
Abstract
Actinidia species are fruit trees with various functions, such as providing edible fruit, serving as ornamental plants, and having medicinal benefits. However, the taxonomy of Actinidia species is controversial due to widespread hybridization, the history of divergence and polyploid speciation among Actinidia species also remains unclear. In this study, we conducted comparative analyses of the chloroplast genomes and ploidy among multiple Actinidia species. The genes clpP, infA, ndhD, ndhK, and rpl20 were absent from these chloroplast genomes. The ycf2 and rpl20 genes in the Actinidia species were under positive selection. Several regions (rps16–trnQ-UUG, trnS-GCU–trnR-UCU, ndhC–trnV-UAC, rbcL–accD, rps12–psbB, trnN-GUU–ndhF, ycf1–trnN-GUU, and trnH-GUG–psbA) and genes (ycf1, ycf2, accD, rpl20) exhibited high variability, which could potentially serve as molecular markers in species delineation and other phylogenetic studies. Through divergence time estimation, the Actinidia genus originated 23 million years ago (Ma), and experienced a tetraploidization event in ~ 20 Ma. Subsequently, Actinidia has undergone extensive diploidization. Our findings will provide valuable information in species identification, breeding programs, and conservation efforts for Actinidia species.
Similar content being viewed by others


Introduction
Actinidia species are among the most significant fruit trees globally, prized for their distinctive flavor and high vitamin C content1, as well as their ornamental and medicinal value2. The genus comprises over 50 species, with the majority being endemic to China3. The rich genetic resources in China offer a unique opportunity for studying the evolution of Actinidia and enhancing breeding programs4. However, only A. chinensis and A. deliciosa are widely cultivated for commercial purposes, while A. arguta and A. kolomikta are grown on a more limited scale5.
Actinidia species are assumed to exhibit extensive natural hybridization, sharing a variety of morphological traits that facilitate this process6. The genus also displays considerable chromosomal ploidy variation, suggesting a history of paleopolyploidization7. While many Actinidia species exist in the wild, their distribution is largely confined to mountainous regions due to human activities and deforestation3. To facilitate the conservation, development, and improvement of wild Actinidia species, further research is needed to clarify species taxonomy, investigate genetic diversity, and explore the role of chromosomal ploidy in domesticated varieties.
The taxonomy of the Actinidia genus has been the subject of ongoing debate, largely due to the widespread occurrence of natural hybridization8. Generally, based on morphological characteristics, the genus has been divided into four sections: Sect. Leiocarpae, Sect. Maculatae, Sect. Stellatae, and Sect. Strigosae9. However, molecular systematics have suggested that the sections Maculatae, Stellatae, and Strigosae may not represent natural groups, proposing instead that the genus should be divided into Sect. Leiocarpae and Sect. Maculatae3,10,11. Nevertheless, these molecular phylogenetic trees still exhibit low support values, indicating the need for further investigation into the phylogenetic relationships within the Actinidia genus. On the other hand, most Actinidia species are diploid, and several are polyploid, which could contribute to the functional diversity of Actinidia genes7. Studying ploidy variation can provide valuable insights into the evolutionary history of Actinidia species.
The chloroplast (cp) is the organelle responsible for photosynthesis in plant cells and contains relatively independent genetic information12. Comparative analysis of cp genomes from different plant species helps to elucidate genetic variation and the evolutionary processes underlying plant diversity13. Because of its uniparental inheritance and abundant informative loci, the complete cp genome is often used in phylogenetic and taxonomic studies14. Moreover, cp genomes have been employed to trace the maternal and paternal ancestry of polyploid crops, estimate species divergence times, and assess the extent of domestication in wild species15. Therefore, the complete cp genome is an ideal resource for studying the phylogenetic relationships and divergence times of polyploid Actinidia species.
To enhance the availability of complete cp genome data for Actinidia, we sequenced the full cp genome of A. lijiangensis, a species closely related to A. chinensis10, suggesting its significant potential for future exploitation. In this study, we conducted a comparative analysis of the cp genome of A. lijiangensis with other Actinidia species from the NCBI database and assessed the ploidy levels of these species. Additionally, we explored the phylogenetic relationships and divergence times within the Actinidia genus based on the complete cp genomes. Our study aimed to: (a) identify divergences and similarities in the cp genomes of diverse Actinidia species; (b) elucidate the phylogenetic relationships within the Actinidia genus based on cp genomes; and (c) clarify the divergence times of Actinidia species with different ploidy levels, providing valuable insights for future breeding efforts. The comprehensive cp genome data generated in this study will serve as a valuable resource for the classification, molecular breeding, and conservation of Actinidia species.
Results
The characteristics of different Actinidia cp genomes
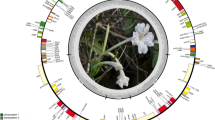
The complete cp genome of A. lijiangensis showed a typical quadripartite structure (Fig. 1). The LSC and SSC regions are located between the two IR regions, namely IRa and IRb. The A. lijiangensis cp genome had a total length of 156,679 bp, which was similar to that of other Actinidia species (155,957–157,659 bp; Table 1).

The circular map of the Actinidia lijiangensis chloroplast genome. Genes drawn inside and outside of the outer ring are transcribed in clockwise and counterclockwise directions, respectively. Genes belonging to different functional groups are color-coded. The GC and AT contents are illustrated with dark green and light green at the inner circles, respectively.
Next, we compared the structural features among different Actinidia cp genomes. The length of the cp genome sequence increases with chromosome ploidy for the same species (A. chinensis and A. deliciosa). The LSC region varied in size across the Actinidia species, ranging from 87,865 bp in A. chinensis (2x) to 90,075 bp in A. cylindrica var. reticulata, while the size of the SSC region ranged from 20,290 bp in A. callosa var. strigillosa to 21,555 bp in A. latifolia. The size of the IR regions varied from 22,776 bp in A. hubeiensis to 24,451 bp in A. deliciosa (6x). The average GC content across all Actinidia species cp genomes was 37.2%, with relatively little variation among species. The GC content of the IR regions (43.0%) was higher compared to the LSC (35.5%) and SSC (31.1%) regions (Table 1).
Most Actinidia species contain a total of 130 genes in their cp genomes, including 83 protein-coding genes, 39 tRNA genes, and 8 rRNA genes (Table 1). The cp genomes of Actinidia species exhibited gene loss (Table 2). Specifically, the infA gene was absent in A. styracifolia and A. zhejiangensis, the ndhD gene was not found in A. callosa var. strigillosa and A. cylindrica, the ndhK gene was lost in A. rubus, and the psbA gene was missing in A. callosa var. henryi and A. callosa var. strigillosa. Additionally, the rpl20 gene was lost in A. zhejiangensis. Notably, Actinidia species frequently lost the clpP gene as well as one copy of the ycf1 genes.
The contraction and expansion of IR regions
The variation in the contraction and expansion of IR regions was analyzed among the cp genomes of different Actinidia species. Many genes, including rpl23, trnH, trnN, ycf1, ndhF, rps15, trnI, trnK, and psbA, are located at the junctions LSC/IRb (JLB), IRb/SSC (JSB), SSC/IRa (JSA), and IRa/LSC (JLA) (Fig. 2). Based on the deletion of the ycf1 copy gene, Actinidia species can roughly be classified into three groups. The first group of Actinidia species lost their ycf1 copy gene at the JSA junction (including A. lijiangensis, A. arguta var. giraldii, A. callosa var. henryi, A. callosa var. strigillosa, A. eriantha, A. kolomikta, A. lanceolata, and A. zhejiangensis). Then, the second group of Actinidia species lost their ycf1 copy gene at the JSB junction (including A. chinensis (2x), A. chinensis (4x), A. cylindrica, A. deliciosa (4x), A. deliciosa (6x), A. fulvicoma, A. hemsleyana, A. hubeiensis, A. latifolia, A. macrosperma, A. melanandra, A. polygama, A. rubus, A. setosa, A. tetramera, A. valvata, and A. venosa). Thirdly, the last group of Actinidia species (A. arguta, A. cylindrica var. reticulata, A. indochinensis, and A. rufa) showed no loss of the ycf1 gene. The ycf1 gene copies in A. styracifolia were absent. In addition, a trnH copy gene was lost in A. callosa var. henryi and A. venosa.

The contraction and expansion of the inverted repeat or single copy (IR/SC) junctions. We compared the junctions among the large single copy (LSC), short single copy (SSC), and inverted repeats (IR) regions in the chloroplast genomes of Actinidia species. JLB (IRb/LSC), JSB (IRb/SSC), JSA (SSC/IRa) and JLA (IRa/LSC) denote the junction sites between each corresponding region in the genome.
Selective pressure and codon usage bias analysis
To analyze the molecular evolution, the selection pressure of protein-coding genes from Actinidia species was assessed based on the Ka/Ks values (Fig. 3). The Actinidia cp genomes showed the diversity of the Ka/Ks values in the range of 0.00 to 2.35 (Table S1). The Ka/Ks values of most genes are less than 1, suggesting the purifying selection. The ccsA, ycf1, rps18, rps4, rps2, rps19, ycf3, atpF, matK, rbcL, ndhI, rpoA, rpoC2, rpl20, ycf2, accD, rps3 genes were clustered together because of Ka/Ks values greater than 0.50 in most Actinidia species. The greatest Ka/Ks value, 2.35, was found in the accD gene of A. styracifolia and A. indochinensis. Notably, the Ka/Ks values of the rpl20, ycf2, accD, and rps3 genes were greater than 1 in most Actinidia species, showing positive selection.

The selective pressure analysis of Actinidia species. Outside of the circle are protein-coding genes of Actinidia species. The values in the grid indicate Ka/Ks values. The color closer to darker green represents the higher Ka/Ks ratio.
The cp genomes of different Actinidia species showed similar codon preferences (Fig. 4). The 64 codons were divided into two groups based on their RSCU values. The 27 codons with RSCU values larger than 1.00 fell into one group, and the remaining codons fell into another. There were two codons (UUG and AGA) with RSCU values above 1.85 (Table S2). RSCU values of codons ending in U or A tended to be above 1.00, while those of codons ending in G or C typically were below 1.00. Notably, the codon usage preferences of A. chinensis (2x) and A. chinensis (4x), as well as A. deliciosa (4x) and A. deliciosa (6x), exhibited a high degree of similarity.

The heat map for the RSCU analysis of Actinidia species. We clustered the RSCU values of 64 codons. The color closer to red indicates the higher RSCU value. Each column represents the various codons.
Divergence analysis of cp genomes in Actinidia species
Multiple alignments of diverse Actinidia cp genomes were conducted using mVISTA, with the annotated A. tetramera genome sequence as the reference (Fig. 5). The LSC and SSC regions were found to be more divergent than the two IR regions. In addition, the noncoding regions showed a greater degree of divergence than the coding regions. The genes ycf1, ycf2, accD, and rpl20 were the main sites of divergence in the coding regions from these cp genomes. We also identified some high divergence in intergenic regions, including rps16–trnQ-UUG, trnS-GCU–trnR-UCU, ndhC–trnV-UAC, rbcL–accD, rps12–psbB, trnN-GUU–ndhF, ycf1–trnN-GUU, and trnH-GUG–psbA.

Alignments of the complete chloroplast genomes of Actinidia species. Using A. tetramera as the reference sequence. The identity percentages (50%–100%) are displayed on the Y-axis. Each arrow indicates the genes and ranscription direction in the reference genome. Colored block represent the different sequence types, purple: exon, blue: untranslated region (UTR), pink: Non-coding sequences (NCS), gray: mRNA .
To examine sequence divergence hotspots, Pi values were estimated through an 800 bp window (Fig. 6). The Pi values of all Actinidia cp genomes ranged from 0 to 0.02993. The average Pi value was 0.00560 (Table S3). The SSC and LSC regions exhibited significantly greater divergence compared to the IR regions, which had notably lower Pi values. Additionally, Pi values for several highly variable regions and genes, including rps16–trnQ-UUG, trnT-UGU–trnL-UAA, petA–psbJ, rps12–psbB, and gene ycf1, were greater than 0.017.

Nucleotide diversity (Pi) analysis. X-axis, the base point along the sequences,Y-axis,the Pi value (window length: 800 bp, step size: 200 bp).
Phylogenetic analysis and divergence time estimation
Phylogenetic trees of Actinidia species were constructed using the Maximum Likelihood (ML) and Bayesian Inference (BI) methods based on the complete cp genomes. Both methods produced trees with the same structure and high support values (Fig. 7). The phylogenetic topology revealed that several species complexes, including the A. chinensis complex, A. rufa complex, A. tetramera complex, A. polygama complex, and A. arguta complex, each formed distinct monophyletic groups. Notably, the commercial cultivars A. kolomikta and A. arguta, which belong to the A. arguta complex, were found at the earliest branching point, while A. chinensis and A. deliciosa from the A. chinensis complex are located at the most recent branching point within the A. chinensis complex. However, the phylogenetic relationships of other Actinidia complexes, including A. lanceolata, A. venosa, and others, remain unclear (Fig. 7).

Divergence time estimation based on chloroplast genome sequences. Phylogenetic trees were constructed using the maximum likelihood (ML) and Bayesian inference (BI) approaches employing the complete chloroplast genomes of Actinidia species. Clematoclethra scandens and Saurauia napaulensis were employed as outgroups. The blue bars reflect the 95% greatest posterior density interval for each node age. The different colored stars on the nodes indicate the support values. The background color for the branch represents the species complex. The first panel on the right side of the tree means the chromosome ploidy, the second panel means the different sections in Actinidia genus. Plio: Pliocene; Quat: Quaternary.
The divergence times of the Actinidia genus were estimated using the phylogenetic tree described above. The results indicate that the crown group of the Actinidia genus emerged around 23.57 Ma, during the Miocene epoch (Fig. 7). The common ancestor of the A. polygama complex is estimated to have appeared around 19.91 Ma, with the notable feature that all members of this complex are tetraploid. The common ancestor of the A. chinensis complex began to diverge at around 4.34 Ma. Of particular note are the earliest diverging diploid and tetraploid Actinidia species on the phylogeny: A. tetramera (about 19.90 Ma) and A. macrosperma (about 19.91 Ma), respectively.
Discussion
Divergences and similarities in diverse Actinidia cp genomes
In this study, we characterized the complete cp genome of A. lijiangensis and compared it with other Actinidia species (Figs. 1, 2, 3 and 4). The length of the cp genomes varied across species (Table 1), likely influenced by factors such as changes in chromosome ploidy, gene loss, divergence in intergenic regions, and the contraction or expansion of IR regions16. Despite these length differences, the cp genomes of Actinidia species showed high similarity in terms of gene composition, gene order, and GC content (Table 1), likely reflecting the generally conservative evolution of cp genomes in this genus17.
Notably, the loss of certain genes in the cp genomes of Actinidia species did not appear to adversely affect their functionality (Table 2). For example, the clpP gene, absent in all Actinidia species, may have been transferred to the nuclear genome during chloroplast evolution17. Similarly, the infA gene is missing in A. styracifolia and A. zhejiangensis, a deletion also observed in the cp genomes of other fruit trees such as Citrus species13. Some Actinidia species also lack certain NDH (NADH dehydrogenase-like) genes, which may not be essential under favorable environmental conditions18. Additionally, we identified a deletion of the rpl20 gene in A. zhejiangensis, which may have been replaced by a nuclear-encoded, mitochondria-targeted rpl20 gene with a similar structure to the cp rpl20 gene19.
We also observed structural divergences and sequence mutations in the cp genomes of Actinidia species. Specifically, we found IR region divergence near the boundary of the SSC region (Fig. 2), particularly at the JSA and JSB junctions. This may be due to a contraction of the IR regions and an expansion of the SSC region13. Furthermore, the ycf2 and rpl20 genes were found to be under positive selection (Fig. 3), suggesting that these genes are rapidly evolving and play an important role in the development of Actinidia species. Our study also highlighted the conservation of codon preference in Actinidia species with varying ploidy levels, such as A. chinensis and A. deliciosa (Fig. 4), indicating the conservation of gene expression and evolution within the genus20. Overall, our results demonstrate the diversity of cp genome structures in Actinidia.
Determination of highly variable regions
The highly variable regions of the cp genome are valuable for generating potential phylogenetic markers. Previous studies have utilized molecular fragments such as matK, psbC–trnS, rbcL, and trnL–trnF for the phylogenetic analysis of Actinidia species21. In this study, we explored additional highly variable intergenic regions of Actinidia species, based on all available complete cp genomes, including rps16–trnQ-UUG, trnS-GCU–trnR-UCU, ndhC–trnV-UAC, rbcL–accD, rps12–psbB, trnN-GUU–ndhF, ycf1–trnN-GUU, and trnH-GUG–psbA (Fig. 5). Of these, the rps16–trnQ-UUG and ndhC–trnV-UAC regions have been previously used in Actinidia phylogenetic studies8. We also found significant variations in the ycf1, ycf2, accD, and rpl20 genes. The estimation of Pi values indicated notable nucleotide diversity in some of these regions, further highlighting their potential for use as phylogenetic markers (Fig. 6). In summary, our study identifies additional highly variable loci in Actinidia cp genomes, expanding the pool of potential phylogenetic markers. However, further research is needed to determine the utility of these regions for phylogenetic and species identification purposes in Actinidia.
Phylogenetic relationships and divergence history of Actinidia species
This study aimed to investigate the phylogenetic relationships within the Actinidia genus using cp genomes. Previous studies have indicated that Actinidia species exhibit a distinct patrilineal mode of cpDNA inheritance, accounting for approximately 75% of both inter- and intraspecific crosses22, which provides a substantial number of informative sites for robust phylogenetic analysis23. Therefore, chloroplast genome-based phylogenetic studies theoretically offer insights into the clustering relationships among Actinidia species. However, our well-supported phylogenetic results, based on cp genomes, are still largely consistent with topologies constructed using multiple molecular markers8,11,21. Specifically, the four subgenera within Actinidia did not form monophyletic groups (Fig. 7), further illustrating the complex, divergent nature of the genus and indicating that the infrageneric classification of Actinidia remains unresolved.
In detail, our results revealed that A. kolomikta, a hardy species, is closely related to A. arguta, which results from hybridization between A. arguta and other Actinidia species8. Three species within the A. polygama complex clustered together, suggesting that these species may not be hybrids but have undergone introgression11,21. On the other hand, previous studies have suggested that Sect. Maculatae, Sect. Stellatae, and Sect. Strigosae (Fig. 7) should be merged into Sect. Maculatae8,10,11. This complexity in taxonomy and systematics may be attributed to reticulate evolution, which is hypothesized to result from homoploid hybrid speciation24.
Previous studies have shown that differences in chloroplast ploidy can influence the construction of phylogenetic relationships among species. Polyploidization events, which may arise from hybridization between distinct chloroplast lineages or genome duplication, complicate the resolution of evolutionary relationships among species25,26,27,28. In this study, A. deliciosa, with differing ploidy levels (4× or 6×), displayed different clustering results in the phylogenetic tree (Fig. 7), suggesting that chloroplast ploidy plays a role in genetic differentiation.
Divergence time estimation provided insights into the evolutionary history of Actinidia species. Our results indicate that the genus originated approximately 23.57 Ma during the Miocene epoch, coinciding with significant climatic changes, including the expansion of temperate regions and the contraction of tropical regions24,29. The A. arguta complex was the first to diverge from other Actinidia species (23.57 Ma), with its distribution primarily in the temperate regions of northern China3. Subsequently, Actinidia species underwent a diversification event around 21.10 Ma, and tetraploid species emerged at this time, contributing to increased species diversity.
The A. rufa complex may have diverged due to geographic isolation, as A. rufa is primarily distributed in Japan and Taiwan30. The A. chinensis complex later diverged and underwent hybridization events. Around 2.29 Ma, species such as A. chinensis and A. deliciosa began to emerge31. Finally, our study revealed that many species, including A. chinensis, A. deliciosa, and A. eriantha, underwent recent differentiation (< 2 Ma), further emphasizing the rapid diversification of the Actinidia genus (Fig. 7).
Although we explored the phylogeny of the Actinidia genus based on cp genomes and paternal inheritance patterns, our results still fall short of fully elucidating the relationships among the four sections of Actinidia. Based on current mainstream algorithms for tree-building and divergence time estimation, we provide a foundational understanding of the evolutionary history and speciation processes of Actinidia. However, the methodologies and assumptions of phylogenetics are often rooted in the neutral theory. The controversial neutral theory framework, which has been increasingly challenged in recent years32,33,34, may not fully account for the genetic diversity and the complexity of evolutionary processes. Therefore, our conclusions must remain tentative. Future studies will require more comprehensive molecular markers, optimized algorithms, and models to further investigate the phylogenetic relationships, hybridization events, and polyploidization mechanisms within the genus.
Conclusions
In this study, we characterized the genomic features, sequence divergences, and mutation patterns in the chloroplast genomes of 32 Actinidia species. Comparative chloroplast genomics has provided valuable insights into the phylogenetic relationships and polyploid speciation within the Actinidia genus. Our findings reveal a well-supported phylogenetic tree for Actinidia, while also highlighting the non-monophyletic nature of the four sections. These results suggest that further, more comprehensive molecular data will be necessary to refine the taxonomy and elucidate the systematic evolution of the genus. We also inferred the evolutionary history of Actinidia species through divergence time estimation. In conclusion, our study provides a foundational understanding for future horticultural breeding programs and will contribute to the improvement of novel traits in Actinidia species.
Methods
Plant materials, DNA extraction, and sequencing
Leaf samples of Actinidia lijiangensis were collected from Guilin City, Guangxi Zhuang Autonomous Region, China (25.2 N, 110.4 E). In accordance with local laws and regulations, leaves from one representative individual were collected as samples. A voucher specimen (QUST20230817) was identifed and deposited at the Qingdao University of Science and Technology (QUST) by Qian-Hui Lin & Wen-Bo Shi. The fresh leaves of A. lijiangensis that have light-white stellate velutinous were chosen for extracting DNA. A previously reported modified high salt method was utilized to extract cpDNA35. The quality of the extracted DNA sample was evaluated by 1% agarose gel electrophoresis. The cp genomic sequencing of A. lijiangensis was finished following the manufacturer’s protocol on the Illumina Hiseq 4000 platform (Illumina, San Diego, CA, USA) by Novogene (Beijing, China), with 150 bp paired-end raw reads generated.
De novo assembly and annotation of the cp genome
Approximately 3 GB of clean reads of A. lijiangensis were produced by eliminating adapters and low-quality reads using Trimmomatic v0.4036. The quality of clean reads was further evaluated using FastQC v0.11.937. De novo assembly of the A. lijiangensis cp genome was performed by NOVOPlasty v4.3.138. GeSeq v139 and CPGAVAS v240 were used to annotate the cp genome. The two annotation results were compared and manually modified using Sequin v1641, with A. eriantha (GenBank Accession no. NC_034914) as the reference species. In addition, the cp genome of A. styracifolia (GenBank Accession no. MN627226), which was marked as “unverified” in the NCBI database, has been re-annotated using the same method. The assembled cp genome of A. lijiangensis has been deposited in GenBank with accession no. NC_066502.
Actinidia species cp genome characteristics and chromosome ploidy statistics
A statistical analysis for cp genomes of various Actinidia species was conducted using Geneious v9.0.242. The characteristics of cp genomes in Actinidia species include the length of the total sequence, large single-copy (LSC), small single-copy (SSC), and inverted repeat (IR) regions, as well as the total number of genes, protein-coding genes, tRNA and rRNA genes, and the GC content of each region (Table 1). The circular genomic map of A. lijiangensis was created in Chloroplot v0.2.443. Chromosomal ploidy statistics for Actinidia species were primarily derived from previous studies44. Additionally, chromosomal ploidy for A. chinensis (GenBank Accession no. NC_026690 and KP297243), A. deliciosa (GenBank Accession no. NC_026691 and KP297245), and A. arguta (GenBank Accession no. MG744576) was determined based on previous reports45.
Evolutionary analysis of various Actinidia cp genomes
The contraction and expansion analysis of IR regions was conducted using CPJSdraw v1.1 on the JSHYCloud platform46. To analyze selective pressure and codon preference, protein-coding genes from diverse complete cp genomes of Actinidia species were extracted in Geneious v9.0.2. The protein-coding genes among various Actinidia species were aligned using MACSE v247. The ratio of non-synonymous (Ka) to synonymous (Ks) substitution rates was calculated in KaKs_Calculator v248, with A. tetramera (NC_031187) as the reference species. The Ka/Ks values of various Actinidia species were shown using Chiplot (https://www.chiplot.online/). The codon usage frequency and relative synonymous codon usage (RSCU) for various Actinidia species were determined by analyzing their protein-coding genes through MEGA v1149. The RSCU values were visualized in TBtools v1.09850.
Comparative analysis of various Actinidia cp genomes
The cp genome variations among the Actinidia species were examined and displayed by the mVISTA program51, with the LAGAN model. A. tetramera (NC_031187) was used as a reference species. The complete cp genomes of all Actinidia species were aligned in MAFFT v7.30852. Pi values were analyzed using a DNA polymorphism module in DnaSP v6.12.353, with a step size of 200 sites and a window length of 800 sites.
Phylogenetic analysis
Phylogenetic analysis was conducted using all available complete cp genomes of Actinidia species obtained from the NCBI database (accession numbers provided in Table 1). The dataset included tetraploid and hexaploid A. deliciosa (NC_026691 and KP297245) as well as diploid and tetraploid A. chinensis (NC_026690 and KP297243). Clematoclethra scandens (NC_069196) and Saurauia napaulensis (NC_069064) were designated as outgroups. A total of 32 complete cp genomes were utilized for subsequent analysis. Multiple sequence alignment of the complete cp genome sequences was performed using MAFFT v7.308 with global model and other default parameters. To remove locations containing comparison gaps, the blurred areas were modified using Gblocks software54.
We employed both Maximum Likelihood (ML) and Bayesian Inference (BI) methods to reconstruct the phylogenetic trees. The optimal evolutionary model was selected using jModeltest v255 based on the Bayesian Information Criterion (BIC). The ML phylogenetic tree was reconstructed using IQ-TREE v2.1.2.56 under the TVM + F + I + G4 model, with branch support values evaluated through 1000 bootstrap replicates. For BI, the phylogenetic topology was inferred using MrBayes v3.2.757 under the HKY85 model. The BI analysis utilized the Markov Chain Monte Carlo (MCMC) algorithm, running for 2 million generations with four independent MCMC chains. Trees were sampled every 1000 generations, with the first 25% of generations discarded as burn-in to ensure convergence.
Divergence time estimation
To estimate the divergence time of genus Actinidia, we used the ML tree inferred from nucleotide alignments previously and the MCMCtree framework58 in PAML v4.959. Three calibration points were selected with the log-normal model: the root node of the Actinidia phylogeny, which was calibrated to 83.5–89 Ma60,61; the crown group of Actinidia species, calibrated to 20–26 Ma60,62; and the divergence time for Clematoclethra scandens and Saurauia napaulensis, set at 82 Ma based on TimeTree63. A strict molecular clock was implemented to accommodate rate variation across branches64. The final consensus timetree was constructed from 25,000 post-burn-in samples, with nodal support values representing posterior probabilities. Convergence of the MCMC chains was assessed using Tracer v1.765, with effective sample sizes (ESS) for all parameters exceeding 200, indicating stable time estimates.
Data availability
The sequence data of Actinidia lijiangensis in this study are deposited in the GenBank database of NCBI (https://www.ncbi.nlm.nih.gov/) under the accession number NC_066502.
References
Wang, F. M. et al. Evaluation of the wild Actinidia germplasm for resistance to Pseudomonas syringae pv. actinidiae. Plant Pathol. 69, 979–989 (2020).
Pinto, D., Delerue-Matos, C. & Rodrigues, F. Bioactivity, phytochemical profile and pro-healthy properties of Actinidia arguta: a review. Food Res. Int. 136, 109449 (2020).
Huang, H. & Ferguson, A. Actinidia in China: natural diversity, phylogeographical evolution, interspecific gene flow and kiwifruit cultivar improvement. Acta Hort. 753, 31–40 (2007).
Liu, Y., Liu, Y. & Huang, H. Genetic variation and natural hybridization among sympatric Actinidia species and the implications for introgression breeding of kiwifruit. Tree Genet. Genomes 6, 801–813 (2010).
Mai, Y.-H. et al. Ultrasound-assisted extraction, identification, and quantification of antioxidants from ‘Jinfeng’ Kiwifruit. In Foods (Basel, Switzerland) Vol. 11 827 (2022).
Li, D. et al. Genetic diversity in kiwifruit polyploid complexes: insights into cultivar evaluation, conservation, and utilization. Tree Genet. Genomes 10, 1451–1463 (2014).
Shi, T., Huang, H. & Barker, M. S. Ancient genome duplications during the evolution of kiwifruit (Actinidia) and related Ericales. Ann. Bot. 106, 497–504 (2010).
Tang, P., Xu, Q., Shen, R. & Yao, X. Phylogenetic relationship in Actinidia (Actinidiaceae) based on four noncoding chloroplast DNA sequences. Plant Syst. Evol. 305, 787–796 (2019).
Li, H.-L. A taxonomic review of the genus Actinidia. J. Arnold Arboretum 33, 1–61 (1952).
Huang, H. et al. Phylogenetic relationships in Actinidia as revealed by RAPD analysis. J. Am. Soc. Hort. Sci. 127, 759–766759 (2002).
Li, J., Huang, H. & Sang, T. Molecular phylogeny and infrageneric classification of Actinidia (Actinidiaceae). Syst. Bot. 27, 408–415 (2009).
Allen, J. F. The function of genomes in bioenergetic organelles. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 358, 19–37 (2003).
Shi, W., Song, W., Liu, J., Shi, C. & Wang, S. Comparative chloroplast genome analysis of Citrus (Rutaceae) species: Insights into genomic characterization, phylogenetic relationships, and discrimination of subgenera. Sci. Hortic. 313, 111909 (2023).
Nock, C. J. et al. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 9, 328–333 (2011).
Brock, J. R., Mandáková, T., McKain, M., Lysak, M. A. & Olsen, K. M. Chloroplast phylogenomics in Camelina (Brassicaceae) reveals multiple origins of polyploid species and the maternal lineage of C. sativa. Horticult. Res. 9, uhab050 (2022).
Uozu, S. et al. Repetitive sequences: cause for variation in genome size and chromosome morphology in the genus Oryza. Plant Mol. Biol. 35, 791–799 (1997).
Wang, W., Chen, S.-Y. & Zhang, X. Chloroplast Genome Evolution in Actinidiaceae: clpP Loss, Heterogenous Divergence and Phylogenomic Practice. PLoS ONE 11, e0162324 (2016).
Ruhlman, T. A. et al. NDH expression marks major transitions in plant evolution and reveals coordinate intracellular gene loss. BMC Plant Biol. 15, 100 (2015).
Shrestha, B., Gilbert, L. E., Ruhlman, T. A. & Jansen, R. K. Rampant nuclear transfer and substitutions of plastid genes in Passiflora. Genome Biol. Evol. 12, 1313–1329 (2020).
Holm, L. Codon usage and gene expression. Nucleic Acids Res. 14, 3075–3087 (1986).
Chat, J., Jáuregui, B., Petit, R. & Nadot, S. Reticulate evolution in kiwifruit (Actinidia, Actinidiaceae) identified by comparing their maternal and paternal phylogenies. Am. J. Bot. 91, 736–747 (2004).
Li, D. et al. Maternal inheritance of mitochondrial genomes and complex inheritance of chloroplast genomes in Actinidia Lind.: evidences from interspecific crosses. Mol. Genet. Genom. 288, 101–110 (2013).
Cipriani, G., Testolin, R. & Morgante, M. Paternal inheritance of plastids in interspecific hybrids of the genus Actinidia revealed by PCR-amplification of chloroplast DNA fragments. Mol. Gen. Genet. MGG 247, 693–697 (1995).
Liu, Y. et al. Rapid radiations of both kiwifruit hybrid lineages and their parents shed light on a two-layer mode of species diversification. The New phytologist 215, 877 (2017).
Alix, K., Gérard, P. R., Schwarzacher, T. & Heslop-Harrison, J. S. Polyploidy and interspecific hybridization: partners for adaptation, speciation and evolution in plants. Ann. Bot. 120, 183–194 (2017).
Albert, V. et al. Polyploidy and angiosperm diversification. Am. J. Bot. 96, 336–348 (2009).
Vamosi, J. et al. Polyploidy and diversification: a phylogenetic investigation in Rosaceae. Int. J. Plant Sci. 167, 349–358 (2006).
Song, Y. et al. The population genomic analyses of chloroplast genomes shed new insights on the complicated ploidy and evolutionary history in Fragaria. Front. Plant Sci. 13, 569 (2023).
Heaney, L. R. A synopsis of climatic and vegetational change in Southeast Asia. Clim. Change 19, 53–61 (1991).
Kim, S.-C., Lee, J.-W., Baek, S.-H., Lee, M.-W. & Kang, Y.-J. The complete chloroplast genome sequence of Actinidia rufa (Actinidiaceae). Mitochondrial DNA Part B 3, 564–565 (2018).
Wang, Z. et al. Cytotype distribution and chloroplast phylogeography of the Actinidia chinensis complex. BMC Plant Biol. 21, 325 (2021).
Lynch, M., Wei, W., Ye, Z. & Pfrender, M. The genome-wide signature of short-term temporal selection. Proc. Natl. Acad. Sci. 121, e2307107121 (2024).
Lake, N. J. et al. Quantifying constraint in the human mitochondrial genome. Nature 635, 390–397 (2024).
Horton, C. et al. Short tandem repeats bind transcription factors to tune eukaryotic gene expression (2022).
Shi, C. et al. An improved chloroplast DNA extraction procedure for whole plastid genome sequencing. PLoS ONE 7, e31468 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Brown, J., Pirrung, M. & Mccue, L. FQC Dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinform. Oxf. Engl 33, 145 (2017).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18–e18 (2017).
Tillich, M. et al. GeSeq—versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11 (2017).
Shi, L. et al. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47, W65–W73 (2019).
Lehwark, P. & Greiner, S. GB2sequin—a file converter preparing custom GenBank files for database submission. Genomics 111, 759–761 (2019).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Zheng, S., Poczai, P., Hyvönen, J., Tang, J. & Amiryousefi, A. Chloroplot: an online program for the versatile plotting of organelle genomes. Front. Genet. 11, 563 (2020).
Huang, H. Kiwifruit: The Genus ACTINIDIA (2024).
Lin, M. et al. The complete chloroplast genome sequence of Actinidia arguta using the PacBio RS II platform. PLoS ONE 13, e0197393 (2018).
Li, Y. et al. The complete chloroplast genome sequences of three lilies: genome structure, comparative genomic and phylogenetic analyses. J. Plant. Res. 135, 723–737 (2022).
Ranwez, V., Douzery, E. J. P., Cambon, C., Chantret, N. & Delsuc, F. MACSE v2: toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Mol. Biol. Evol. 35, 2582–2584 (2018).
Wang, D., Zhang, Y., Zhang, Z., Zhu, J. & Yu, J. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 8, 77–80 (2010).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Chen, C. et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202 (2020).
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M. & Dubchak, I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279 (2004).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302 (2017).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772–772 (2012).
Trifinopoulos, J., Nguyen, L.-T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235 (2016).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001).
Puttick, M. N. MCMCtreeR: functions to prepare MCMCtree analyses and visualize posterior ages on trees. Bioinformatics 35, 5321–5322 (2019).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Pilkington, S. M. et al. Genetic and cytological analyses reveal the recombination landscape of a partially differentiated plant sex chromosome in kiwifruit. BMC Plant Biol. 19, 172 (2019).
Keller, J. A., Herendeen, P. S. & Crane, P. R. Fossil Flowers and Fruits of the Actinidiaceae from the Campanian (Late Cretaceous) of Georgia. Am. J. Bot. 83, 528–541 (1996).
Cui, Z., Huang, H. & Xiao, X. Actinidia in China (China agricultural science and technology press, 2002).
Hedges, S. B., Dudley, J. & Kumar, S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22, 2971–2972 (2006).
Lepage, T., Bryant, D., Philippe, H. & Lartillot, N. A general comparison of relaxed molecular clock models. Mol. Biol. Evol. 24, 2669–2680 (2007).
Rambaut, A., Drummond, A., Xie, D., Baele, G. & Suchard, M. Posterior summarization in bayesian phylogenetics using tracer 1.7. System. Biol. 67, 901 (2018).
Acknowledgements
The authors would like to sincerely acknowledge Editor and anonymous reviewers for their comments and hard work.
Funding
This work was supported by the National Natural Science Foundation of China (NO. 32370244 and NO. 32400371), and Shandong Province Natural Science Foundation of China (NO. ZR2019BC094).
Author information
Authors and Affiliations
Contributions
Conceptualization: QHL and SW; Data curation: QHL, WBS and BYZ; Formal analysis: QHL, SQH and YHH; Funding acquisition: SW and BYZ; Investigation: QHL, SQH and ZHW; Methodology: QHL, ZHW and BYZ; Project administration: QHL, SW and BYZ; Software, visualization, and writing-review and editing: QHL, SQH, YHH, WBS, ZHW and BYZ; Supervision: SW and BYZ; Validation: SW and BYZ; Writing-original draft: QHL, WBS and BYZ. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, Q., Hu, S., Wu, Z. et al. Comparative chloroplast genomics provides insights into the phylogenetic relationships and evolutionary history for Actinidia species. Sci Rep 15, 13291 (2025). https://doi.org/10.1038/s41598-025-95789-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-95789-y
