
Abstract
The finger millet (Eleusine coracana (L.) Gaertn) genome, comprised 166 conserved microRNAs (miRNAs) belonging to 39 families and three novel miRNAs. The miR169 is one of the most conserved miRNA families, while Eco_N1 is a species-specific miRNA prevalent in finger millet. Its members regulate the expression of genes encoding the Nuclear Factor-Y subunit A (NF-YA) via transcript cleavage. However, the role of miRNA genes in regulating the expression of NF-YA transcription factors in finger millet needs to be deciphered. The present study characterized 166 conserved and novel miRNAs (Eco_N1, Eco_N2 and Eco_N3). Further, secondary structures were predicted, and the potential miR genes targeting the NF-YA transcription factors regulating abiotic stress tolerance were analysed. Twenty-three Eco-miR169 members and one Eco_N1 miRNA targeting EcNF-YA13 were identified in the finger millet genome. The presence of relevant cis-elements such as ABRE (abscisic acid-responsive elements), DRE (dehydration-responsive element), and MYB (myeloblastosis) indicates that the target of Eco-miR169 might be involved in abiotic stress responses. The tissue-specific RNA-seq transcriptomic expression pattern of Eco-miR169 showed variable fold of expression in seedlings compared to the control. At the same time, the expression of EcNF-YA13 (target genes of Eco-miR169 members and Eco_N1) presented a downregulated trend under salinity and dehydration conditions compared to the control. Tissue-specific RNA-seq followed by expression analysis confirmed the antagonistic effect of Eco-miR genes on EcNF-YA13. In a nutshell, the results of this study could be utilized as a platform for further exploration and characterization of finger millet Eco-miR169-EcNF-YA13gene regulatory network.
Similar content being viewed by others

Introduction
MicroRNAs (miRNAs) are small (18–24 nucleotides), single-stranded noncoding RNAs. Genes encoded by miRNA are considered independent transcriptional units, which are transcribed into long primary transcripts by the RNA polymerase II enzyme, referred to as precursor microRNAs (pre-miRNAs)1. It is believed that miRNAs regulate target genes by cleaving the target mRNA and repressing translation. Plants have most miRNAs that are perfectly or near perfectly complementary to their target mRNAs and cleave them2. Plants contain several miRNAs that regulate stress responses, signalling pathways, and growth and development1,3.
Agriculture is seriously threatened by abiotic stresses such as drought, salinity, heavy metals toxicity, nutrient deficiency, and extreme temperatures, which can adversely affect the environment and plant growth4,5,6,7. Among the principal abiotic stresses plants encounter, are drought and salinity, which are becoming increasingly prevalent throughout the globe. In response to abiotic stress, plants express many genes and biochemical molecules. However, little has been explored about the role of miRNAs in regulating abiotic stress response1,8,9. Historically, finger millet (Eleusine coracana L.) has been cultivated in harsh environments of drylands and semi-arid regions of the world and is considered a naturally stress-tolerant crop10. With insect resistance properties, gluten-free protein, and high micronutrient density, finger millet grains could serve as an alternative to rice and maize meal for hundreds of millions of poor people11,12,13. As a result of these characteristics, finger millet is an ideal crop for climate-smart agriculture. Even though finger millet is comparatively tolerant to abiotic stresses14, little is known about the genetic elements conferring abiotic stress tolerance, particularly the miRNAs-NF-Ys gene regulatory network.
Previous work conducted by RT-qPCR and Northern blot by Usha et.al., 2015 and Nageshbabu e.al., 201315,16 for validation of novel miRNAs as well as randomly selected conserved miRNAs in finger millet tissues indicated that miR156, miR157, miR393, miR159, miR169, miR396, miR397, and miR398 are expressed under drought stress and salinity stress. Several novel miRNAs have also been observed in finger millet (Eco_N1, Eco_N2, and Eco_N3) with the same expression patterns15. Sequence analysis of finger millet Eco-miR genes (referred to as Eco-miR) revealed two proximate Dehydration-Responsive Elements (DREs) within the promoter region of miR-169, indicating that miR-169 directly regulates the CCAAT binding factor (CBF) under abiotic stress15. Researchers have recently focused on transcription factors and miRNAs that regulate genes during stressful conditions17. Still, none of the Nuclear Factor-Y (NF-Y) genes were reported as a target for Eco-miR genes of finger millet, which might be associated with several abiotic stresses.
Thirty percent of eukaryotic genes contain the CCAAT box. The NF-Y TF, also termed the CBF, has been reported in plants, animals, and fungi7,18. This TF consists of three subunits NF-YA, NF-YB, and NF-YC7,19. Plants are unique, as multiple genes are known to code for each subfamily of NF-Ys, while a single gene codes for each subfamily of NF-Y in the case of yeast and mammals20,21. Several roles unique to plants, including response to abiotic stresses, have been assigned to the identified NF-Ys7,22. Finger millet genome mining revealed 18EcNF-YA genes, 23EcNF-YB genes, and 18 EcNF-YC genes19. To our knowledge, no reports are available for the NF-Y-miRNA gene regulatory network in finger millet, which can be integrated with various abiotic stress responses.
Previous studies on finger millet used non-finger millet genomes to identify miRNAs, but the chromosome-specific miRNA count was not identified because the genome was distributed in the scaffolds23,24. In this study, the recently available well-assembled reference genome (Devos et al., 2023) was utilized for mining all the miRNAs, characterization of secondary structure by in-silico analysis, and identification of their conserved domains in finger millet. Further, the target mRNAs regulated by the miRNA169 genes were identified and functional genes and TF were also annotated. It was also elucidated that the NF-YA transcription factor of finger millet, EcNF-YA13, is a target gene of these dehydration-inducible Eco-miR169 members and Eco_N1. The miRNA of finger millet might be cleaving mRNA (EcNF-YA, a target of miRNA169) under upregulation by high salinity and dehydration treatment. The study on the integration of the EcNF-YA-Eco-miRNA module will be able to address several regulatory phenomena of stress signaling pathways and specific miRNAs regulating the expression of potential EcNF-Y genes in response to dehydration and salinity stress. These potential candidates can be utilized to enhance the productivity of finger millet under harsh environmental conditions. Moreover, this study will also support to initiation of further functional characterization of miRNA genes in finger millet by yeast complementation assay or genome editing approaches or RNAi for crop improvement programs.
Materials and methods
Plant materials
The seeds of the finger millet MR1 genotype (drought tolerant) were obtained from ICAR-Vivekananda Institute of Hill Agriculture, Almora, Uttarakhand, India and grown in a plant growth chamber as per the reported protocols of Puranik et.al 201325. After 21 days, seedling was uprooted and washed under running tap water to remove soil adhering to it26. Then for stress imposition, seedlings were placed into 20% PEG-6000 (dehydration stress), and 250 mM NaCl (salinity stress) and normal condition (control) for 6 h and 12 h to detect the expression pattern19. The tissue from each group was harvested separately at 0 h (control), 6 h (early stress), and 12 h (late stress) time points. Then the collected samples were immediately frozen in liquid nitrogen and stored at − 80 °C for RNA isolation and qRT-PCR-based expression analysis of EcNF-YA genes. All the samples were collected in triplicate to maintain the accuracy and reliability of the data.
Sequence collection
The nucleotide sequences of 39 miRNA families encoding 166 miRNA loci were retrieved from the finger millet genome (Eleusine coracana v1) using the Plant miRNA ENcyclopedia (PmiREN2.0) database (https://www.pmiren.com/). Based on the findings of (Usha et al., 2015), three miRNAs of finger millet designated as novel miRNAs were identified and included in this study. The BLASTN module in the PmiREN2.0 database was used to align all the pooled novel Eco-miRNA sequences from the literature to avoid duplicity. The BLASTN was performed on the PmiREN database with the expectation threshold of 10 and maximum target sequence of 250; the match-mismatch score was set to 2, and the gap cost was set to 5/2. The alignment results of finger millet miRNA mature sequences in PmiREN2.0 lack any of the selected novel Eco-miR genes. Therefore, sequences were verified as novel and not duplicates and were referred to by names as reported earlier (i.e., Eco_N1, Eco_N2, and Eco_N3). These retrieved sequences (169 candidate genes (known as Eco-miR (Eleusine coracana miRNA) have been compiled in Supplementary File S1. The nucleotide sequence of finger millet EcNF-Y genes was retrieved from the NCBI database with accession numbers BK063099, BK063100, BK063101, BK063102, BK063103, BK063104, BK063105, BK063106, BK063107, BK063108, BK063109, BK063110, BK063111, BK063112, BK063113, BK063114, BK063115, BK063116 provided in Supplementary File S219,27.
Target gene prediction and gene ontology (GO) analysis
Finger millet miRNA and mRNA sequence complementarity were used to identify potential gene targets. The targets against all Eco-miR genes were identified by using a plant small RNA Target server (psRNA Target) (https://www.zhaolab.org/psRNATarget/)28. EcNF-Y genes of finger millet were taken as inputs at seed region of 2 to 13 nt, with an expected value of 3 to 5. In addition, up to two consecutive mismatches were permitted between mature miRNA and potential targets29,30. As the miRNA target genes were identified, QuickGo (https://www.ebi.ac.uk/QuickGO/), was used to annotate Eco-miR169 and Eco_N1 target genes in finger millet plant slim (http://www.omicshare.com/tools) was applied and limited to the taxon finger millet, with a significance threshold (p < 0.05) used to obtain annotations with statistical significance in comparison with the background data.
Chromosomal localization and prediction of secondary structure
MapGene2Chromosome V2.1 software (http://mg2c.iask.in/mg2c_v2.0/) was used to map the chromosomal localization of Eco-miR169 and Eco_N1. The precursor sequences of Eco-miR169 gene families were obtained from PmiREN2.0. Based on default parameters, the Unified Nucleic Acid Folding (UNAFold) web server and Zuker folding (Zuker, 2003) were used to predict the secondary structures and Minimal Folding Energy (MFE) of pre-Eco miRNA169s. The Minimum Folding Free Energy Index (MFEI) was calculated using MFEI = [(MFE/length of the RNA sequence)*100]/(G + C)%31,32. The structures expected to have the lowest free energy were inspected physically. The identified miRNAs were subjected to a manual screening procedure to look for mature sequences that are ten base pairs or fewer than the source miRNAs involved in Watson–Crick (G/U model) matching between the mature miRNA and its opposing strand (miRNA*) in the duplex.
Phylogenetic analysis
The mature sequences of miR169 members from four species, including rice (Oryza sativa), maize (Zea mays), foxtail millet (Seteria italica), and sorghum (Sorghum bicolor), were retrieved from the PmiREN2.0 database. Phylogenetic trees were constructed with finger millet Eco-miR169 members and Eco_N1 with other Poaceae family members using ClustalW33 via MEGA V11.0 software34. The phylogenetic tree was constructed at 1000 bootstraps using the maximum likelihood method and iTOL v.6 (https://itol.embl.de/upload.cgi) for interactive tree visualization.
Identification of Stress-related cis-regulatory elements (CREs) in promoter of Eco-miR169 genes and EcNF-YA13
The 2 kb upstream promoter region of Eco-miR169s , Eco_N1 and EcNF-YA13 was retrieved from NCBI database using the genome data viewer limiting to assembly (Eleusine_coracana_v1.0 (GCA_032690845.1)). Afterwards, CREs present in this region were identified using the PlantCARE database (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/). TB tool was used for visual analysis of CREs.
Digital expression profiling of Eco-miR169 gene family
The transcriptomic dataset of the Eco_miR169 gene family and Eco_N1 genes in 3 different tissues (seedling, leaf, and floret) was obtained from PmiREN2.0 database35. Further, it was cross-validated with the transcriptomic data set (SRR1832812) to analyse the expression patterns of all of the Eco-miR169s and Eco_N1 miRNA under drought and control conditions of finger millet15. Gene transcript levels were calculated using reads per million after filtering the data by BiGGEsTS software (http://kdbio.inesc-id.pt/software/biggests/)36. The analysis was performed by using parameters of gene filtering, for filtering genes with missing values, then normalize genes to 0 and std. 1 with discretization technique by opting gene expression average. This results into transformation of each element of the expression matrix into a downregulated (D), if its absolute value is lower than the mean, or into a upregulated (U) otherwise. The gene expression of miRNAs was deduced in the form of a heat map using log fold change based on RPM data using TBtools software37. Using qRT-PCR, the differentially expressed genes (DEGs) Eco-miR169a-i (nine members) and Eco_N1 were further cross-validated using the relative expression levels previously reported by Nageshbabu et. al. 201316 and Usha et. al 201515 under salt stress and dehydration stress respectively.
qRT-PCR based expression of EcNF-YA genes of finger millet
For the expression analysis of finger millet Eco-miR169 and Eco_N1 target, i.e., EcNF-YA genes, total RNA was isolated from control and stress-treated whole seedlings, as per the reported protocol of Rani et. al., 202419 followed by Quantitative real-time PCR analysis (Applied Biosystems 7500 real-time PCR system) by using SYBR Premix (TaKaRa Code: DRR041A, Japan). Each sample was analyzed in a final volume of 10 µl with three biological replicates and three technical replicates. NCBI Primer-BLAST was used to verify the EcNF-YA gene-specific primers, and β-Tubulin (β-TUB) served as an internal control during the expression profiling process38. The relative expression level of each genes was analysed using the 2-ΔΔCT method39.
Interaction network of Eco-miR169s and Eco_N1 target gene
The targets of Eco-miR genes and their interacting partners were analysed by using STRING v.12.0 with Arabidopsis as a reference organism. Genes at a high confidence level of 0.70 with the highest bit score were utilized to construct protein networks. Further, the functional annotation of these targets was performed through interacting network and gene enrichment analysis by using Cytoscape 3.2 software40.
Statistical analysis
For each treatment three biological and three technical replicates were used. Data were evaluated using OriginPro 2021software (https://www.originlab.com/origin) by using ANOVA, opting Tukey test for multiple comparisons to confirm the significant differences (p ≤ 0.05) between the gene expression profiles and the respective treatments.
Results
miRNA identification, characterization, and target prediction in finger millet genome
A total of 169 miRNA loci, belonging to 40 different families (conserved and novel), were identified, and characterized in the finger millet genome (Table1); among these Eco-miRNA families, 18 Eco-miRNA families of finger millet contain more than two members, as shown in (Fig. 1). The psRNAtarget reveals that among all Eco-miR families, members of Eco-miR169s (Conserved) and Eco_N1 (Novel) microRNA targeted finger millet EcNF-YA13 gene with the best exception below 3 through cleavage activity. Remaining Eco-miRNA targeting different members of EcNF-Y at exception levels of 4 to 5 (Table 2). Therefore, this analysis did not include those targets to avoid false predictions. Thus, further structural characterization and validations were carried out for only the Eco-miR169s and Eco_N1 genes of finger millet. Twenty-three members of the Eco-miR169 family were identified in the finger millet genome and designated from Eco-miR169a to Eco-miR169w. Across all the mined miRNA genes, there are differences in lengths of precursor sequences (64 to 150 nt) and miRNA sequences (20 to 22 nt), as represented in Table 3. Members of Eco-miR169s and Eco_N1 were distributed at chromosomes 2, 3, 4, 6 and 8 in the finger millet genome (Fig. 2), whereas the target of Eco-miR genes, i.e., EcNF-YA13 were separately located at Chr 7B. The multiple sequence alignment (MSA) reveals that mature sequences of (Eco-miR169a/b/e/h/i/m/n/o/p/q/r/s/u), (Eco-miR169c/d/f/i/j/k/t) and Eco-(miR169v/w) were highly consistent and possess more conserved nucleotides. In contrast, the mature sequence of Eco-miR169g and Eco-miR_N1 showed divergence from other Eco-mir169 of finger millet (Fig. 3).

(A) Abundance of conserved and novel miR genes in finger millet. The star represents number of members in each family. The Fig. reveals that miR165 family followed by miR169 contain highest miR genes (B) Chromosome abundance of all the 166-miRNA loci of finger millet revealing Chromosome 2 containing multiple miRNA loci in finger millet.

Graphical (scaled) representation of physical locations for each Eco-miR169 gene on finger millet chromosomes (numbered 1–9). Eco-miR169 members represent by red, Eco_N1 by green and EcNF-YA13 genes which is the target of Eco-mioR169 members are represented by blue and located on separate chromosome 7B. Chromosomal distances are given in Mb. The physical location of miR genes on the respective chromosome represented in right indicates the chromosomal location (in Mbp) and the respective miRNA IDs are provided on the left.

Multiple sequence alignment of members of Eco-miR169 and Eco_N1, created by using MEGAv.11 software, revealing conserved and differences in nucleotide and length in finger millet. The residues were labeled with orange boxes, showing differences in amino acid residue with conserved miR169 members. Meanwhile, the pointed arrow indicates a difference in nucleotide among members of the miR169 family of finger millet.
Secondary structure of Eco-miR169s of finger millet
The secondary structure of all the Eco-miR169 members were constructed using the UNA Fold web server. It was found that these pre-Eco-miR169 sequences (Supplementary File S3) formed a typical stem-loop structure. The MFE has been considered an important parameter for determining the secondary structure of pre-miRNA and the identification of miRNA. The MFE (ΔG kcal/mol) ranged from − 56.8 kcal/mol (pre-Eco-miR169s) to − 30.40 kcal/mol (pre-Eco miR169h and Eco-miR169u) (Fig. 4). The MEFI, which differentiates pre-miRNA from other coding and non-coding RNAs and RNA fragments, ranged from 1.05 (Eco-miR169c) to 0.74 (Eco-miR169g) (-kcal/mol). Each of the miRNAs was explored for its loop structure and revealed that the number of sub-loops ranged from 2 (pre-Eco-miR169h and pre-Eco-miR169u) to 8 (pre-Eco-miR169s) (Fig. 4). Further, the AU, GC, and nucleotide of pre-Eco-miRNA169s were analysed. The AU contents ranged from 32.9% (pre-Eco-miR169f.) to 54.4% (pre-Eco-miR169n), while GC content ranged from 45.5% (pre-Eco-miR169n). to 67.05%(pre-Eco-miR169f.). Across the predicted miRNA families, there was a variation in nucleotide content. Different attributes of Eco-miR169s are summarized in Table 3.

Secondary structure of Eco-miR169 members and Eco_N1. In the stem loop structures, the mature sequences are shown in red whereas, the star sequence is shown in green in the stem section with little bulges.
Phylogenetic analysis
Comparative phylogenetic analysis was performed to get an insight into evolutionary relationship among NF-YA genes and miR169s of finger millet (Eco-miR169a-w and Eco_N1), rice (Osa-miR169a-p), maize (Zma-miR169a-r), foxtail millet (Sit-miR169a-o) and sorghum (Sbi-miR169a-o). A total of 64 NF-YA genes together with 88 of miR169 members were clustered into five clades (Fig. 5). Clade I, II and IV consist of a single conserved miRNA of maize (Zma-miR169f.), novel miRNA (Eco_N1) of finger millet and Osa-miR169f. of rice respectively, revealing substantial variations than other members of miR169 family. Clade V contains rest of the eighty-five members of miR169 family of rice, maize, finger millet, sorghum, and foxtail millet. These members showed uniform occurrence of Eco-miR169w/v/t/k/j/i/f/d/c and close similarity among them while EcomiR169g, appeared in different subclades and shared a close genetic relation with sorghum Sbi-miR169c. Moreover, EcomiR169a/l/m/o/p/q/r/s showed high sequence similarity with maize (Zma-miR169i/j), foxtail millet (Sit-miR169g/h/i/j/l/m/n), sorghum (Sbi-miR169l/h/g/f), and rice (Osa-miR169h/i/j/k/l/m), revealing similar evolutionary processes. Eco-miR169u/h and Eco-miR169 e/b were closely associated with each other and distantly related to sorghum Sbi-miR169n/m and rice Osa-miR169g.

A comparative evolutionary analysis of NF-YA and miR169 genes of finger millet together with rice, maize, sorghum and foxtail millet. The miR gene sequences of all these plants Eco-miR169a-w and Eco_N1, Osa-miR169a-p, Zma-miR169a-r, Sit-miR169a-o and Sbi-miR169a-o. are retrieved from pmiREN database whereas, NF-YA genes were retrived from Plant Transcription Factor database, . The tree was constructed using MEGA v.11 software using the NJ method. The phylogenetic tree was divided into five clades. The novel miR gene Eco-NI is specific to clade I. Other members of the conserved miR169 family of finger millet and other plants of the Poaceae family are distributed among II, III, and V clades of this phylogenetic tree. whereas clade IV consists NF-YA genes. Accession number of NF-YA genes of rice, maize, sorghum and foxtail millet used in construction of phylogenetic tree is given in supplementary file S7.
Clade III consists of 64 NF-YA genes of rice, maize, finger millet, foxtail millet, and sorghum. Among these genes, EcNF-YA15 and EcNF-YA7 are closely clustered with each other and distantly related to ZmNF-YA12, associated with drought resistance and growth recovery41. This suggests that EcNF-YA15 and EcNF-YA7 might be associated with drought resistance and growth recoverability in finger millet. EcNF-YA6/9/12 and EcNF-YA5/8/18 are evolutionarily related to rice OsNF-YA6 and OsNF-YA10, which have a tolerance against viral infection42. Additionally, EcNF-YA2 is closely clustered with foxtail millet NF-YA10, sorghum NF-YA3, and maize NF-YA3. Based on the qRT-PCR expression analysis along with yeast one-hybrid assay and electrophoretic mobility shift assay (EMSA), it has been reported that maize NF-YA3 is involved in early flowering, drought tolerance, and high-temperature tolerance43. Similar studies need to be carried out in finger millet and other cereals for functional characterization of NF-Y A subunits.
Identification of Stress-related cis-regulatory elements (CRE)
Plant CARE database was used to obtain an insight into the putative roles of EcNF-Y A13 and Eco-miR169 members, particularly for stress tolerance. A number of cis-elements (MYC, CAAT box, GC motif, AuxRR core, as-1, TGACG motif, GT1 motif, ARE, ABRE motif, TATA motif, STRE motif, G motif, ATCT motif) were identified, that are known for their functional roles in stress responses, hormonal signaling, and plant development. (Table 4 and Supplementary file S4). Moreover, the analysis of promoter region of Eco-miR169’s (Fig. 6) reveals presence of more than 200 CREs (Fig. 6F) that have been categorized into four categories related to their functions namely light, stress, hormones, growth, and development, shown in (Fig. 6A) and Supplementary File S4. Except for the core promoters and enhancers (Fig. 6E), stress responsive elements (Fig. 6C) were quite diverse and in abundance, followed by hormone (Fig. 6D) and light responsive cis-regulatory elements (Fig. 6B). A total of six types of biotic/abiotic stress response elements were identified (Fig. 6C), including W-box (pathogen responsive), MYB, MYC (drought and salt-responsive) (TC-rich repeats (defense responsive) and DRE (dehydration responsive). Further, ABRE and TGACG-motif, were also observed, associated with Abscisic acid and MeJA-responsiveness. The promoters of Eco-miR169s were also identified revealing around 360 growth and development response elements (Fig. 6E), which are primarily divided into three types, including the O2 sites (zein metabolism regulation), circadian control and CCAAT-box, involved in meristem expression, and NF-YA binding sites. These analyses indicate that Eco-miR169 members are widely associated with stress responses, plant growth and development. Figure 6G, illustrates the organization of the major stress-responsive element in the 2 kb upstream of the promoter region of Eco-miR169 members.

Cis-regulatory elements (CRE) in the 2 kb upstream of the promoter region of Eco-miR169 and Eco_N1. A. Different classes of cis-regulatory elements B. Light Responsive CREs, C. Stress Responsive CREs D. Hormone responsive cis-regulatory element, E. Plant growth and development related CREs F. Abundance of CREs in 2 kb upstream of EcomiR169 members G. organization of significant stress-responsive CREs in 2 kb promoter region of finger millet.
Digital expression profiling of Eco-miR 169 gene family
Finger millet miR169 members are perfect homologs of maize miR169 members (Table 1), so expression data of 24 homolog genes (23 Eco-miR169a-w and one Eco_N1) was retrieved from seedling, leaf, and floret tissues of finger millet in response to drought stress from PmiREN2.0 database by comparing it with maize sRNA dataset (seedling (GSM1529456_SRR161888), leaf (GSM1782954_SRR2086139) and floret (GSM433621_SRR034098). Further, the digital expression profiling of Eco-mir169a-i was cross-validated with qRT-PCR based expression data reported by Usha et al. in 2015 (SRA: SRS863641)15. Interestingly, many Eco-miR169 members were induced by drought stress in the seedling and leaf tissues of finger millet (Fig. 7A). For example, nine Eco-miR169 members viz. Eco-miR169c/d/f/i/j/k/t/v/w were highly up-regulated (> two fold) in finger millet leaf tissue under drought stress compared to seedling and floret tissues. However, all nine genes were downregulated in seedlings and floret tissues of finger millet. Similarly, fourteen Eco-miR169 members viz. Eco-miR169a/b/e/h/l/m/n/o/p/q/r/s/u/g were found to be induced by drought stress by > three fold in seedling than leaf and floret tissue of finger millet. Moreover, under drought stress, Eco_N1 expression was significantly higher by more than three-fold within the floret tissue of finger millet compared to the other two tissues.

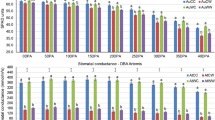
(A) Heat-map representing expression analysis of Eco-miRNA169a-w and Eco_N1 in leaf, seedling and floret tissue under dehydration stress as compared to control. The colour scale indicates the expression value (pink indicate higher expression value, cyan indicates lower gene expression values). (B) Eco-miR169 members and Eco_N1 targeting finger millet EcNF-YA13 through cleavage activity. Yellow highlights represent the novel miR gene of finger millet, purple colour represents conserved miR genes and cyan highlights targetedEcNF-YA13 gene. Green line shows miR genes interacting with cleavage activity, whereas, red points represent targated gene. (C) Graph illustrating a significant downregulation of the EcNF-YA13 gene of finger millet under dehydration stress and salinity stress at time points 6 h and 12 h. The EcNF-YA16 gene, however, exhibits significantly greater expression levels (> tenfold) under dehydration stress conditions as well as (> 20 fold) under salinity stress conditions, followed by EcNF-YA5 under dehydration stress (> five fold) and salinity stress (> four fold). The values presented above the bar following a different letter are significantly different from one another at P ≤ 0.05.
qRT-PCR based expression profiling of the target genes
The psRNA target prediction, revealed that 24 Eco-miR genes of finger millet targeting solely one NF-Y transcription factor, i.e., EcNF-YA13 out of 18 members of EcNF-YA genes (Fig. 7B) (Table 2). The qRT-PCR-based expression pattern of EcNF-YA13, together with other members of EcNF-YA subunits (EcNFYA1/5/9/13/15/16), was attempted to check the expression pattern of those genes that are not targeted by any other miR genes of finger millet (Supplementary Table S5).
All the EcNF-Y genes viz. EcNF-YA1/5/9/15 and 16, except EcNF-YA13, showed more than two-fold upregulated expression at 6 h of dehydration and salinity stress, whereas all these genes, including EcNF-YA13, exhibited downregulated expression patterns at 12 h stress treatment. Two transcription factors (EcNF-YA5 and EcNF-YA9) were highly induced (> 1.5 fold) by 6 h of dehydration stress than by 12 h of dehydration stress (Fig. 7C). EcNF-YA13 showed higher expression in 6 h and 12 h salinity stress compared to 6 h and 12 h dehydration stress. Still, there were no significant differences in expression level between the 6 h and 12 h salinity stress (Fig. 7C). The EcNF-YA15 showed uniform expression in 6 h of dehydration and 6 h of salinity stress, but this gene showed a better response in 12 h of salinity stress when compared to 12 h of drought stress. Among all the six transcription factors, EcNF-YA16 was found to be the key transcription factor, as its expression level was more than fivefold higher in 6 h dehydration stress and 6 h and 12 h salinity stress (Fig. 7C). EcNF-YA1, EcNF-YA5, EcNF-YA9, and EcNF-YA16 showed higher expression under 6 h dehydration stress compared to the 12 h dehydration stress. Similarly, the expression levels of EcNF-YA1, EcNF-YA5, EcNF-YA15, and EcNF-YA16 were higher in 6 h salinity stress compared to 12 h salinity stress. This result indicated that those EcNF-YA genes, which were not targeted by any members of Eco-miRNA169s, expressed well under early dehydration and salinity stresses than late dehydration and salinity stresses, whereas Eco-miR169a-w and Eco_N1, which targets EcNF-YA13 through cleavage activity, might be associated with downregulation of its expression pattern in both dehydration and salinity stress at both times point of 6 h and 12 h (Fig. 7C).
Network analysis of the EcNF-YA13 gene of finger millet
To explore the interactions of Eco-miRNA target genes with other proteins and to understand the regulatory mechanisms that may be associated with abiotic stress tolerance, an interaction network of the target genes of Eco-miRNA was constructed. The network analysis reveals that, the EcNF-YA13 interacting with multiple genes of Arabidopsis by using 74 nodes and 480 edges (Fig. 8). With the MCODE Cytoscape App2.0, the best-interacting proteins were filtered out at the highest score (Fig. 7). According to the study, EcNF-YA13, homolog of AtNF-YA5 was closely associated with nine NF-Y genes of Arabidopsis (AtNF-YA2/3, AtNF-YB4/5/6/7, AtNF-YC1/2/4), which indicates their biological function in regulating responses to water deprivation, ABA signalling pathway, somatic embryogenesis, and transcription regulation. The GO annotation and enrichment analysis of the whole network revealed the target gene’s functions in four categories: biological processes, cellular components, molecule functions, and role in the KEEG pathway (Fig. 9). Analysis of GO processes and enrichment score (Supplementary Table S6) revealed that most interacting partners were associated with cell biosynthesis, regulation of primary metabolic processes, gene expression, transcription, CCAAT binding, and catalytic process.

Protein–protein interaction of Ec-NFYA13 gene of finger millet formed by String and visualized by Cytoscape. The interior pink line represents the p-p interaction, whereas, the node represents orthologs of finger millet EcNF-YA13 gene in Arabidopsis for functional annotation. The yellow highlighted genes indicate best interacting partner.

The bubble chart shows the top 50 results for GO enrichment of the target genes of 23 members of Eco-miR169 and Eco_N1 in finger millet. GO terms as estimated at an FDR cutoff of 0.05. Enrichment p values are shown on right bar. The size of the bubble represents the number of core targets involved in the term; and the color from green to red indicates the FDR value from small to large, that is, the higher the intensity, the higher the significance of the term. Four bar plots are shown for the common enriched GO term. BP stand for biological Process; MF stands for Molecular Function; CC stands for Cell Compartment and KEGG stands for KEGG pathways.

Finger millet has been shown to activate Eco-miR169 members and EcNF-YA genes under drought and salinity stress. It has been shown that the interaction between Eco-miR169 members and EcNF-YA13 cleaves its expression and results in a plant that is sensitive to abiotic stress. A miR169 mimicry approach can assist in making finger millet more tolerance to abiotic stress in this direction. In response to the abiotic stress (drought and salinity), the receptors on the cell membrane of finger millet detect the stress signal and generate rapid responses in the form of cytoplasmic cascades. This leads to the activation of NF-YA13 gene and miRNA169. Due to the overexpression of miRNA169, the activity of NF-YA13 get ceased, inhibiting further expression of mRNA and making finger millet susceptible to drought and salinity. MIM169 can be utilized for mimicking miRNA169 and inhibiting its activity, which in turn allows the activation of NF-YA13 gene, which activates plethora of molecular and biological functions and makes finger millet tolerance to drought and salinity stress,
Discussion
The microRNAs have a critical role in plant development and adaptation to stress tolerance, hence substantial studies have been made in various crops like Arabidopsis44, wheat45, rice46, maize47, and foxtail millet48. Still, several underutilized millets, like finger millet, have not yet been thoroughly explored for the specific roles of micro RNAs in imparting abiotic stress tolerance An extensive survey of the available literature through Google Scholar and Pubmed (till 19.08.24) revealed that in finger millet, few Eco-miR169s gene related study have been conducted but the chromosome-specific miRNA count could not be tagged due to distribution of genomes in scaffolds 15,16,23,24. In order to precisely annotate Eco-miR genes and functionally anticipate their targets, the finger millet reference genome E.coracana v.1 (taxon id 4511)49 was mined for this investigation. Currently, the PmiREN database is the only microRNA database that has sufficient information regarding the finger millet miRNA35. Therefore, the current study was initiated with PmiREN database, the obtained sequences were filtered for the tRNA and ribosomal RNA contamination using the Rfam database (UNAFold) web server and Zuker folding (Zuker, 2003). In the present investigation, 23 members of the finger millet miR169 gene family were identified based on the assembled and fully annotated genome sequence of finger millet49. In contrast, the previous studies15,23,24 reported only nine members of the finger millet miR169 family. This may be the result of fragmented reference genome sequences of finger millet used in previous studies.
Rice, maize, Arabidopsis, and foxtail millet have been extensively investigated for miR169 genes30,48,50,51,52. Under biotic and abiotic stress, some members of the miR169 gene family have been shown to regulate NF-YA transcription factors’ expression patterns30,50,53. It may be due to the presence of CCAAT boxes in the promoters of most miR169 (Fig. 6E ). The expression profiling of EcNF-Y genes, revealed that EcNF-Y genes were upregulated in the tolerant genotype under salinity and dehydration stress. However, downregulation of the expression levels of some members of the EcNF-YA subfamily was also observed despite the presence of stress-responsive cis regulatory elements such as DRE, MYB, and MYC19,27.
Keeping this in view, we attempted to decipher all Eco-miR genes, both conserved and novel, which might be associated with regulating the expression level of finger millet NF-Y genes responsive to abiotic stress tolerance in finger millet7,54, especially in response to drought and salt stress. A total of 23 members of Eco-miR169a-w in finger millet were retrieved from the PmiREN database, and Eco_N1 novel microRNA was collected from the previous report15. These 24 Eco-miR genes were characterized for several attributes, such as chromosomal distribution, secondary structure, evolutionary relationship, and gene expression profiles in different tissues subjected to drought and salinity stress. The expression levels of the Eco-miR169a-i family members have been verified from the earlier reports of Usha et. al, 2015 for finger millet under drought and control conditions15. In the present study, it was observed that all the 23 members of Eco-miR169 were upregulated under drought condition. Similarly, the qRT-PCR-based experiments of Eco-miR169 a-i genes confirmed that miR169 is responsive to drought and salinity stress in several species of millet, including finger millet15.
In this study, several putative Eco-miRNAs were identified, with their corresponding precursors, whose sequences were highly conserved compared to monocot and dicot orthologs (Table 1). This finding suggests that miRNAs may perform similar physiological functions in monocots as well as dicots, which accounts for their universal conservation55. The finger millet miRNA precursors that were predicted here showed an extensive range of sizes, from 60 to 151nt, and stable stem-loop secondary structures, which is in line with findings from rice50, maize30 and foxtail millet48. There is a strong correlation between the MFE and the thermodynamic stability of secondary structures, and in general, the lower the values of MFE, the greater the stability of secondary structures56. Most of the sequences analysed had low MFE values (-56.8 kcal/mol to -30.40 kcal/mol) and MFEI (-1.05 to -0.74); hence are thermodynamically stable. The MFEI values of tRNAs (-0.64 kcal/mol), rRNAs (-0.59 kcal/mol), and mRNA are (0.62–0.66 (-kcal/mol)), respectively57, significantly lower than the finger millet miRNAs reported in the present study. This reveals that the putative finger millet Eco-miRNAs are true micro-RNAs. Additionally, uracil is significantly more prevalent at the beginning of the predicted Eco-miRNAs. As a consequence, the present studies can be regarded as reliable and are in alignment with previous findings, emphasizing the crucial role that uracil plays at the beginning of a mature miRNA in regulating miRNA-mediated regulation mechanisms58,59. Further, the analysis of all secondary structure criteria for Eco_N1 performed in the present study is similar to what has been reported earlier (Usha et al., 2015)15.
When plants are exposed to high salinity levels, miRNAs are expressed (upregulated), resulting in a negative effect on target mRNAs that encode proteins. Contrary to this, downregulation of miRNAs promotes the expression of target mRNAs, thereby contributing to stress adaptation in positive manner60. Therefore, by regulating the expression of target genes, miRNA contributes to the growth and development of plants. The 23 members of Eco-miR169, together with Eco_N1, targeting the NF-YA13 gene, have been substantially characterized in the present study. Similarly, Eco_N1 and conserved miR169 genes of finger millet targeting uncharacterized nuclear factor Y transcription factors have been reported by Usha et al., 2015, though could not elucidate their function and change in expression pattern. The genome mining of finger millet revealed presence of 18 EcNF-YA, genes19. With these results, efforts were made to decipher the regulatory mechanism of Eco-miR169a-w and EcNF-Y gene network in response to abiotic stresses in finger millet.
NF-Y-mediated drought and salt tolerance with ABA-dependent or independent signalling based on the type of cis-elements in the promoter of NF-Ys genes have been investigated by Maheshwari et al. (2019); Kavi et al. (2022)22,61. The PlantCARE database analysis in the present study revealed presence of majority of CREs, in promoter of EcNF-YA13 including MYC, CAAT box, GC motif, as-1, ARE, ABRE, STRE, and G-box (Table 4). Among these regulatory elements, the G-box containing nucleotide CACGTC is known to be involved in ROS regulation or H2O2 activation62, while the remaining CREs are associated with hormonal, plant development and stress signalling pathways. This indicates the possibility that the down regulation of EcNF-YA13 gene might influence several genes involved in stress signalling pathways.
The analysis of psRNA Target revealed that Eco-miR169a-w and Eco_N1 primarily target EcNF-YA13, which is the only subunit of the NF-Y complex that is regulated post-transcriptionally by miR16950. In stressed plants, EcNF-YA13 transcript abundances were negatively correlated with Eco-miR169 expression levels, which contain key stress-responsive regulatory elements like MYC, as-1, CCAAT, and ABRE in promoter region27. Which reveals that, Eco-miR169a-w targeting EcNF-YA13 might suppress stress tolerance by affecting the CCAAT binding factor. Further, miR169 has been implicated in regulating dehydration and salinity stress15,16. The promoter regions of Eco_N1 and miR169 genes of finger millet are enriched with CREs (Fig. 6), known to regulate abiotic stresses via plant signal transduction pathways63,64. The presence of common CREs MYC, ABRE, and CCAAT regulatory elements in promoters of Eco-miR169 indicate significant cross-talk with EcNF-YA13 during drought and salinity stress.
Further, sRNA-seq studies, on the dataset (GSM1529456_SRR161888), (GSM1782954_SRR2086139) and (GSM433621_SRR034098) taken from seedlings, leaf and floret tissue grown under water withhold conditions for 4 days revealed the upregulation of Eco-miR169 members under dehydration stress compared to control. Similar findings of miR169 have been reported through qRT-PCR expression analysis in finger millet for Eco-miR169a-h24. Plant growth and development are controlled by miRNAs, which inhibit the expression of their target genes65. Several studies have demonstrated that At-miR169a and Gm-miR169c negatively regulate AtNF-YA5, which is responsible for making Arabidopsis susceptible to drought stress66,67. Similarly, in maize, most of the targeted ZmNF-YA genes exhibited a reverse correlation with zma-miR169 gene expression over short-term and long-term stress treatment68. The rice miR169g/n/o reported as high salinity-responsive miRNAs in rice, are known to inhibit NF-YA members50. These expression and overexpression studies revealed that NF-YA TF is negatively regulated by miRNA169 genes, which has been explored in the present study in finger millet. The study conducted by Rani et al., 202419 showed that most of the NF-YA/B/C members were highly upregulated under drought and salinity stress, though few were downregulated. This led to deciphering the involvement of finger millet Eco-miRNA in inhibiting NF-YA genes. In the MR1 genotype, where expression analysis of miR169 genes was validated (Usha et al., 2015), in the same cultivar, the expression of NF-YA genes was checked under salt or dehydration stress for a period of 6 h and 12 h. It has been observed that EcNF-YA13 is targeted by 24 Eco-miR genes of finger millet. A total of fourteen Eco-miR genes (Eco-miR169a/b/e/h/i/m/n/o/p/q/r/s/u/g) were found to be induced by drought stress by > threefold in seedling tissues of finger millet and similar tissue were showing inhibition of expression pattern of NF-YA13 under both dehydration and salinity stress. This indicates that Eco-miR169 in finger millet might be negatively regulating the EcNF-YA13 gene under abiotic stress. This could be further validated by overexpression and RACE techniques. It is hypothesized that by using a mimicry strategy for Eco-miR169 by developing Eco MIM169 (mimicry miRNA169), the negative regulation of EcNF-YA13 genes in response to abiotic stress could be suppressed (Fig. 10). In that case, the Eco-MIM169 will act as a sponge for the accumulation of all overexpressed miR169, due to which inhibition of the EcNF-YA13 gene will be prevented, and the NF-YA13 gene will be able to show tolerance towards abiotic stresses, which could be confirmed through overexpression analysis and can be utilized for crop improvement.
Conclusion
This is probably the first report of comprehensive studies on the association of Eco-miRNAs, Eco-miR169, Eco_N1 (novel microRNA), and EcNF-YA13 transcription factors in regulating abiotic stresses, especially dehydration and salt. The entire set of Eco-miR genes targeting EcNF-YA13 gene has been substantially characterized. The high level of conservation among Eco-miR169 members revealed the predominance of tandem duplication events during evolution. The presence of MYB, MYC, ABRE, STRE, and WUN-motifs and CCAAT boxes in the promoter of Eco-miR169 provides insight into how the expression of microRNAs is dynamically regulated. The EcNF-A13 has been deciphered as a potential target of Eco-miR169 members with diverse biological functions. Further, gene annotations showed the presence of conserved and novel miRNAs, which might play an essential role in growth, development, and stress responses. The present study confirmed that under abiotic stress, the expression of Eco-miR169 members and EcNF-YA13 in finger millet shows a negative relationship; the expression of EcNF-YA13 is repressed with enhanced expression of Eco-miR169 genes. The transcription factor EcNF-YA13 of finger millet possesses cis-regulatory elements for salinity and dehydration tolerance, but their expression is inhibited due to the cleavage activity of finger millet Eco-miR169genes. Ultimately, the research concluded that if Eco-miR169 expression could be controlled, then inhibition of the EcNF-YA13 gene could be prevented, and the EcNF-YA13 gene could show tolerance to abiotic stress. Further studies need to be carried out to reveal the molecular basis of the EcNF-YA13-Eco-miR169 module under diverse abiotic stresses. This could be then translated for crop improvement programs.
Data availability
The datasets generated and/or analysed during the current study for EcNF-YA genes are available in the NCBI database under GenBank Accession Number TPA: BK063099-BK063116 and for microRNA BioSample: SAMN03388565; Sample name: Finger millet Drought; SRA: SRS863641 were used. All data generated or analysed during this study are also included in this article (and its supplementary files).
References
Samynathan, R., Venkidasamy, B., Shanmugam, A., Ramalingam, S. & Thiruvengadam, M. Functional role of microRNA in the regulation of biotic and abiotic stress in agronomic plants. Front. Genet. 14, 1–16 (2023).
Brousse, C. et al. A non-canonical plant microRNA target site. Nucleic Acids Res. 42, 5270–5279 (2014).
Raza, A. et al. miRNAs for crop improvement. Plant Physiol. Biochem. 201, 107857 (2023).
Maharajan, T., Palayullaparambil, T. & Krishna, A. Role of genome sequences of major and minor millets in strengthening food and nutritional security for future generations. Agriculture 14(5), 670 (2024).
Maharajan, D., Ceasar, S. A. & Krishna, T. Harnessing the transcriptomic resources of millets to decipher climate resilience and nutrient enrichment traits. CRC. Crit. Rev. Plant Sci. https://doi.org/10.1080/07352689.2024.2354981 (2024).
Ceasar, S. A. The role of millets in attaining United Nation‘s sustainable developmental goals. Plants, People, Planet https://doi.org/10.1002/ppp3.10254 (2022).
Rani, V., Joshi, D. C., Joshi, P., Singh, R. & Yadav, D. “Millet Models” for harnessing nuclear factor-Y transcription factors to engineer stress tolerance in plants: current knowledge and emerging paradigms. Planta 258, 29 (2023).
de Cardoso, T. C. S. et al. New insights into tomato microRNAs. Sci. Rep. 8, 16069 (2018).
Li, Q., Shen, H., Yuan, S., Dai, X. & Yang, C. miRNAs and lncRNAs in tomato: Roles in biotic and abiotic stress responses. Front. Plant Sci. 13, 1–13 (2023).
Sood, S., Joshi, D. C., Kumar, A. & Anil, C. Phenomics and genomics of finger millet : Current status and future prospects. Planta 250, 731–751 (2019).
Maharajan, T., Ceasar, S. A. & Ajeesh Krishna, T. P. Finger millet (Eleusine coracana (L.) Gaertn): Nutritional importance and nutrient transporters. CRC. Crit. Rev. Plant Sci. 41, 1–31 (2022).
Kumar, A. et al. Nutraceutical value of finger millet [Eleusine coracana (L.) Gaertn.], and their improvement using omics approaches. Front. Plant Sci. 7, 1–14 (2016).
Sood, S., Babu, B. K. Joshi, D. History, Botanical and Taxonomic Description, Domestication, and Spread BT The Finger Millet Genome. in (eds. Kumar, A., Sood, S., Babu, B. K., Gupta, S. M. Rao, B. D.) 1–12 (Springer International Publishing). https://doi.org/10.1007/978-3-031-00868-9_1 (2022)
Ceasar, A. Genome-editing in millets: current knowledge and future perspectives. Mol. Biol. Rep. 49, 773–781 (2022).
Usha, S. et al. Identification of microRNAs and their targets in finger millet by high throughput sequencing. Gene 574, 210–216 (2015).
Nageshbabu, R., Jyothi, M. N., Sharadamma, N., Rai, D. V. & Devaraj, V. R. Expression of miRNAs confers enhanced tolerance to drought and salt stress in finger millet (Eleusine coracona). J. Stress Physiol. Biochem. 9, 220–231 (2013).
Sunkar, R., Chinnusamy, V., Zhu, J. & Zhu, J.-K. Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends. Plant Sci. 12, 301–309 (2007).
Zhao, H. et al. The arabidopsis thaliana nuclear factor Y transcription factors. Front. Plant Sci. 7, 1–11 (2017).
Rani, V., Rana, S., Muthamilarasan, M., Joshi, D. C. & Yadav, D. Expression profiling of nuclear factor-Y (NF-Y) transcription factors during dehydration and salt stress in finger millet reveals potential candidate genes for multiple stress tolerance. Planta 259, 136 (2024).
Li, X. Y. et al. Evolutionary variation of the CCAAT-binding transcription factor NF-Y. Nucleic Acids Res. 20, 1087–1091 (1992).
Petroni, K. et al. The promiscuous life of plant NUCLEAR FACTOR Y transcription factors. Plant Cell 24, 4777–4792 (2013).
Kavi, P. B., Showkat, K., Ganie, A., Wani, S. H. & Guddimalli, R. Nuclear Factor - Y ( NF - Y ): Developmental and stress - responsive roles in the plant lineage. J. Plant Growth Regul. https://doi.org/10.1007/s00344-022-10739-6 (2022).
Chakraborty, A., Viswanath, A., Malipatil, R., Rathore, A. & Thirunavukkarasu, N. Structural and functional characteristics of miRNAs in five strategic millet species and their utility in drought tolerance. Front. Genet. 11, 1–15 (2020).
Usha, S. et al. Computational identification of microRNAs and their targets from finger millet (Eleusine coracana). Interdiscip. Sci. – Comput. Life Sci. 9, 72–79 (2017).
Puranik, S. et al. Comprehensive genome-wide survey, genomic constitution and expression profiling of the NAC transcription factor family in foxtail millet (Setaria italica L.). PLoS ONE https://doi.org/10.1371/journal.pone.0064594 (2013).
Suresh, B. V. et al. De novo transcriptome analysis identifies key genes involved in dehydration stress response in kodo millet (Paspalum scrobiculatum L.). Genomics 114, 110347 (2022).
Rani, V., Singh, V. K., Joshi, D. C., Singh, R. & Yadav, D. Genome-Wide identification of nuclear factor -Y (NF-Y) transcription factors family in finger millet reveals structural and functional diversity. Heliyon https://doi.org/10.1016/j.heliyon.2024.e36370 (2024).
Dai, X. & Zhao, P. X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159 (2011).
Chai, J. et al. Bioinformatic identification and expression analysis of banana MicroRNAs and their targets. PLoS ONE 10, 1–15 (2015).
Luan, M. et al. Expression of zma-miR169 miRNAs and their target ZmNF-YA genes in response to abiotic stress in maize leaves. Gene 555, 178–185 (2015).
Bravo-vázquez, L. A. et al. Identification and expression profiling of microRNAs in leaf tissues of Foeniculum vulgare Mill . under salinity stress. Plant Signal. Behav. 19, 2361174 (2024).
Vignesh, D. & Parameswari, P. MiRPI : Portable software to identify conserved miRNAs, targets and to MiRPI : Portable software to identify conserved miRNAs. Targets Calc. Precursor Stats. https://doi.org/10.5808/GI.2011.9.1.039 (2011).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Guo, Z. et al. PmiREN: A comprehensive encyclopedia of plant miRNAs. Nucleic Acids Res. 48, D1114–D1121 (2020).
Gonçalves, J. P., Madeira, S. C. & Oliveira, A. L. BiGGEsTS: Integrated environment for biclustering analysis of time series gene expression data. BMC Res. Notes 2, 1–11 (2009).
Chen, C. et al. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202 (2020).
Reddy, P. S. et al. Comprehensive evaluation of candidate reference genes for real-time quantitative PCR (RT-qPCR) data normalization in nutri-cereal finger millet [Eleusine Coracana (L.)]. PLoS ONE 13, 1–17 (2018).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Shannon, P. et al. Cytoscape: A software environment for integrated models. Genome Res. 13, 426 (1971).
Cao, L. et al. Genome-wide identification of NF-Y gene family in maize (Zea mays L.) and the positive role of ZmNF-YC12 in drought resistance and recovery ability. Front. Plant Sci. https://doi.org/10.3389/fpls.2023.1159955 (2023).
Tan, X. et al. NF-YA transcription factors suppress jasmonic acid-mediated antiviral defense and facilitate viral infection in rice. PLoS Pathog. 18, 1–21 (2022).
Su, H. et al. Dual functions of ZmNF-YA3 in photoperiod-dependent flowering and abiotic stress responses in maize. J. Exp. Bot. 69, 5177–5189 (2018).
Ding, J., Li, D., Ohler, U., Guan, J. & Zhou, S. Genome-wide search for miRNA-target interactions in Arabidopsis thaliana with an integrated approach. BMC Genomics https://doi.org/10.1186/1471-2164-13-S3-S3 (2012).
Sun, F. et al. Whole-genome discovery of miRNAs and their targets in wheat (Triticum aestivum L.). BMC Plant Biol. 14, 142 (2014).
Zhu, Q.-H. et al. A diverse set of microRNAs and microRNA-like small RNAs in developing rice grains. Genome Res. 18, 1456–1465 (2008).
Tang, Q. et al. Characteristics of microRNAs and target genes in maize root under drought stress. Int. J. Mol. Sci. https://doi.org/10.3390/ijms23094968 (2022).
Yi, F., Xie, S., Liu, Y., Qi, X. & Yu, J. Genome-wide characterization of microRNA in foxtail millet (Setaria italica). BMC Plant Biol. https://doi.org/10.1186/1471-2229-13-212 (2013).
Devos, K. M. et al. Genome analyses reveal population structure and a purple stigma color gene candidate in finger millet. Nat. Commun. https://doi.org/10.1038/s41467-023-38915-6 (2023).
Zhao, B. et al. Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Mol. Biol. https://doi.org/10.1186/1471-2199-10-29 (2009).
Xie, Z. et al. Expression of arabidopsis MIRNA genes. Plant Physiol. 138, 2145–2154 (2005).
Sorin, C. et al. A miR169 isoform regulates specific NF-YA targets and root architecture in arabidopsis. New Phytol. 202, 1197–1211 (2014).
Yu, C. et al. Overexpression of miR169o, an overlapping MicroRNA in response to both nitrogen limitation and bacterial infection, promotes nitrogen use efficiency and susceptibility to bacterial blight in rice. Plant Cell Physiol. 59, 1234–1247 (2018).
Rani, V., Singh, V. K., Joshi, D. C., Singh, R. & Yadav, D. Molecular docking insights into nuclear factor Y (NF-Y) transcription factor and pyrabactin resistance 1 (PYL) receptor proteins reveal abiotic stress regulation in finger millet. Crop Des. 3, 100051 (2024).
Choudhary, A., Kumar, A., Kaur, H. & Kaur, N. MiRNA: the taskmaster of plant world. Biologia (Bratisl). 76, 1551–1567 (2021).
Prabu, G. R. & Mandal, A. K. A. Computational identification of miRNAs and their target genes from expressed sequence tags of tea (Camellia sinensis). Genom., Proteom. Bioinform. 8, 113–121 (2010).
Zhang, B. H., Pan, X. P., Cox, S. B., Cobb, G. P. & Anderson, T. A. Evidence that miRNAs are different from other RNAs. Cell. Mol. Life Sci. 63, 246–254 (2006).
Bhavsar, M., Mangukia, N., Patel, S., Rawal, R. & Mankad, A. Unraveling the miRnome of Nicotiana rustica (Aztec tobacco) - A Genomewide computational assessment. Plant Gene 32, 100378 (2022).
Xing, H. et al. Genome-wide investigation of microRNAs and expression profiles during rhizome development in ginger (Zingiber officinale Roscoe). BMC Genomics 23, 1–13 (2022).
Lotfi, A. et al. Role of microRNAs and their target genes in salinity response in plants. Plant Growth Regul. 82, 377–390 (2017).
Maheshwari, P. et al. Genome-wide identification and expression profile analysis of nuclear factor Y family genes in Sorghum bicolor L. (Moench). PLoS ONE 14, 1–27 (2019).
Petrov, V. et al. Identification of cis-regulatory elements specific for different types of reactive oxygen species in arabidopsis thaliana. Gene 499, 52–60 (2012).
Doornbos, R. F., Geraats, B. P. J., Kuramae, E. E., Van Loon, L. C. & Bakker, P. A. H. M. Effects of jasmonic acid, ethylene, and salicylic acid signaling on the rhizosphere bacterial community of arabidopsis thaliana. Mol. Plant. Microbe Interact. 24, 395–407 (2011).
Rivas-San, M. V. & Plasencia, J. Salicylic acid beyond defence: its role in plant growth and development. J. Exp. Bot. 62, 3321–3338 (2011).
Lu, L. et al. Identification and characterization of Csa-miR395s reveal their involvements in fruit expansion and abiotic stresses in cucumber. Front. Plant Sci. 13, 1–12 (2022).
Li, W. X. et al. The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 20, 2238–2251 (2008).
Yu, Y. et al. Overexpression of soybean miR169c confers increased drought stress sensitivity in transgenic Arabidopsis thaliana. Plant Sci. 285, 68–78 (2019).
Luan, M. et al. Family-wide survey of miR169s and NF-YAs and their expression profiles response to abiotic stress in maize roots. PLoS ONE 9, e91369 (2014).
Acknowledgements
The financial support by the U.P. Council of Agriculture Research (UPCAR), Lucknow India (1241/06/RG/Millet/2023-24, dated 19/12/2023 to DY and RG is sincerely acknowledged. The authors also wish to acknowledge the Head, Department of Biotechnology, D.D.U Gorakhpur University, Gorakhpur for providing the infrastructural facilities.
Author information
Authors and Affiliations
Contributions
VR designed the analysis, performed the research work, and drafted the manuscript. SR helped in the experimental setup and analysis of qRT-PCR. MM verified the result and improved the language of the manuscript. DC-J reviewed and edited the manuscript, and improved the language of the manuscript. RG and RS edited the manuscript. DY conceived the idea of research work, supervised, and finalized the final draft of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rani, V., Rana, S., Muthamilarasan, M. et al. Identification and characterization of Eco-miR 169-EcNF-YA13 gene regulatory network reveal their role in conferring tolerance to dehydration and salinity stress in finger millet. Sci Rep 15, 12338 (2025). https://doi.org/10.1038/s41598-025-96233-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-96233-x
