. Prenatal stress decreases sperm quality, mature follicles and fertility in rats. Syst. Biol. Reprod. Med. 65, 223–235 (2019).
Bajaj, B. & Kapoor, G. Sperm DNA fragmentation and reproductive outcomes. Fertil. Sci. Res. 9, 10–15 (2022).
Della Togna, G. et al. Influence of extracellular environment on the motility and structural properties of spermatozoa collected from hormonally stimulated Panamanian Golden Frog (Atelopus zeteki). Theriogenology 108, 153–160 (2018).
Funding
This research received no external funding.
Author information
Authors and Affiliations
Contributions
Conceptualization, Z.T.K. and S.V.; Investigation, methodology, Z.T.K., S.V., A.G., P.H.Z., Z.K and; formal analysis, supervision, writing-original draft, Z.T.K., S.V. and; Writing—review & editing, S.V., A, G., P.H.Z., Z.K and Z.T.K. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
All methods in this study were carried out in accordance with relevant guidelines and regulations.
Informed consent
The informed consent was obtained from all subjects and/or their legal guardian(s).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Taheri-Khas, Z., Gharzi, A., Vaissi, S. et al. Advanced sperm preservation techniques in yellow spotted mountain newts Neurergus derjugini enhance genetic management and conservation efforts. Sci Rep 15, 9334 (2025). https://doi.org/10.1038/s41598-025-93284-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-93284-y
