
Abstract
Cancer-related pain is prevalent and severely impairs patients’ quality of life. However, conventional cancer therapies primarily target tumor cell destruction, often overlooking the management of cancer pain. Thus, there is an immediate necessity to develop therapeutic agents that can both suppress tumor growth and alleviate cancer pain. In this study, we report a celastrol (CEL)-based nanocomposites (PDA-BSA-MnO2-CEL) for pain-less cancer immunotherapy. Results from in vitro and in vivo experiments demonstrate the efficacy and mechanism of the nanocomposites in pain-less immunotherapy. MnO2 and CEL induce immunogenic cell death (ICD), mediating immunotherapy. Additionally, CEL significantly reduces the secretion of the immunosuppressive factor Yes-associated protein (YAP) within the tumor microenvironment, thereby enhancing the efficacy of immunotherapy. The downregulation of YAP leads to reduced expression of vascular endothelial growth factor (VEGF), inhibiting tumor growth and decreasing activation of the pain-associated VEGF receptor 1 (VEGFR1), thus providing an analgesic effect. Moreover, CEL reduces inflammatory pain by lowering levels of inflammatory factors in tumors. The design of this nanocomposites system integrates immunotherapy with cancer pain inhibition, offering a novel approach to patient-centered tumor therapy.
Similar content being viewed by others

Introduction
Cancer represents a substantial threat to human health, owing to its high mortality rate1,2. Cancer-related pain is a common complication that severely impacts patients’ quality of life. Therefore, the development of treatment agents capable of simultaneously inhibiting tumor growth and alleviating cancer pain is essential1,3,4. Immunotherapy is considered one of the most promising therapeutic approaches for tumors, owing to its high efficacy, minimal side effects, and potential to stimulate both innate and adaptive immunity5,6,7,8,9,10,11,12,13,14. However, the presence of immunosuppressive factors within the tumor microenvironment hinders adequate T lymphocyte (T cell) infiltration, limits tumor antigens, disrupts antigen presentation, and results in suboptimal T cell activation in the tumor region15,16. Thus, the key to effective immunotherapy lies in activating the immune response while simultaneously eliminating immunosuppressive factors present in the tumor microenvironment17,18.
Yes-associated protein (YAP) is recognized as an immunosuppressive factor that contributes to immune evasion and promotes tumor metastasis. Therefore, down-regulating YAP may alleviate immunosuppression and enhance the efficacy of immunotherapy19,20,21,22. Additionally, vascular endothelial growth factor (VEGF), a crucial mediator in facilitating tumor growth and metastasis, shows a positive correlation with YAP23,24 The down-regulation of YAP reduces VEGF expression within the tumor microenvironment, thereby inhibiting tumor growth. Furthermore, as previously reported, the activation of VEGFR1 (a VEGF-related receptor) can result in cancer pain through the facilitation of abnormal neurodevelopment in tumor regions and activation of the pain receptor TRPV125,26,27,28. Therefore, inhibiting YAP in the tumor microenvironment can not only mitigate immunosuppression but also reduce VEGFR1 activation by decreasing VEGF expression, ultimately enhancing immunotherapy and alleviating cancer pain.
Celastrol (CEL), a quinone methyl triterpenoid compound derived from the root bark of Tripterygium wilfordii, has attracted significant attention for its diverse pharmacological activities, including anti-inflammatory, neuroprotective, and antitumor effects. Modern pharmacological studies have particularly highlighted CEL’s broad-spectrum anticancer activity across various cancer types29,30. Moreover, recent research has identified CEL as a natural inhibitor of YAP, effectively reducing YAP expression in the tumor microenvironment31. However, the intrinsic properties of CEL, such as limited water solubility and reduced bioavailability, significantly hinder its therapeutic efficacy in cancer treatment32,33. Therefore, incorporating CEL into an appropriate nano-delivery system may enhance its effectiveness.
Polydopamine (PDA)/bovine serum albumin (BSA)/manganese dioxide composite nanoparticles (PM) are regarded as a promising drug delivery platform for cancer treatment. The presence of hydrophobic cavities in BSA facilitates the effective loading of hydrophobic drugs34. CEL and MnO2 can induce immunogenic cell death (ICD) in tumor cells while also eliciting a sustained anti-tumor immune response35,36. PDA boasts remarkable biocompatibility and adhesive properties, significantly enhancing the stability of these composite nanoparticles (NPs)37.
In this study, we developed a multifunctional nanoplatform (PDA-BSA-MnO2-CEL, PM-CEL) aimed at simultaneously inhibiting tumor growth and alleviating cancer pain through the encapsulation of CEL within PDA-BSA-MnO2 (PM) composite NPs. A cancer pain model was established for evaluation purposes. Upon intratumoral injection, PM-CEL decomposes within the tumor microenvironment, leading to the release of CEL. Both CEL and manganese oxide induce ICD and promote the apoptosis of tumor cells38,39. Concurrently, CEL reduces the levels of YAP and pro-inflammatory cytokines, including tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6), in the tumor microenvironment, thus alleviating immunosuppression and local inflammation. Furthermore, CEL has been demonstrated to downregulate the expression of VEGF, thereby mitigating hyperalgesia and achieving sustained analgesic effects. This innovative approach offers a new perspective for future patient-centered tumor therapies1,2.
Materials and methods
Materials
All experimental materials utilized in this study are provided in the Supplementary Information section.
Cell culture
HEK293T cells are human embryonic kidney cells 293 purchased from the ATCC Cell Bank of the Chinese Academy of Sciences, Shanghai. The 4T1 (mouse breast cancer) cell line used in this experiment was from Procell Co. Ltd (Wuhan, China). 293T cells were cultured in DMEM medium (GIBCO), whereas 4T1 cells were cultured in RPMI 1640 medium (GIBCO). All the cells were incubated at 37 °C with 5% CO2 with the above-mentioned media supplemented with fetal bovine serum (10%, BI), penicillin (100.0 U·mL-1), and streptomycin (100.0 µg·mL-1).
Synthesis and characterisation of PM-CEL NPs
PM and PM-CEL were prepared according to a previously reported method with minor modifications37,40. KMnO4 was used as the oxidizing agent, BSA served as the template, and PM was synthesized by oxidizing dopamine in the presence of KMnO4. Initially, PM NPs were prepared. Briefly, 2 mL of aqueous KMnO4 solution (15 mg/mL) was added dropwise to 100 mL of pure water containing 200 mg of BSA and 100 mg of dopamine. The mixture was magnetically stirred for 2 h at room temperature. Subsequently, the PM NPs were purified by centrifugation at 5000 rpm for 5 min. For CEL loading, 3 mg of CEL was dissolved in 1 mL of anhydrous ethanol. This solution was then mixed with 1 mL of a 3 mg/mL PM aqueous solution and magnetically stirred for 24 h at room temperature. The resultant products were obtained by centrifugation (5000 rpm for 5 min) and washed twice with anhydrous ethanol. Finally, the synthesized PM and PM-CEL were stored at 4 °C for subsequent experiments.
Drug release of PM-CEL
PM-CEL (containing 2.0 mg of CEL) was dissolved in 10 mL of phosphate-buffered saline (PBS) at pH values of 7.4 and 5.5, respectively. The solutions were then incubated with continuous stirring at 37 °C. At specified time points, the supernatant was collected following centrifugation, after which an equivalent volume of PBS was added. UV-vis spectrophotometry was utilized to quantify the release of CEL.
Assessment of cellular internalization efficiency
Rhodamine B (RhB)-labeled PM-CEL NPs were employed to investigate cellular uptake in 4T1 cells. The detailed procedures are as follows: 1 mL of RhB-PM-CEL NPs at a concentration of 2 µg/mL were co-cultured with 4T1 cells for 0.5, 2, and 4 h, respectively. The medium was subsequently removed, and the cells were washed three times with PBS. To fix the cells, 4% paraformaldehyde was added for 15 min, followed by the addition of 150 µL of 5 mg/mL DAPI solution for staining. Images were captured using confocal laser scanning microscopy (CLSM).
In vitro cytotoxicity assessment
The efficacy of PM, CEL, and PM-CEL in inhibiting tumor growth in vitro was assessed in 4T1 cells. The MTT assay was performed according to standard protocols. In summary, 4T1 cells were cultured in the presence of PM, CEL, and PM-CEL. After 24 h, MTT was added and the cells were incubated for an additional 4 h. Cell cytotoxicity was quantified using a microplate reader (model DMI3000, Leica Microsystems, Germany). A similar methodology was employed to evaluate the viability of 293T cells, Macrophages and DCs. Live/dead staining experiments were conducted for a more intuitive assessment of cell viability. 4T1 cells, seeded in 6-well plates, were incubated with PBS, PM, CEL, and PM-CEL for 24 h. After three washes with PBS, the cells were stained with Calcein-AM/PI, and their viability was assessed using a fluorescence microscope (Leica DM IL LED, Germany). Apoptosis in the cells was determined using flow cytometry, with a detailed procedure provided in the Supporting Information section.
Evaluation of mitochondrial damage
Mitochondrial damage was evaluated by detecting changes in mitochondrial membrane potential. Briefly, 4T1 cells at a density of 2.0 × 105 cells were seeded in a confocal dish and incubated for 24 h. The medium was then replaced with 2.0 mL of FBS-DMEM supplemented with PBS, PM, CEL, and PM-CEL, respectively. Following a 12-hour incubation, the cells were rinsed three times with PBS and subsequently stained with a solution of 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) (10.0 µg/mL). Cellular images were acquired using CLSM.
ICD assay in vitro
The overexpression of calreticulin (CRT) was evaluated through immunofluorescence staining. 4T1 cells were incubated with PM-CEL for 24 h, followed by washing with PBS-Tween 20 (PBST) and fixation with 4% formaldehyde. The cells were then incubated with 5% FBS for 30 min. Subsequently, a rabbit anti-CRT antibody was applied and incubated at 4 °C for 12 h. The cells were then co-stained with DyLight 549 and DAPI. CLSM was utilized to assess the expression level of CRT41.
HMGB1 and ATP release levels were quantified using the HMGB1 ELISA kit and the chemiluminescence ATP kit. In summary, PM-CEL was incubated with 4T1 cells for 24 h. Following this, the expression levels of HMGB1 and ATP were determined according to the manufacturer’s protocols for the HMGB1 and ATP kits, respectively.
Western blot analysis
Western blot (WB) analysis was conducted to evaluate the secretion levels of YAP, VEGF, and VEGFR1 in vivo. After the behavioral experiments, the mice were deeply anesthetized with 0.3% sodium pentobarbital via intraperitoneal injection. Subsequently, the tumors were excised and homogenized. The expression levels of YAP, VEGF, and VEGFR1 in the samples were subsequently analyzed using WB techniques.
Evaluation of antitumor and immune responses in vivo
Female BALB/c mice (4–5 weeks old) were purchased from Pengyue Co., LTD. (Jinan, China). 4T1 cells (5.0 × 106 cells suspended in 20.0 µL of 1640 medium) were administered into the hind paw of 4- to 5-week-old BALB/c mice to establish a cancer pain model. When the tumors reached an approximate volume of 100.0 mm³, the mice were randomly assigned into four groups, each containing five mice: PBS (100.0 µL), PM (165 µg in 100.0 µL of PBS), CEL (45 µg in 100.0 µL of PBS), and PM-CEL (210 µg of PM and 45 µg of CEL). Treatments were delivered through intratumoral injections. Body weight and tumor size of the mice were measured every other day throughout the 15-day treatment period using an electronic scale and a vernier caliper. A comprehensive measurement methodology can be found in the Supplementary Materials.
Upon completion of the treatment, euthanasia was performed via cervical dislocation. Subsequently, tumor tissues and major organs (heart, spleen, liver, kidney, and lung) were harvested for hematoxylin and eosin (H&E) staining.
To evaluate T cell infiltration, the single-cell of spleen was prepared using mechanical pulverization, and mixed with anti-CD3-APC, anti-CD4-PE, and anti-CD8-FITC antibodies for staining. The results were analyzed via flow cytometry. Additionally, the percentage of regulatory T cells (Tregs) was determined through flow cytometry after staining splenic single-cell suspensions with anti-CD25-PE, anti-CD4-FITC, and anti-FOXP3 antibodies. To assess dendritic cell (DC) maturation status after various treatments, single-cell suspensions isolated from lymph nodes were stained with anti-CD80-PE/Cy5.5, anti-CD11C-PE, and anti-CD86-APC antibodies for subsequent flow cytometric analysis. A similar flow cytometry was employed to tumor tissues.
For tumor immunohistochemistry, tumor tissue slices were first incubated overnight at 4 °C with primary antibodies against FOXP3, CD8, and CD4. Following this, biotin-conjugated secondary antibodies were added and incubated for one hour. After staining with 3,3’-diaminobenzidine (DAB) and hematoxylin, the sections were analyzed under a microscope.
All animal procedures were conducted in strict adherence to the guidelines outlined in the National Research Council’s Guide for the Care and Use of Laboratory Animals. The study received approval from the Institutional Animal Care and Use Committee (IACUC) at Binzhou Medical University, under protocol number BZMU-2022-096. This study adheres to the ARRIVE guidelines for reporting in vivo experiments.
Pain behavior evaluations
Mice were administered treatments on alternate days over a 15-day period, with the maximum withdrawal time (MWT) and tail withdrawal latency (TWL) assessed 24 h after each treatment. Detailed procedures for behavioral testing are provided in the supplementary information.
Immunofluorescence assay
To examine pain-related factors, tumor sections were stained with antibodies targeting VEGF, VEGFR1, TRPV1, and PGP9.5, as well as rabbit anti-mouse IgG conjugated to FITC. To examine the in vivo mechanisms of ICD, tumor sections were stained using antibodies to CRT and HMGB1, as well as FITC-conjugated rabbit anti-mouse IgG. Fluorescence imaging was performed using an inverted microscope. Comprehensive experimental procedures can be found in the Supporting information.
Statistical analysis
Each experiment was performed at least three times. Data analysis was conducted using Origin 2019b 64-bit software. Numerical results are reported as the mean ± standard deviation (SD). The Student’s t-test was utilized to assess statistical significance, with the following notation for p-values: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Results and discussion
Synthesis and characterization of PM NPs
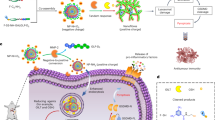
Figure 1 illustrates the preparation procedure of PM-CEL and its mechanisms for antitumor effects and alleviation of cancer pain. The multifunctional PM NPs (PM and PM-CEL) were synthesized according to a previously reported strategy37. Transmission electron microscopy (TEM) confirmed the spherical shape and excellent dispersion characteristics of the PM NPs. As shown in Fig. 2a, the average diameters of the PM and PM-CEL NPs were measured to be 53 ± 6.03 nm and 180.8 ± 14.02 nm, respectively.

Schematic diagram of the fabrication procedure for PM-CEL and the mechanism involved in anti-tumor growth and cancer pain inhibition.

TEM images of PM and PM-CEL (a). Elemental mappings (b) and EDS analysis (c) of PM-CEL. (d) Survey XPS spectrum of PM. Particle size (e) and zeta potential (f) of PM and PM-CEL. (g) UV-vis spectra of PM, CEL, and PM-CEL. (h) Release profiles of PM-CEL at pH 5.5 and 7.4. Data are presented as mean ± SD (n = 3).
The C, N, O, and Mn elements exhibited a uniform distribution within the PM-CEL, as illustrated in Fig. 2b. Additionally, the elemental composition of the NPs was quantitatively analyzed using EDS, with the results presented in Fig. 2c. The elemental composition of the PM was further assessed using XPS. As shown in Fig. 2d, the findings confirm the presence of C, O, N, and Mn elements in the PM NPs.
Dynamic light scattering (DLS) was employed to determine the particle sizes of PM and PM-CEL. As depicted in Fig. 2e, the average diameters were found to be 60.3 ± 2.11 nm and 200 ± 15.44 nm, respectively. The observed increase in particle size for PM-CEL can likely be attributed to the efficient encapsulation of CEL. Furthermore, the zeta potentials for PM and PM-CEL were − 14.33 ± 0.57 mV and − 18.67 ± 0.31 mV, respectively. The non-significant zeta potential change (Fig. 2f) may be attributed to the fact that most CEL molecules are incorporated into the inner cavity without being adsorbated to the surface of PM42.
In addition, UV spectroscopy was employed to quantitatively assess drug loading efficiency (DLE). As shown in Fig. 2g, an absorption peak at 425 nm, attributed to CEL, was observed in PM-CEL, confirming the successful incorporation of CEL into PM. The drug loading efficiency (DLE) and drug loading content (DLC) were determined to be 70.9% and 21.6%, respectively. Detailed methodologies for these calculations are provided in the Supporting Information.
To characterize the combination of CEL and PM, Fourier Transform Infrared (FTIR) spectroscopy analyses were performed. The FTIR analysis of PM-CEL revealed the presence of a hydroxyl group vibration at 3300 cm− 1, characteristic of PDA, and the amide II band vibration at 1540 cm− 1, characteristic of BSA. This indicates the successful incorporation of PDA and BSA into PM-CEL. Additionally, the characteristic peak of CEL at 1450 cm− 1 was retained in PM-CEL, further confirming the successful introduction of CEL (Figure S1)43.
The stability of NPs is crucial for ensuring the safety and efficacy of therapeutic applications. We evaluated the stability of PM-CEL NPs in FBS, medium, and PBS at 37 °C. As presented in Figure S2, the NPs exhibited no significant changes in size over a 48-hour period in all solutions, indicating the absence of agglomeration or decomposition. These results verify the excellent stability of the NPs. To assess the release behavior of CEL from PM-CEL, the NPs were immersed in PBS at pH 7.4 or 5.5 and maintained at 37 °C for 24 h. The amount of released CEL was then quantified using UV-vis spectrophotometry. As illustrated in Fig. 2h, CEL demonstrated the fastest release rate at pH 5.5, with a cumulative release of 70.15% within only 6 h, which was significantly higher compared to the release at pH 7.4. These findings suggest efficient release of PM-CEL in an acidic tumor microenvironment44.
Evaluation of antitumor efficacy in vitro
Efficient cellular internalization is crucial for antitumor efficacy. Therefore, the cellular uptake of RhB-labeled PM-CEL in 4T1 cells was first evaluated. When PM-CEL (CEL concentration of 2 µg·mL− 1) was co-incubated with 4T1 cells for 0.5, 2, and 4 h, the intracellular red fluorescence intensity increased progressively with longer incubation times. These results demonstrate that the cellular uptake of the NPs is time-dependent (Fig. 3a).

(a) Cellular uptake images of 4T1 cells following treatment with PM-CEL for 0.5, 2, and 4 h. (b) Cell viability of 293T incubated with PM-CEL for 24 h. (c) Cell viability, (d) dead/living cell fluorescence, and (e) apoptosis analysis of 4T1 cells after being treated with PM, CEL and PM-CEL for 24 h. (*p < 0.05, **p < 0.01 and ***p < 0.001)
Building on these findings, a comprehensive in vitro antitumor evaluation of the PM NPs was conducted. The biosafety of PM-CEL was assessed using a cell viability assay after a 24-hour co-incubation of PM-CEL with normal renal epithelial cells (293T). As shown in Fig. 3b, the cell survival rate remained high at 88.25 ± 7.2% when the CEL concentration reached 5 µg/mL. MTT assay was then performed to evaluated the biosafety of PM-CEL on immune cells after incubation with mouse mononuclear macrophages (RAW264.7) and dendritic cells (DCs) for 24 h. As showed in Figure S3, even PM-CEL concentration was as high as 5 µg/mL, the survival rate of both cells remained above 89 ± 7.3%%, confirming the excellent biocompatibility of PM-CEL to immune cells. The antitumor efficacy of PM-CEL was evaluated using the MTT assay. As illustrated in Fig. 3c, PM-CEL (13.99 ± 2.04%) exhibited a greater inhibitory effect on 4T1 cell viability than free CEL (15.23 ± 22.12%) and was significantly more effective than the PM group (88.59 ± 1.92%). This suggests that the enhanced antitumor activity of PM-CEL is attributable to the potent activity of CEL. The selective killing mechanism of PM-CEL on 4T1 cancer cells may be attributed to several factors. Since microtubules in cancer cells are in a highly dynamic state relative to normal cells, Celastrol damages cells by disrupting tubulin heterodimers.The hyperdynamic microtubules of the tumor were more sensitive to Celastrol45. In addition, another potential reason could be attributed to the acid-responsive degradation behavior of PM-CEL. Within the acidic tumor microenvironment, PM-CEL undergoes degradation to release CEL, which exerts its cytotoxic effects by damaging mitochondria and inducing apoptosis in tumor cells46. Considering tumor cell killing and biosafety, we finally selected the concentration of PM-CEL as 9.5 µg/mL (CEL: 2 µg/ml) for subsequent experiments.
For a more visual assessment of cell viability, a live/dead staining assay was performed. Calcein-AM (green) and propidium iodide (PI) (red) were used to stain live and dead cells, respectively. As shown in Fig. 3d, the PM-CEL group exhibited significantly brighter red fluorescence compared to the other groups, further confirming the superior antitumor efficacy of PM-CEL.
Additionally, cellular apoptosis was quantitatively analyzed using Annexin V-FITC/PI staining. As shown in Figs. 3e and S4, the apoptosis rates were 1.91%, 9.82%, 37.87%, and 41.14% for the PBS, PM, CEL, and PM-CEL groups, respectively. These results corroborate the findings from the MTT and live/dead cell staining assays. To evaluate the potential of PM-CEL as an immunotherapeutic agent, YAP expression was measured. ELISA analysis revealed that PM-CEL significantly downregulated YAP expression in 4T1 cells compared to other groups (Figure S5). This observation aligns with previous reports indicating that CEL inhibits YAP expression47.
These results suggest that PM-CEL not only demonstrates potent tumor inhibition but also has the potential to serve as an immunotherapeutic agent by reducing the immunosuppressive factor YAP within the tumor microenvironment.
Evaluation of mitochondrial membrane potential
Mitochondrial damage is known to induce ICD48,49. CEL is capable of targeting mitochondria, leading to mitochondrial dysfunction50. To determine if PM-CEL similarly affects mitochondrial integrity, the mitochondrial membrane potential was evaluated using the JC-1 staining method.
JC-1 is a widely recognized marker for mitochondrial membrane potential, forming aggregates and emitting red fluorescence in healthy cells. In cells with damaged mitochondria, JC-1 exists as monomers, producing green fluorescence51. In the control group, cells exhibited intense red fluorescence, with no significant green signal detected (Fig. 4a). In contrast, PM-CEL-treated cells showed a marked reduction in red fluorescence intensity and a substantial increase in green fluorescence intensity. Quantitative analysis revealed that the green/red fluorescence ratio in the PBS group was 0.22, which increased to 1.06 after treatment with PM-CEL (Figure S6). These findings indicate that PM-CEL accumulates in mitochondria and induces mitochondrial dysfunction.

(a) Mitochondrial membrane potentials of 4T1 cells after incubated with PM, CEL and PM-CEL for 12 h. (b) CRT release and (c) mean fluorescence intensity (MFI) from 4T1 tumor cells after various treatments. ELISA analysis of intracellular release of (d) ATP and (e) HMGB1. (*p < 0.05, **p < 0.01 and ***p < 0.001)
DAMP release in vitro
To examine anti-tumor immune activation following various treatments, we assessed ICD by measuring damage-associated molecular patterns (DAMPs) in vitro. DAMPs are integral to the induction of ICD, with specific molecules such as CRT, adenosine triphosphate (ATP), and high mobility group box 1 (HMGB1) facilitating DC maturation and enhancing antigen presentation by exposing these markers on the surface of tumor cells. Notably, CRT exposure serves as a crucial mediator of immunogenicity, delivering a strong “phagocytose me” signal52.
As demonstrated in Fig. 4b and c, the quantity of CRT secreted from the cell surface in the CEL and PM-CEL groups was significantly higher compared to those in the PBS and PM groups. This result can be attributed to CEL’s potent ability to induce ICD. Remarkably, the CRT levels in the PM-CEL group exhibited the highest degree of CRT exposure, likely due to the enhancement of ICD induction by Mn2+ ions. ATP released from apoptotic cells triggers DCs to generate cytokines53,54,55, while HMGB1 released from dying cells can elicit an inflammatory response, recruit a variety of immune cells, and promote DC maturation56,57. The release levels of ATP and HMGB1 in the cells were measured using ATP and HMGB1 ELISA kits.
As illustrated in Fig. 4d and e, the intracellular levels of HMGB1 and ATP were significantly reduced in the CEL and PM-CEL groups compared to the PBS and PM groups, with PM-CEL displaying the most substantial decrease. These findings confirm that both CEL and PM-CEL can effectively induce ICD, suggesting that PM-CEL can provoke intense ICD in 4T1 cells through mitochondrial damage, thereby triggering an anti-tumor immune response.
Tumor growth Inhibition in vivo
We developed a cancer-related pain mice model to assess tumor inhibition and the efficacy of pain relief from PM NPs in vivo58,59. Subsequently, tumor therapy experiments were conducted following the established experimental protocol (Fig. 5a). Treatments with PBS, PM, CEL, and PM-CEL were administered once the tumor volume increased to 100.0 mm3.
The tumor size was measured with vernier calipers every two days. As depicted in Fig. 5b, the PM-CEL group exhibited the most pronounced inhibitory effect on tumor growth. In contrast, the PM and CEL groups demonstrated only a modest antitumor effect.
Upon completion of treatment, the mice were euthanized, and the tumors were subsequently removed, photographed, and weighed (Fig. 5c). Experimental results indicated that PM-CEL achieved the most significant tumor growth inhibition, with tumor size representing only 22.2% of that in the PBS group (Fig. 5d). Furthermore, histological analysis revealed notable nuclear shrinkage and reduced cell numbers in the PM-CEL group compared to the PM, CEL, and PBS groups, suggesting an increased incidence of apoptosis or necrosis (Fig. 5e). These findings validate the optimal antitumor efficacy of PM-CEL.

Assessment of tumor growth inhibition of PM NPs in vivo. (a) Standardized protocol for tumor therapy experimentation. (b) Tumor growth curves. (c) Tumor photos (d) Tumor weights, and (e) H&E staining images after various treatments. (*p < 0.05, **p < 0.01 and ***p < 0.001)
PM-CEL induced immune response
To evaluate the immune activation capability of PM-CEL, we initially detected DAMP expression in tumor tissues using immunofluorescence staining. The results demonstrated that CRT and HMGB1 levels were elevated to varying extents across different treatment groups. Specifically, the PM-CEL group exhibited a twofold enhancement in CRT fluorescence intensity and a marked increase in HMGB1 expression relative to the control group.(Figure S7). Subsequently, we collected splenic lymphocytes from mice subjected to various treatments. The percentage of CD8+ cytotoxic T lymphocytes (CTL) in the PM group (12.9%) was significantly higher than in the PBS group (10.8%). The proportions in the CEL and PM-CEL groups were 24.3% and 28.7%, respectively. A similar increase in CD4+ T cells was observed (Fig. 6a). These results indicate a notable immune activation effect in the CEL and PM-CEL groups, as evidenced by the substantial increases in both CD4+ and CD8+ T cell populations60.

(a) Flow cytometric analysis of CD4+ and CD8+ T cells in the spleen, and (b) Tregs identified by CD4+, CD25+, and FOXP3+ markers. (c) Mature DCs, defined by the expression of CD11c, CD80, and CD86, within the draining lymph nodes. (d) Immunohistochemical staining for CD4+ T cells and (e) CD8+ T cells, along with (f) FOXP3+ Tregs, within the tumor microenvironment.
The mechanism of immune activation was further investigated by assessing YAP levels in tumor tissue. As shown in Figure S8, the CEL group exhibited significantly reduced YAP concentrations compared to the PBS and PM groups. The PM-CEL group showed the lowest YAP levels, suggesting a more pronounced effect.
Reduced YAP expression in the tumor microenvironment is known to decrease the number of immunosuppressive Treg cells, thereby enhancing the efficacy of immunotherapy22,61,62,63,64. To assess Treg cell populations, flow cytometry was performed to measure the expression of CD25 and FOXP3 on CD4+ T cells isolated from spleen tissue. The levels of Treg cells in the PM-CEL group was 2.74%, which was onbiously lower than that in both the PBS group (11%) and the PM group (11.2%) (Fig. 6b).
Additionally, the levels of mature DCs in tumor-draining lymph nodes were analyzed. The PM-CEL group exhibited a significantly higher proportion of mature DCs (28.9%) compared to the PBS (4.88%) and PM groups (6.27%) (Fig. 6c). Mature DCs enhance CD4+ and CD8+ T lymphocyte activation through antigen presentation, further supporting the immune activation observed with PM-CEL treatment. Statistical analysis confirmed the effective activation of mature DCs, CD4+ T cells, and CD8+ T cells in both the draining lymph nodes and spleen (Figure S9).
These findings were further confirmed by immunohistochemistry (IHC) of tumor sections as well as flow cytometry (FCM) analysis of the tumor area. The PM-CEL group showed the highest levels of CD4+ and CD8+ T cell infiltration while demonstrating the lowest levels of FOXP3+ Treg cell infiltration. The result indicate that PM-CEL effectively activates the immune system, thereby contributing to tumor inhibition (Fig. 6d-f, Figure S10 and Figure S11).
Analgesic evaluation
The analgesic efficacy of PM-CEL was evaluated using mechanical and thermal hyperalgesia tests47,65,66. The experimental protocol is depicted in Fig. 5a. Treatments were administered once the tumor volume reached 100.0 mm3. MWT and TWL were measured to assess pain levels after various treatments. In the model group, MWT and TWL decreased to approximately 5 g and 8s, respectively, compared to the Normal group (9 g, 13s), indicating the induction of hyperalgesia (Fig. 7a, b).
Following treatment, MWT and TWL were assessed 4 h post-treatment. As shown in Fig. 7a and b, the MWT and TWL in the PM group were similar to those in the PBS group. In contrast, the CEL and PM-CEL groups showed significantly higher MWT and TWL compared to the PBS and PM groups, demonstrating the effective cancer pain inhibition of CEL29.
However, the effect in the CEL group diminished after approximately eight days, likely due to the poor water solubility of free CEL, resulting in reduced bioavailability. Notably, in the PM-CEL group, the sensitivity to thermal and mechanical stimulation continued to decrease, and both MWT and TWL remained elevated. These results indicate that PM-CEL provides more sustained analgesic effects compared to other treatments, offering effective pain relief.

Behavioural test and analysis of analgesic mechanisms. The curves of the mechanical withdrawal threshold (a) and thermal withdrawal latency (b) throughout the treatment period. Measurements were taken 8 h post-treatment after each treatment. ELISA assays of cytokine levels: (c) IL-6, (d) TNF-α. (e) Immunofluorescent staining of VEGF, VEGFR1 and PGP9.5 in the tumor after treatment. (f) Western blot assay of YAP, VEGF, and VEGFR1 proteins in tumor following the completion of treatment. (*p < 0.05, **p < 0.01 and ***p < 0.001).
Evaluation of analgesic mechanisms
To assess the mechanisms underlying the analgesic effects of PM-CEL, we measured the levels of several inflammation-related cytokines, including IL-6 and TNF-α, in tumor tissues following various treatments. The contents of these cytokines were obviously reduced in the PM-CEL group (Fig. 7c, d), indicating the effectiveness of PM-CEL in modulating the tumor inflammatory microenvironment.
Tumor-derived VEGF is known to increase pain sensitivity by activating VEGF receptor 1 (VEGFR1), which is present on sensory neurons23,24,25,26. Overexpression of VEGF in the tumor region binds to VEGFR1, leading to the sensitization of peripheral pain receptors, including the sprouting of peripheral sensory nerves23,24,25,26,64,65. To investigate the mechanism by which PM-CEL modulates cancer pain, immunofluorescence analysis was performed in the tumor area to evaluate the levels of Protein Gene Product 9.5 (PGP9.5, a neuronal and nerve fiber marker), VEGF, and VEGFR1. As shown in Fig. 7e and Figure S12, the PM-CEL group exhibited lower protein expression levels of both VEGF and VEGFR1 compared to the other groups, accompanied by a reduction in the number of corresponding nerve fibers. Furthermore, WB analysis confirmed that the PM-CEL group had lower expression levels of YAP, VEGF, and VEGFR1 in tumor tissues compared to other groups (Fig. 7f, S13). Consistent trends were observed in behavioral assessments. These results suggest that PM-CEL inhibits VEGFR1-mediated peripheral nerve sensitization by reducing VEGF expression in the tumor microenvironment, contributing to the alleviation of cancer pain.
Biosafety assessment
To evaluate the biosafety of PM-CEL, major organs, including the heart, liver, kidney, lung, and spleen, were harvested after treatment. Histological analysis of these organs revealed no significant pathological changes in the PM-CEL group, indicating normal cellular morphology and confirming its good biocompatibility (Figure S14).
Blood compatibility was assessed through in vitro hemolysis assays, with PBS serving as the negative control and distilled water as the positive control. The supernatant of the distilled water group exhibited bright red coloration due to significant hemolysis, while the supernatants of the PM, CEL, and PM-CEL groups were yellowish, similar to the PBS group (Figure S15a). Hemolysis rates in all three treatment groups were below 5% (Figure S15b). Furthermore, no significant weight loss was observed in the PM-CEL group compared to the PBS group (Figure S16), further confirming the reliable biosafety of PM-CEL.
Conclusion
In this study, we developed a PM-CEL nanoplatform for tumor therapy and cancer pain relief. CEL and MnO2 were found to induce severe mitochondrial damage, leading to ICD. During treatment, CEL reduced the levels of immunosuppressive factors, such as YAP and Treg cells, in the tumor microenvironment, effectively alleviating its immunosuppressive state. The reduction in YAP expression not only enhanced the immune response but also decreased VEGF and VEGFR1 expression, which contributed to the alleviation of hyperalgesia and provided lasting analgesia. These findings demonstrate that PM-CEL effectively inhibits tumor growth and relieves cancer pain, offering new insights for the development of patient-centered cancer treatment systems.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author and will be provided on reasonable request.
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263 (2024).
Zeng, L. et al. Poly(phenylalanine) and poly(3,4-dihydroxy-L-phenylalanine): promising biomedical materials for Building stimuli-responsive nanocarriers. J. Control Release. 372, 810–828 (2024).
Bhansali, D. et al. Nanotechnology for pain management: current and future therapeutic interventions. Nano Today 39, 101223 (2021).
Zhang, P. et al. Cancer nanomedicine toward clinical translation: obstacles, opportunities, and future prospects. Med 4, 147–167 (2023).
Soler, M. F., Abaurrea, A., Azcoaga, P., Araujo, A. M. & Caffarel, M. M. New perspectives in cancer immunotherapy: targeting IL-6 cytokine family. J. Immunother Cancer 11 (2023).
Qin, S., He, G. & Yang, J. Nanomaterial combined engineered bacteria for intelligent tumor immunotherapy. J. Mater. Chem. B. 12, 9795–9820 (2024).
Wu, J. et al. Targeted glycan degradation potentiates cellular immunotherapy for solid tumors. Proc. Natl. Acad. Sci. U S A. 120, e2300366120 (2023).
Heras-Murillo, I., Adán-Barrientos, I., Galán, M., Wculek, S. K. & Sancho, D. Dendritic cells as orchestrators of anticancer immunity and immunotherapy. Nat. Rev. Clin. Oncol. 21, 257–277 (2024).
Meng, L. et al. Emerging immunotherapy approaches for advanced clear cell renal cell carcinoma. Cells 13 (2023).
Tang, L., Huang, Z., Mei, H. & Hu, Y. Immunotherapy in hematologic malignancies: achievements, challenges and future prospects. Signal. Transduct. Target. Ther. 8, 306 (2023).
Casirati, G. et al. Epitope editing enables targeted immunotherapy of acute myeloid leukaemia. Nature 621, 404–414 (2023).
Leake, I. Hyperprogression during immunotherapy. Nat. Cancer. 4, 1640 (2023).
Yang, K., Halima, A. & Chan, T. A. Antigen presentation in cancer - mechanisms and clinical implications for immunotherapy. Na T Rev. Clin. Oncol. 20, 604–623 (2023).
Abramson, H. N. Immunotherapy of multiple myeloma: Current status as prologue to the future. Int. J. Mol. Sci. 24 (2023).
Janes, P. W., Vail, M. E., Ernst, M. & Scott, A. M. Eph receptors in the immunosuppressive tumor microenvironment. Cancer Res. 81, 801–805 (2021).
Cao, L. L. & Kagan, J. C. Targeting innate immune pathways for cancer immunotherapy. Immunity 56, 2206–2217 (2023).
Xue, X. et al. A transformable nanoplatform with multiple therapeutic and immunostimulatory properties for treatment of advanced cancers. Biomaterials 299, 122145 (2023).
Cao, Z. et al. Lactate oxidase nanocapsules boost T cell immunity and efficacy of cancer immunotherapy. Sci. Transl Med. 15, eadd2712 (2023).
Ho, W. S. et al. PP2Ac/STRN4 negatively regulates STING-type I IFN signaling in tumor-associated macrophages. J. Clin. Invest. 133 (2023).
Stampouloglou, E. et al. Yap suppresses T-cell function and infiltration in the tumor microenvironment. PLoS Biol. 18, e3000591 (2020).
Zanconato, F., Cordenonsi, M. & Piccolo, S. YAP/TAZ at the roots of cancer. Cancer Cell. 29, 783–803 (2016).
Ni, X. et al. YAP is essential for treg-mediated suppression of antitumor immunity. Cancer Discov. 8, 1026–1043 (2018).
Elaimy, A. L. & Mercurio, A. M. Convergence of VEGF and YAP/TAZ signaling: Implications for angiogenesis and cancer biology. Sci. Signal. 11 (2018).
Hicklin, D. J. & Ellis, L. M. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 23, 1011–1027 (2005).
Ma, K. et al. Nanoparticle-based Inhibition of vascular endothelial growth factor receptors alleviates osteoarthritis pain and cartilage damage. Sci. Adv. 10, eadi5501 (2024).
Yang, Y. et al. Decreased abundance of TRESK two-pore domain potassium channels in sensory neurons underlies the pain associated with bone metastasis. Sci. Signal. 11 (2018).
Zhang, Z. et al. Microglial Annexin A3 downregulation alleviates bone cancer-induced pain through inhibiting the Hif-1α/vascular endothelial growth factor signaling pathway. Pain 161, 2750–2762 (2020).
Selvaraj, D. et al. A functional role for VEGFR1 expressed in peripheral sensory neurons in cancer pain. Cancer Cell. 27, 780–796 (2015).
Lim, H. Y. et al. Celastrol in cancer therapy: recent developments, challenges and prospects. Cancer Lett. 521, 252–267 (2021).
Wang, C. et al. Celastrol as an emerging anticancer agent: current status, challenges and therapeutic strategies. Biomed. Pharmacother. 163, 114882 (2023).
Chitturi, P., Xu, S. & Abdi, A. Tripterygium wilfordii derivative Celastrol, a YAP inhibitor, has antifibrotic effects in systemic sclerosis. Ann. Rheum. Dis. 82, 1191–1204 (2023).
Zhou, R. et al. Biotin decorated celastrol-loaded ZIF-8 nano-drug delivery system targeted epithelial ovarian cancer therapy. Biomed. Pharmacother. 167, 115573 (2023).
Cheng, S. et al. Molecular mechanism underlying the action of a celastrol-loaded layered double hydroxide-coated magnesium alloy in osteosarcoma Inhibition and bone regeneration. ACS Biomater. Sci. Eng. 9, 4940–4952 (2023).
Zhong, R. et al. Discrete nanoparticle-BSA conjugates manipulated by hydrophobic interaction. ACS Appl. Mater. Interfaces. 6, 19465–19470 (2014).
Duan, X., Chan, C. & Lin, W. Nanoparticle-nediated Immunogenic cell death enables and potentiates cancer immunotherapy. Angew Chem. Int. Ed. Engl. 58, 670–680 (2019).
Ding, B., Yue, J., Zheng, P., Ma, P. & Lin, J. Manganese oxide nanomaterials boost cancer immunotherapy. J. Mater. Chem. B. 9, 7117–7131 (2021).
Xiao, B. et al. Integration of polymerization and biomineralization as a strategy to facilely synthesize nanotheranostic agents. ACS Nano. 12, 12682–12691 (2018).
Qiu, N. et al. Celastrol nanoemulsion induces immunogenicity and downregulates PD-L1 to boost abscopal effect in melanoma therapy. Biomaterials 277, 121121 (2021).
Lu, S., Li, Y. & Yu, Y. Glutathione-scavenging celastrol-Cu nanoparticles induce self-amplified Cuproptosis for augmented cancer immunotherapy. Adv. Mater. 36, e2404971 (2024).
Guo, L. et al. Glomerulus-targeted ROS-responsive polymeric nanoparticles for effective membranous nephropathy therapy. ACS Appl. Mater. Interfaces. 16, 35447–35462 (2024).
Li, W. et al. Targeting photodynamic and photothermal therapy to the Endoplasmic reticulum enhances Immunogenic cancer cell death. Nat. Commun. 10, 3349 (2019).
Colombo, M. et al. Specific immunosuppressive role of nanodrugs targeting calcineurin in innate myeloid cells. iScience 25, 105042 (2022).
Zeng, X. et al. Celastrol-conjugated Chitosan oligosaccharide for the treatment of pancreatic cancer. Drug Deliv. 29, 89–98 (2022).
Li, H. J. et al. Stimuli-responsive clustered nanoparticles for improved tumor penetration and therapeutic efficacy. Proc. Natl. Acad. Sci. U S A. 113, 4164–4169 (2016).
Jo, H. et al. Natural product Celastrol destabilizes tubulin heterodimer and facilitates mitotic cell death triggered by microtubule-targeting anti-cancer drugs. PLoS One. 5, e10318 (2010).
Chen, X. et al. Celastrol induces ROS-mediated apoptosis via directly targeting peroxiredoxin-2 in gastric cancer cells. Theranostics 10, 10290–10308 (2020).
Peng, F. et al. Nanocrystals slow-releasing ropivacaine and doxorubicin to synergistically suppress tumor recurrence and relieve postoperative pain. ACS Nano. 17, 20135–20152 (2023).
Sacks, D. et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke. Int. J. Stroke. 13, 612–632 (2018).
Wang, Y. et al. In situ vaccination with mitochondria-targeting Immunogenic death inducer elicits CD8(+) T cell-dependent antitumor immunity to boost tumor immunotherapy. Adv. Sci. (Weinh). 10, e2300286 (2023).
Hu, M. et al. Celastrol-induced Nur77 interaction with TRAF2 alleviates inflammation by promoting mitochondrial ubiquitination and autophagy. Mol. Cell. 66, 141–153e146 (2017).
Cossarizza, A., Baccarani-Contri, M., Kalashnikova, G. & Franceschi, C. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolcarbocyanine iodide (JC-1). Biochem. Biophys. Res. Commun. 197, 40–45 (1993).
Mannion, J. et al. A RIPK1-specific PROTAC degrader achieves potent antitumor activity by enhancing Immunogenic cell death. Immunity 57, 1514–1532e1515 (2024).
Mishchenko, T. A. et al. Targeting Immunogenic cell death for glioma immunotherapy. Trends Cancer. 10, 8–11 (2024).
Demuynck, R. et al. Nanomedicine to aid Immunogenic cell death (ICD)-based anticancer therapy. Trends Cancer. 10, 486–489 (2024).
Liu, X., Lu, Y., Li, X., Luo, L. & You, J. Nanoplatform-enhanced photodynamic therapy for the induction of Immunogenic cell death. J. Control Release. 365, 1058–1073 (2024).
Qian, X. et al. Celecoxib augments paclitaxel-induced Immunogenic cell death in triple-negative breast cancer. ACS Nano. 18, 15864–15877 (2024).
Liu, X. Z. et al. Bioengineered bacterial membrane vesicles with multifunctional nanoparticles as a versatile platform for cancer immunotherapy. ACS Appl. Mater. Interfaces. 15, 3744–3759 (2023).
Petrellis, M. C. et al. Laser-photobiomodulation on experimental cancer pain model in walker tumor-256. J. Photochem. Photobiol B. 210, 111979 (2020).
Li, Y. et al. Calcium carbonate/polydopamine composite nanoplatform based on TGF-β Blockade for comfortable cancer immunotherapy. ACS Appl. Mater. Interfaces. 16, 3187–3201 (2024).
Chen, W. et al. A multifunctional CaCO(3) bioreactor coated with coordination polymers enhances cancer immunotherapy. J. Control Release. 368, 780–796 (2024).
Gershoni, A. et al. TAZ facilitates breast tumor growth by promoting an immune-suppressive tumor microenvironment. Mol. Oncol. 17, 2675–2693 (2023).
Moroishi, T. et al. The Hippo pathway kinases LATS1/2 suppress cancer immunity. Cell 167, 1525–1539e1517 (2016).
Wang, Y. et al. MCM6 is a critical transcriptional target of YAP to promote gastric tumorigenesis and serves as a therapeutic target. Theranostics 12, 6509–6526 (2022).
Wang, Z. et al. Extracellular vesicles in fatty liver promote a metastatic tumor microenvironment. Cell. Metab. 35, 1209–1226e1213 (2023).
Zheng, H. et al. Celastrol-encapsulated microspheres prepared by microfluidic electrospray for alleviating inflammatory pain. Biomater. Adv. 149, 213398 (2023).
Hu, X. F. et al. The analgesic effects of triptolide in the bone cancer pain rats via inhibiting the upregulation of HDACs in spinal glial cells. J. Neuroinflammation. 14, 213 (2017).
Acknowledgements
This work was supported by the Education Department of Shandong Province (no. 2022KJ091), State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology (CRE-24-C003), the National Natural Science Foundation of China (grant no. 51903015 and 81772281), Science Fund of Shandong Laboratory of Advanced Materials and Green Manufacturing at Yantai (AMGM2021F03, AMGM2023F16), the Shandong Science and Technology Committee (no. ZR2022LSW002, ZR2020KH015), the Scientific Research Foundation of Binzhou Medical University (no. 50012304274), Shandong Province Taishan Scholar Project (no. ts201712067), ICTS ″NANBIOSIS″, in particular by the Drug Formulation Unit (U10) of the CIBER in Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), and the Basque Foundation for Science.
Author information
Authors and Affiliations
Contributions
Zhaokun Hao and Yuming Zhou contributed equally to this work. Zhaokun Hao: Methodology, Investigation, Writing - Original Draft; Yuming Zhou: Methodology, Investigation; Yuqiang Zhang: Methodology; Danyang Wang and Yiying Wei: Investigation, Formal analysis; Xiaopu Ji: Methodology, Formal analysis; Wan Ru Sun: Investigation, Formal analysis; Pingyu Wang and YouJie Li–Methodology; Irene Bautista Lope: Formal analysis, Writing - Review & Editing; José Luis Pedraz: Writing - Review & Editing; Murugan Ramalingam and Shuyang Xie: Resources, Funding acquisition, Conceptualization, Writing - Review & Editing; Ranran Wang: Conceptualization, Resources, Funding acquisition, Writing - Review & Editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hao, Z., Zhou, Y., Zhang, Y. et al. Celastrol loaded nanocomplex for painless tumor therapy via YAP inhibition. Sci Rep 15, 13133 (2025). https://doi.org/10.1038/s41598-025-97055-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-97055-7
