
Abstract
To evaluate genetic changes in respiratory syncytial virus (RSV) between 2019 and 2023, we analyzed RSV strains from Myanmar before and after the COVID- 19 pandemic. Real-time polymerase chain reaction (RT-PCR) positive samples from children presenting with acute respiratory infections at outpatient clinics in Yangon were sequenced to determine the genotype. Phylogenetic and molecular evolutionary analyses were conducted using the Bayesian Markov Chain Monte Carlo method to construct the time-scale Maximum Clade Credibility tree. Of 1127 samples, 104 (9.2%) RSV-A and 233 (20.7%) RSV-B were positive by RT-PCR. There was an absence of a notable epidemic in 2020, a temporal shift with a surge of RSV-A in the 2021 outbreak, a lack of expected cases in 2022 and a substantial resurgence of RSV-B in 2023. The genotype of RSV-A was mainly A.D.3 lineage through the study period, while RSV-B were B.D.4.1.1 and B.D.E.1. RSV-A showed that the same lineage persisted within Myanmar throughout the pandemic, leading to a large outbreak post-COVID. In contrast, RSV-B strains appear to have temporarily disappeared during the pandemic, but subsequently, globally circulating strains likely entered Myanmar, resulting in a major outbreak in 2023. The estimated evolutionary rate at the G-ectodomain for RSV-A was 7.76 × 10⁻³ and RSV-B was 5.67 × 10⁻³ substitutions/site/year. Strengthening genomic surveillance will likely support comparisons of circulating strains with those in other countries and facilitate the introduction of vaccines and other interventions.
Similar content being viewed by others


Introduction
Respiratory syncytial virus (RSV) is a major cause of serious lower respiratory tract infections among infants and young children1. Globally, it is estimated that there were 36 million hospital admissions and 26,300 in-hospital deaths among children aged 0–60 months associated with RSV in 20192. In low- and middle-income countries (LMICs) settings, approximately 97% of RSV-associated deaths in children under 5 years of age occur, highlighting the substantial disease burden that these countries face2,3,4.
RSV is classified under the genus Orthopneumovirus within the family Pneumoviridae. It is an enveloped, single-stranded negative-sense ribonucleic acid (RNA) virus. The RSV genome is non-segmented, approximately 15.2 kb in length, and encodes 11 proteins in the coding order of nonstructural proteins (NS1 and NS2), nucleocapsid (N), phosphoprotein (P), matrix protein (M), small hydrophobic protein (SH), attachment glycoprotein (G), fusion protein (F), regulatory M2 proteins (M2 - 1, M2 - 2), and large polymerase protein (L)5,6,7,8. G and F proteins are the primary targets of neutralizing antibodies during natural infection. RSV can be classified into two antigenic subgroups, RSV-A and RSV-B, based on the cross-reactivity of antibodies to the G and F proteins5,8. The G gene, which encodes the attachment protein of RSV, is the most variable among the 11 genes of the virus, largely due to the selective pressure exerted by the host immune system9,10. This gene contains two hypervariable regions (HVRs). Traditionally, the genotyping of RSV relied on variations within the second HVR. The second HVR has been utilized to classify RSV-A into genotypes such as GA1 - 7, SAA1, NA1 - 4, and ON1, while RSV-B has been classified into genotypes including GB1 - 5, SAB1 - 4, URU1 - 2, and BA1 - 105,6,9. Currently, the most prevalent genotypes worldwide are ON1 for RSV-A and BA9 for RSV-B, according to this genotyping56. However, due to the absence of a centralized structure to control the classification methods, individual researchers have independently reported new genotypes, leading to a lack of standardization. This has created challenges in comparing RSV genotypes across different studies. In 2020, Goya et al. proposed a classification based on the extracellular domain (550–600 base pairs) of the G gene that defines three major genotypes of RSV-A (GA1-GA3) and seven genotypes of RSV-B (GB1-GB7)6. Further, the HRSV Genotyping Consensus Consortium (RGCC) was established to standardize classification below the subgroup level, applicable to complete genome sequences (~15,000 base pairs)7. It defines 24 major lineages for RSV-A and 16 lineages for RSV-B. This new system enables classification not only based on complete genome sequences but also using G and F gene sequences. They stated that unlike the previous methods that relied on the G gene HVR2 alone, this approach ensured a higher phylogenetic resolution by analyzing mutations across all viral proteins. In addition, since the platform uses Nextstrain, researchers can upload RSV sequences to public genetic databases such as GenBank, allowing them to be reflected on the Nextstrain platform (https://clades.nextstrain.org/ (accessed on 22 November 2024)). This approach offers the advantage of enabling a unified, globally consistent genetic classification.
Although the standardization of genetic classification aids in identifying globally circulating RSV strains, conducting phylogeographic analyses to estimate transmission routes within specific regions still necessitates the use of tailored molecular analysis tools based on viral genetic sequence data. BEAST software is a versatile, cross-platform tool that enables Bayesian analysis of molecular sequences through MCMC methods11. It focuses on rooted, time-measured phylogenies inferred through strict or relaxed molecular clock models12. These analyses can provide insights into viral geographical transmission patterns and genetic changes of RSV over time.
Our group has conducted surveillance of human influenza and other respiratory viruses in Myanmar for nearly two decades5. In the past, we reported genetic variation in the second hypervariable region (HVR) of the G gene in RSV strains collected in Myanmar from 2015 to 20185. The dominant genotypes identified during that period were ON1 for HRSV-A and BA9 for HRSV-B. In recent years, the COVID- 19 pandemic, which began in 2020, has directly or indirectly influenced the RSV epidemic, leading to changes in the size and seasonal patterns of outbreaks13,14,15,16. The primary aim of this study is to investigate changes in RSV circulation patterns and genetic variability in the ectodomain of the RSV G gene in Myanmar over five consecutive epidemic seasons from 2019 to 2023. By upgrading the RSV classification method from the previous HVR-based approach to the G protein ectodomain, we can compare Myanmar RSV strains with those from other countries worldwide. In addition, molecular epidemiologic surveillance conducted before and after the COVID- 19 pandemic in Myanmar provides valuable insights into RSV transmission dynamics, illustrating the impact of public health measures and the subsequent rebound following their relaxation.
Materials and methods
Study location and setting
Myanmar is a lower-middle-income country located in Southeast Asia, with Yangon being the most populated city, housing approximately 54.1 million residents (https://data.who.int/countries/104 (accessed on 22 November 2024)). In Myanmar, it was reported that about a quarter of patients under five years of age with acute respiratory illnesses have tested positive for RSV, indicating a higher burden compared to high-income or industrialized countries2,5,17. RSV displays distinct seasonal epidemic patterns that vary with climate: in temperate regions, outbreaks typically occur during the winter months, while in tropical and subtropical regions, they are more prevalent during the monsoon season5,18. Myanmar’s tropical monsoon climate aligns with these trends, and we previously reported that seasonal peaks in RSV cases occurred during rainy monsoon seasons from September to October 2015–20185.
This study involved pediatric patients presenting with acute respiratory infection symptoms, such as fever (≥ 37.5 °C), coughing, wheezing, rhinorrhea, headache, dyspnea, myalgia, arthralgia, and diarrhea, visited two outpatient clinics (Thingangyun Sanpya General Hospital and Yankin Children’s Hospital) in Yangon, Myanmar, during the period from February 2019 to December 2023 (Fig. 1). In this study, written informed consent was obtained from patients or their guardians before enrolment. Then, clinicians recorded patient information such as gender, age, clinical symptoms (fever, coughing, wheezing, rhinorrhea, headache, dyspnea, myalgia, arthralgia, and diarrhea), and RDT results for RSV.

Geographic map of Yangon, Myanmar, showing the locations of Yankin Children’s Hospital and Thingangyun General Hospital (red crosses) where samples were collected. This study used Myanmar digital administrative boundaries map from Myanmar Information and Management Unit (MIMU), Myanmar (https://www.themimu.info/search/external? field_search=Myanmar% 20State% 20and% 20Region% 20Boundaries% 20MIMU% 20v9.4 (accessed on 25 February 2025). Yankin Children’s Hospital (N16.7883˚ E96.1364˚) and Thingangyun General Hospital (N16.8327˚, E96.192˚) are located in Yankin and Thingangyun areas in Yangon, Myanmar, respectively.
Collection of clinical samples and data
Nasal swabs were collected twice from each participant: one sample was used to perform a rapid diagnostic test (RDT) (Quick-Navi™ Flu + RSV or Quick-Navi™ RSV2; Denka, Tokyo, Japan) for RSV screening, while the other sample was placed in viral transport medium, frozen at − 20 °C at the study site, and sent to the National Health Laboratory in Yangon within a few weeks of collection, where it was stored at − 80 °C. The samples were then transported by international courier to the Division of International Health (Public Health) at Niigata University (Niigata, Japan) and stored at − 80 °C for further laboratory examination.
Sample screening
For RDT-positive samples, viral RNA was extracted from 140 µL of clinical specimens using the QIAamp Viral RNA Mini Kit (QIAGEN, Hilden, Germany) following the manufacturer’s instructions. Reverse transcription was conducted using random primers and M-MLV reverse transcriptase (Invitrogen Corporation, Carlsbad, CA, USA) to generate complementary DNA (cDNA) through incubation at 37 °C for 1 h. The resulting cDNA samples were subjected to a cycling probe real-time polymerase chain reaction (RT-PCR) assay using a probe set targeting the RSV membrane fusion protein (F) to determine subtype (RSV-A or -B) as reported previously19. Positive cycle threshold (Ct) values were defined as ≤ 40 cycles, while the curves exceeding 40 cycles were considered negative. The Cycling Probe RT-PCR method was used as a qualitative test because only a single positive control is included per PCR run. As a result, a standard curve cannot be generated, making it impossible to determine the viral RNA load quantitatively.
RSV G gene sequencing
After subgrouping by RT-PCR, RSV-positive samples underwent conventional PCR targeting the glycoprotein (G) ectodomain region, utilizing the PrimeScript™ II High Fidelity One-Step RT-PCR Kit (TaKaRa Bio Inc., Shiga, Japan) and specific primers (Table S1)20,21. The amplified PCR product was purified using the QIAquick PCR Purification Kit (QIAGEN). The purified products were labeled with the BigDye Terminator Cycle Sequencing Kit version 3.1 (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturer’s instructions. Sequencing was performed on an ABI Prism 3130xl Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA, USA) using different primer sets based on the subgroup (Table S1)20. The primers used in RT-PCR and sequencing were adapted from those originally reported by Goya et al.6. The generated sequences were assembled using the Lasergene SeqMan Pro package 17.5.0 (DNASTAR, Madison, WI, USA). Sequences were aligned using the ClustalW algorithm within the BioEdit software version 7.2.5 (https://bioedit.software.informer.com/7.2/ (accessed on 22 November 2024)).
Genotyping using nextclade and registration numbers of RSV strains
We first utilized the Nextclade web tool (https://clades.nextstrain.org/(accessed on 14 February 2025)) to identify the lineage and amino acid substitutions of the RSV strains obtained in this study6,7. The G gene ectodomain region nucleotide sequences generated in this study have been submitted to GenBank under accession numbers LC846997-LC847045 for RSV-A and LC847046-LC847090 for RSV-B (Tables S2 and S3).
Phylogenetic and molecular evolutionary Analysis using Bayesian Markov chain Monte Carlo (MCMC) method.
To understand the genetic relationship between the strains obtained in this study and those circulating worldwide, we first performed a Basic Local Alignment Search Tool (BLAST) (https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 20 June 2024)) search in the GenBank database and selected the top five matched strains from other countries (RSV-A; 74, RSV-B; 74) that showed more than 98% similarity with each sequence from Myanmar (Tables S2 and S3). Additionally, we added some strains from Japan (RSV-A; 70, RSV-B; 62) obtained in our previous study as a part of worldwide strains (Tables S2 and S3)20. Then, phylogenetic and molecular evolutionary analyses using Bayesian Markov Chain Monte Carlo (MCMC) methods were performed using BEAST software for the strains from Myanmar and other countries11. Sequences were aligned using ClustalW in MEGA version 6, and the most appropriate nucleotide substitution models were selected22. The Tracer was used to visualize and analyze the MCMC trace files generated through Bayesian phylogenetic inference23. The TreeAnnotator was used to infer the maximum clade credibility tree. Finally, FigTree was used to view and edit trees produced by TreeAnnotator and produce figures.
Time-calibrated phylogenetic analyses were conducted using the MCMC method implemented in BEAST version 1.8.4 under the strict clock and exponential growth demographic models24. The best-fit model of nucleotide substitution, GTR + G for RSV-A and TN93 + G for RSV-B, was selected using MEGA6. MCMC was run for 500 million steps, with 10% of the burn-in discarded in each run with 50 million steps. To calibrate the molecular clock, we used prior evolutionary rates reported in our previous study on the HVR of the G gene from Myanmar RSV strains: 1.39 × 10−2 substitutions/site/year (95% highest posterior density [HPD], 6.03 × 10−3 – 2.12 × 10−2) for RSV-A and 2.12 × 10−2 substitutions/site/year (95% HPD, 8.53 × 10−3 – 3.63 × 10−2) for RSV-B5. The appropriate model (clock and demographic) was selected based on an effective sample size (ESS) of ≥ 200. Convergence was assessed using Tracer version 1.7.2 (https://github.com/beast-dev/tracer/releases/latest), ensuring effective sample size values > 200 for each parameter in each run. Subsequently, Tree Annotator version 1.8.3 was employed to construct the time-calibrated phylogenetic tree (Maximum Evolutionary Branch Confidence tree, MCC tree), which was edited using FigTree version 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 22 November 2024)). We used the Nextclade web tool (https://clades.nextstrain.org/ (accessed on 19 February 2025)) to find unique amino acid substitutions in Myanmar strains. All amino acid substitutions are relative to the default reference sequence (RSV-A: hRSV/A/England/397/2017 (EPI_ISL_412866); RSV-B: hRSV/B/Australia/VIC-RCH056/2019 (EPI_ISL_1653999)).
To estimate the evolutionary rates of RSV-A and RSV-B during the 2019–2023 period, the molecular evolutionary analyses were conducted exclusively on RSV strains collected in Myanmar, using the G-ectodomain methods which was employed in China and Taiwan region studies25,26. Phylogenetic and molecular evolutionary analyses were performed using the Bayesian MCMC method implemented in BEAST version 1.8.4. The analysis was performed using the strict clock and exponential growth demographic models. The best-fit nucleotide substitution model of the G-ectodomain region was GTR + G for RSV-A and RSV-B. TN93 + G and TN93 + I were the best-fit nucleotide substitution models for RSV-A and RSV-B in the second HVR, respectively. MCMC was run for 200 million steps, with 10% of the burn-in discarded in each run with 20 million steps. Prior evolutionary rates of 1.39 × 10−2 and 2.12 × 10−2 substitutions/site/year for RSV-A and RSV-B were utilized to calibrate the molecular clock. Convergence was assessed using Tracer version 1.7.2, ensuring effective sample size values > 200 for each parameter in each run and that estimated evolutionary rates were obtained. Notably, the evolutionary rate was estimated using all sequences from the entire five-year study period (2019–2023) were used as a single dataset, rather than dividing the analysis into pre-pandemic and post-pandemic periods. This approach was chosen because the number of RSV strains collected each year was limited, and separate estimations for different time periods would have resulted in lower statistical confidence in the estimates of the evolutionary rate.
Ethical consents
All samples and patient information were collected after written informed consent from the parents or caregivers of the study participants. This study received approval from the Niigata University Ethical Committee (Nos. 2015–2533 and 2020 − 0155) and the Review Committee of the Department of Medical Research, Ministry of Health, Myanmar (Nos. 016516 and 2023 - 16).
Results
Clinical characteristics of patients with RSV in Myanmar
A total of 1,127 nasal swab samples were collected from outpatients with acute respiratory infection symptoms in Myanmar during the five-year study period (2019–2023) (Table 1). Of these, 444 (39.3%) samples tested positive for RSV by RDT. Subsequent analysis of these 444 samples confirmed RSV positivity in 337 samples through RT-PCR. The clinical characteristics of the patients in these 337 samples are shown in Table 2. During the entire study period, the median age of the patients was 1.0 years (interquartile range (IQR); 0.5, 2.0 years). Notable differences in the median age were observed across the years, with 2020 (3.1 years) and 2021 (1.6 years) having higher median ages compared to 2019 (1.0 years), 2022 (0.9 years), and 2023 (1.0 years) (p < 0.001, by Mann–Whitney U test). The overall infection rate was higher in males (56.7%) than in females (42.4%), excluding patients with unknown gender information (0.9%). The proportion of male patients was higher than that of female patients in most years, except in 2019. Among the patients in our study, RSV-B infections (69.1%) were detected more often than RSV-A infections (30.9%) because of a large outbreak of RSV-B in 2023. The most frequent clinical symptoms were cough (92.9%), followed by rhinorrhea (84.0%), fever (28.5%), and dyspnea (18.7%) throughout the study period. Patients presenting with symptoms of dyspnea were only observed in RSV-B-infected patients in 2023 (63/220, 28.6%). A very small number of patients experienced wheezing, headache, and diarrhea symptoms. Myalgia and arthralgia were not observed in our study participants.
Epidemic curve of RSV in Myanmar from 2019 to 2023
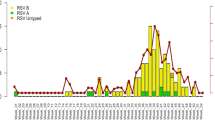
In 2019, epidemics of RSV occurred from July to November, with a peak from August to October (Fig. 2), similar to what we observed in our previous study5. Both RSV-A and RSV-B subgroups co-circulated during the epidemic. In 2020, no apparent RSV epidemics were observed, and only a few RSV-A cases were detected in August and September of 2020. In 2021, the RSV epidemic occurred from November to December, later than in the pre-pandemic seasons. More cases were collected compared with previous years; all were identified as RSV-A. The epidemic in 2022 was smaller than the previous year, with only a small number of RSV-A cases detected in July and August. In contrast, the largest RSV epidemic occurred in 2023, predominantly caused by RSV-B, with more than half of the samples in our study collected in 2023. The epidemic lasted from July to October 2023, peaking in August and almost returning to what was observed before COVID- 19 5.

The monthly distribution epidemic curve of respiratory syncytial virus (RSV) collected in Myanmar from 2019 to 2023. The gray shading represents the number of nasal swab samples collected; the orange bar graph represents the number of RSV-A-positive samples (n = 104) detected using real-time polymerase chain reaction (RT-PCR); the blue bar graph represents the number of RSV-B-positive samples (n = 233) detected using RT-PCR; the gray line represents the number of cases that tested positive using a rapid diagnostic test (RDT).
Lineages and evolutionary rates of RSV-A and -B virus in Myanmar
The G gene ectodomain region was successfully sequenced by Sanger sequencing in 49 of 104 (47.1%) RT-PCR-detected RSV-A and 45 of 233 (19.3%) RSV-B. After confirming with Nextclade (https://clades.nextstrain.org/ (accessed on 7 February 2025)), we obtained the lineages of the Myanmar strain; RSV-A remained consistent as A.D.3 (equivalent to genotype GA2.3.5) lineage except for one strain in 2020 belonging to A.D (GA2.3.5). Although RSV-B was not observed in Myanmar from 2020 to 2022, lineage B.D.4.1.1 (GB5.0.5a) was observed both before and after the pandemic. Lineage B.D.E.1 (GB5.0.5a) emerged in 2023.
Time-calibrated phylogenetic tree analysis of Myanmar RSV-A and RSV-B G-ectodomain sequences was performed separately by BEAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 20 June 2024)), incorporating reference sequences from other countries obtained through BLAST searches on GenBank (Tables S2 and S3) and our previous study in Japan20. The analyzed RSV-A strains were categorized into the following regions: Myanmar (49), North America (15), China (21), Europe (26), Japan (70), Philippines (2), South Korea (1), Vietnam (3), India (1), and Thailand (5). For RSV-B, the strains were grouped as follows: Myanmar (45), North America (6), China (4), Europe (52), Japan (62), Philippines (2), South America (1), and Australia (9). Time-calibrated phylogenetic tree analysis revealed that RSV strains in Myanmar were split into globally and locally circulating groups (Figs. 3 and 4). A globally circulating group was defined as a phylogenetic branch containing strains from multiple countries or regions within the same year. In contrast, a locally circulating group was defined as a branch containing strains from a single country or region within the same year.
Before the pandemic, RSV-A strains in Myanmar belonged to the globally circulating A.D.3 lineage, which was also detected in Europe, China, Japan, the Philippines, and Vietnam (Fig. 3). RSV-A remained predominantly within the A.D.3 lineage in 2020 and beyond; however, strains were found to cluster into both locally circulating and globally circulating groups within A.D.3. Certain locally circulating A.D.3 groups exhibited unique amino acid substitutions (K213 N, Y280H, and T281 K) in the G-gene ectodomain, which were exclusively found in Myanmar strains. The presence of this locally circulating group suggests that RSV-A in Myanmar persisted since before the pandemic and continued to spread domestically even after the pandemic. Additionally, a separate globally circulating group (lineage A.D) was detected in 2020, which appears to have co-circulated in Japan around the same time. The presence of these globally circulating groups indicates possible reintroductions of RSV-A into Myanmar from other countries.

Time-calibrated maximum clade credibility (MCC) tree inferred from RSV-A G-ectodomain sequences in Myanmar and reference sequences from other countries. The branches are on a time scale in years and are color-coded according to the location of the most probable ancestor of descendant nodes. Genetic lineages and unique amino acid substitutions are pointed out using black arrows.
For RSV-B, the strains responsible for the 2019 epidemic belonged to a globally circulating group within lineage B.D.4.1.1, likely originated from China and Europe (Fig. 4). However, this group appeared to have been disrupted, as no RSV-B strains were collected between 2020 and 2022. In 2023, RSV-B strains were classified into three groups: two locally circulating groups within lineage B.D.4.1.1 and one globally circulating group within B.D.E.1. Among the two locally circulating B.D.4.1.1 groups, one exhibited unique amino acid substitutions (Q142L, L284 F, and T300I) and likely originated from RSV-B strains circulating in China before the pandemic (Fig. 4, Figure S2), while the other appeared to have descended from strains that circulated in North America in 2020. The globally circulating group within B.D.E.1, which emerged in 2023, included strains from Europe, Japan, and North America, in addition to those from Myanmar. This pattern suggests that both local persistence and international reintroductions contributed to RSV-B circulation in Myanmar after the pandemic.

Time-calibrated MCC tree inferred from RSV-B G-ectodomain sequences in Myanmar and reference sequences from other countries. The branches are on a time scale in years and are color-coded according to the location of the most probable ancestor of descendant nodes. Genetic clade and unique amino acid substitutions are pointed out using black arrows.
To estimate the evolutionary rate of the G-ectodomain region for RSV-A and RSV-B in Myanmar, phylogenetic trees were constructed using the Bayesian MCMC method. After removing identical sequences, 49 RSV-A and 45 RSV-B strains collected between 2019 and 2023 were included in the analysis. As shown in Table 2, the estimated evolutionary rate of the G-ectodomain region for Myanmar RSV-A strains was 7.76 × 10⁻³ substitutions/site/year (95% HPD: 4.62 × 10⁻³–1.07 × 10⁻²), which was slightly faster than the rate for RSV-B strains, estimated at 5.67 × 10⁻³ substitutions/site/year (95% HPD: 3.70 × 10⁻³–7.89 × 10⁻³).
Discussion
This study depicted variations in the prevalence, subtypes, and lineages of respiratory syncytial virus (RSV) in Myanmar before and after the COVID- 19 pandemic. The years 2019 to 2023 witnessed significant fluctuations in RSV activity in Myanmar, characterized by the absence of a notable epidemic in 2020, a temporal shift with an RSV-A surge during the 2021 outbreak, a lack of expected cases in 2022, and a substantial resurgence of RSV-B in 2023. Our G-gene ectodomain sequencing revealed that RSV-A consistently belonged to lineage A.D.3 (GA2.3.5) both before and after the pandemic, while RSV-B retained the previously circulating lineage B.D.4.1.1 (GB5.0.5a), and introduced a new lineage, B.D.E.1 (GB5.0.5a), in 2023. However, detailed temporal phylogenetic analysis revealed that RSV-A and RSV-B exhibited different surviving patterns in Myanmar. The estimated evolutionary rate at the G-ectodomain for RSV-A was 7.76 × 10⁻³, and RSV-B was 5.67 × 10⁻³ substitutions/site/year.
This study highlights that the infection prevention and control measures implemented during the COVID- 19 pandemic significantly impacted the incidence of RSV cases in Myanmar from 2019 to 2023. While RSV epidemics were observed throughout most of the study period, only sporadic outbreaks occurred in 2020 and 2022. Based on our previous observations, the seasonal pattern of RSV epidemics (RSV-A and RSV-B) in Myanmar typically coincides with the rainy season, spanning July to October, with peak activity occurring in September and October5. In contrast, almost no RSV outbreak was observed in 2020, coinciding with the onset of the COVID- 19 pandemic27. This suppression of the RSV epidemic in 2020 was caused by the COVID- 19-related public health measures implemented in Myanmar28,29.After the first COVID- 19 case was reported in March 2020, the government imposed strict public health measures, including mandatory mask-wearing, social distancing, and travel restrictions28,30,31,32. International borders were closed, domestic travel was restricted, and lockdowns were enforced. Myanmar experienced multiple COVID- 19 waves, with the Delta variant surge in July–August 2021 severely overwhelming the healthcare system. During this time, infectious diseases surveillance was likely limited as hospitals prioritized COVID- 19 patients. Stringent border closures and travel restrictions from 2020 to 2022 minimized RSV and other respiratory virus introductions, while mask mandates, social distancing, and school closures further reduced respiratory pathogen transmission. Widespread RT-PCR testing, contact tracing, and government quarantine programs also contributed to the suppression of respiratory viruses. This trend was similar to that reported in northern hemisphere countries, including the United Kingdom, the United States, and Japan13,20,33,34,35. As a result, the epidemics of respiratory infectious diseases, including RSV and influenza, were suppressed to varying degrees worldwide in 202033,36,42,36,35. A study by Reicherz et al. indicated that antibody titers in samples collected from infants in 2021 were 15-fold lower than those measured in 202043. On the other hand, due to the relaxation of non-pharmaceutical measures during the COVID- 19 pandemic, a significant RSV epidemic was observed in 2021 and 2023. This situation has been referred to as “RSV immunity debt,” attributed to a decline in population immunity following a prolonged period of minimal RSV exposure15,40,41. A similar resurgence was reported in various countries and regions, including Taiwan, the United States, the United Kingdom, Argentina, and Australia16,35,44,45,46. The relaxation of hygiene measures, travel restrictions, and increased respiratory infection screening also contributed to the rise in RSV cases13,33,47.
We observed that the median age of RSV-infected patients was higher during the COVID- 19 pandemic in 2020 and 2021 compared to the post-pandemic period in 2022 and 2023 (Table 1). In pre-pandemic 2019, the median infection age was approximately 1.0 years, consistent with our previous findings from 2015 to 20185. This suggests that older children were more likely to be infected following the COVID- 19 pandemic, potentially due to the absence of RSV epidemics in 2020, which may have delayed exposure to the virus13, resulting in higher infection age.
In terms of gender, the incidence of RSV infection was higher among males than females. Although this difference was insignificant in most years, this result aligns with previous studies conducted in Myanmar and Japan5,19, and a similar study conducted in China48. The most frequent symptoms associated with RSV were cough and rhinorrhea, with percentages of 92.9% and 84.0%, respectively. RSV infection among young children and older adults typically commences with upper respiratory tract illness, including rhinorrhea, nasal congestion, cough, sneezing, and sometimes fever and myalgia5. Symptoms may worsen as the infection progresses to the lower respiratory tract, including a more severe cough, increased respiratory rate, dyspnea, and intercostal muscle retractions5. Notably, dyspnea was observed in a higher proportion of patients in 2023, all of whom were infected with RSV-B, which was also observed in RSV-B patients in our previous study in Myanmar5.
For both RSV-A and RSV-B, the predominant genetic lineages remained largely unchanged even after the pandemic (A.D.1, B.D.4.1.1), except for the appearance of a new lineage for RSV-B in 2023 (B.D.E.1). However, a closer examination revealed that within the same lineage, some groups remained, some groups disappeared, while others were likely introduced from other countries. This suggests the presence of diverse intrinsic groups within each lineage. RSV-A showed that the same lineage persisted within Myanmar throughout the pandemic, leading to a large outbreak post-COVID. In contrast, RSV-B strains appear to have temporarily disappeared during the pandemic, but subsequently, globally circulating strains likely entered Myanmar, resulting in a major outbreak in 2023. This finding suggests that RSV continues to circulate even during periods of very low or no observed incidence in various parts of the world49,50,,51. We also identified unique amino acid substitutions in the G-ectodomain of certain Myanmar RSV strains: K213 N, Y280H, and T281 K in RSV-A, and Q142L, L284 F, and T300I in RSV-B. These unique amino acids circulating in Myanmar indicated that RSV continued to evolve in this location. Similar observations have been reported in countries such as China, Japan, the United States, and various European nations, but with different amino acid substitutions20,52,53,,54. Furthermore, the endemic RSV-B strains in 2019 were closely related to strains from China, consistent with findings from our previous study in Myanmar, where the endemic strain from 2018 also showed similarity to Chinese strains5. This suggests that the 2019 epidemic may have been a continuation of previous outbreaks. Our previous study from 2015 to 2018 observed that the second HVR genotype of RSV-A in Myanmar was classified as ON1 (A.D lineage), while RSV-B was BA9 (B.D to B.D.4.1.1 lineage). However, due to the change in the target region for genetic analysis in this study, we did not make a direct comparison of genotypes and other country strains5.
Our estimated evolutionary rates of the G-gene ectodomain during the pandemic were not significantly different from pre-pandemic rates reported in other countries. A study from China (2016–2019) focusing on the G-ectodomain region reported evolutionary rates consistent with those observed in this study (RSV-A: 1.95 × 10⁻³ substitutions/site/year [95% HPD: 1.45 × 10⁻³–2.48 × 10⁻³], and RSV-B: 3.15 × 10⁻³ substitutions/site/year [95% HPD: 2.45 × 10⁻³–3.95 × 10⁻³])25. Another study from the Taiwan region (2008–2017) also reported similar rates (RSV-A: 2.90 × 10⁻³ substitutions/site/year [95% HPD: 2.26 × 10⁻³–3.59 × 10⁻³], and RSV-B: 5.23 × 10⁻³ substitutions/site/year [95% HPD: 4.14 × 10⁻³–6.37 × 10⁻³])26. This suggests that the evolutionary rate of the G-gene ectodomain was not significantly affected by the interruption of RSV circulation. However, comparable post-pandemic studies have not yet been identified, possibly due to variations in the lengths of G-gene sequences analyzed. Several factors could influence differences in evolutionary rates, including the time frame of analysis, the predominant lineages circulating during the study period, and community herd immunity levels55. Therefore, a definitive conclusion regarding these evolutionary rates requires further investigation.
This study has several limitations. First, samples were collected from Yangon, Myanmar, which did not fully represent the entire country. Second, in this study, we only qualitatively tested for RSV viruses and were unable to determine differences in RSV viral load over time or across populations. Finally, our analysis focused solely on the G-ectodomain region rather than the whole genome. Recent studies have increasingly utilized whole genome analysis for RSV genotyping, as it allows for more comprehensive assessments, such as estimating the effectiveness of immunization and analyzing mutations related to antibody-resistant drugs and vaccines7,51,52. This limitation may impact the comparability of our results with those of other studies.
Conclusions
In summary, this study offers valuable insights into the patterns of RSV circulation in Myanmar from 2019 to 2023, revealing notable variations in epidemic trends, lineages, and clinical presentations. The results emphasize the influence of the COVID- 19 pandemic on RSV epidemiology, including changes in age distribution, genetic survival patterns, and evolution rates. While there are limitations regarding sample representativeness and the focus on the G gene analysis, our findings enhance the understanding of RSV dynamics and its public health implications. Future research to broaden genomic investigations, such as a whole genome sequencing of the RSV, will improve comparability and guide targeted intervention efforts such as the introduction of vaccines.
Data availability
The nucleotide sequences generated in this study have been submitted to GenBank under accession numbers LC846997-LC847090.
References
Obodai, E. et al. The significance of human respiratory syncytial virus (HRSV) in children from Ghana with acute lower respiratory tract infection: A molecular epidemiological analysis, 2006 and 2013–2014. PLoS One. 13, e0203788. https://doi.org/10.1371/journal.pone.0203788 (2018).
Li, Y. et al. Global, regional, and National disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in children younger than 5 years in 2019: a systematic analysis. Lancet 399, 2047–2064. https://doi.org/10.1016/S0140-6736(22)00478-0 (2022).
Wittenauer, R., Pecenka, C. & Baral, R. Cost of childhood RSV management and cost-effectiveness of RSV interventions: a systematic review from a low- and middle-income country perspective. BMC Med. 21, 121. https://doi.org/10.1186/s12916-023-02792-z (2023).
Srikantiah, P., Vora, P. & Klugman, K. P. Assessing the full burden of respiratory syncytial virus in young infants in Low- and Middle-Income countries: the importance of community mortality studies. Clin. Infect. Dis. 73, S177–S179. https://doi.org/10.1093/cid/ciab486 (2021).
Phyu, W. W. et al. Evolutionary analysis of human respiratory syncytial virus collected in Myanmar between 2015 and 2018. Infect. Genet. Evol. 93, 104927. https://doi.org/10.1016/j.meegid.2021.104927 (2021).
Goya, S. et al. Toward unified molecular surveillance of RSV: A proposal for genotype definition. Influenza Other Respir Viruses. 14, 274–285. https://doi.org/10.1111/irv.12715 (2020).
Goya, S. et al. Standardized phylogenetic classification of human respiratory syncytial virus below the subgroup level. Emerg. Infect. Dis. 30, 1631–1641. https://doi.org/10.3201/eid3008.240209 (2024).
Griffiths, C., Drews, S. J. & Marchant, D. J. Respiratory syncytial virus: infection, detection, and new options for prevention and treatment. Clin. Microbiol. Rev. 30, 277–319. https://doi.org/10.1128/CMR.00010-16 (2017).
Tan, L. et al. The comparative genomics of human respiratory syncytial virus subgroups A and B: genetic variability and molecular evolutionary dynamics. J. Virol. 87, 8213–8226. https://doi.org/10.1128/JVI.03278-12 (2013).
Sullender, W. M. Respiratory syncytial virus genetic and antigenic diversity. Clin. Microbiol. Rev. 13, 1–15. https://doi.org/10.1128/cmr.13.1.1 (2000).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016. https://doi.org/10.1093/ve/vey016 (2018).
Drummond, A. J. & Rambaut, A. B. E. A. S. T. Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214. https://doi.org/10.1186/1471-2148-7-214 (2007).
Garg, I., Shekhar, R., Sheikh, A. B. & Pal, S. Impact of COVID-19 on the changing patterns of respiratory syncytial virus infections. Infect. Dis. Rep. 14, 558–568. https://doi.org/10.3390/idr14040059 (2022).
van Summeren, J. et al. Low levels of respiratory syncytial virus activity in Europe during the 2020/21 season: what can we expect in the coming summer and autumn/winter? Euro. Surveill. 26 https://doi.org/10.2807/1560-7917.ES.2021.26.29.2100639 (2021).
Abu-Raya, B., Vineta Paramo, M., Reicherz, F. & Lavoie, P. M. Why has the epidemiology of RSV changed during the COVID-19 pandemic? EClinicalMedicine 61, 102089, (2023). https://doi.org/10.1016/j.eclinm.2023.102089.
Eden, J. S. et al. Off-season RSV epidemics in Australia after easing of COVID-19 restrictions. Nat. Commun. 13, 2884. https://doi.org/10.1038/s41467-022-30485-3 (2022).
Nair, H. et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375, 1545–1555. https://doi.org/10.1016/S0140-6736(10)60206-1 (2010).
White, L. J. et al. Understanding the transmission dynamics of respiratory syncytial virus using multiple time series and nested models. Math. Biosci. 209, 222–239. https://doi.org/10.1016/j.mbs.2006.08.018 (2007).
Hibino, A. et al. Molecular epidemiology of human respiratory syncytial virus among children in Japan during three seasons and hospitalization risk of genotype ON1. PLoS One. 13, e0192085. https://doi.org/10.1371/journal.pone.0192085 (2018).
Yoshioka, S. et al. Molecular Epidemiology of Respiratory Syncytial Virus during 2019–2022 and Surviving Genotypes after the COVID-19 Pandemic in Japan. Viruses 15, (2023). https://doi.org/10.3390/v15122382
Rojo, G. L. et al. Unravelling respiratory syncytial virus outbreaks in Buenos Aires, Argentina: molecular basis of the spatio-temporal transmission. Virology 508, 118–126. https://doi.org/10.1016/j.virol.2017.04.030 (2017).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. https://doi.org/10.1093/molbev/mst197 (2013).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904. https://doi.org/10.1093/sysbio/syy032 (2018).
Rambaut, A., Lam, T. T., Max Carvalho, L. & Pybus, O. G. Exploring the Temporal structure of heterochronous sequences using tempest (formerly Path-O-Gen). Virus Evol. 2, vew007. https://doi.org/10.1093/ve/vew007 (2016).
Sun, Y. P. et al. Molecular evolution of attachment glycoprotein (G) and fusion protein (F) genes of respiratory syncytial virus ON1 and BA9 strains in Xiamen, China. Microbiol. Spectr. 10, e02083–e02021. https://doi.org/10.1128/spectrum.02083-21 (2022).
Lee, C. Y., Fang, Y. P., Wang, L. C., Chou, T. Y. & Liu, H. F. Genetic diversity and molecular epidemiology of Circulating respiratory syncytial virus in central Taiwan, 2008–2017. Viruses 14, 32 (2022).
Wang, C., Horby, P. W., Hayden, F. G. & Gao, G. F. A novel coronavirus outbreak of global health concern. Lancet 395, 470–473. https://doi.org/10.1016/S0140-6736(20)30185-9 (2020).
Phyu, W. W. et al. Epidemiology and Genetic Analysis of SARS-CoV-2 in Myanmar during the Community Outbreaks in 2020. Viruses 14, (2022). https://doi.org/10.3390/v14020259
Phyu, W. W. et al. Evolutionary dynamics of Whole-Genome influenza A/H3N2 viruses isolated in Myanmar from 2015 to 2019. Viruses 14, 2414 (2022).
World Health Organization Country Office for Myanmar. Curriculum on COVID-19 Response for Myanmar Community Health Volunteers, (2022). https://cdn.who.int/media/docs/default-source/searo/myanmar/documents/curriculum-on-covid-19-response-for-chvs-book-(english)_web.pdf?sfvrsn=d2b1d3d0_3&download=true
Chon, I. et al. Whole-Genome analysis of influenza A(H3N2) and B/Victoria viruses detected in Myanmar during the COVID-19 pandemic in 2021. Viruses 15 https://doi.org/10.3390/v15020583 (2023).
Oo, K. Z. et al. Genomic tracking of SARS-CoV-2 variants in Myanmar. Vaccines 11, 6 (2023).
Groves, H. E. et al. The impact of the COVID-19 pandemic on influenza, respiratory syncytial virus, and other seasonal respiratory virus circulation in Canada: A population-based study. Lancet Reg. Health Am. 1, 100015. https://doi.org/10.1016/j.lana.2021.100015 (2021).
Hamid, S. et al. Seasonality of respiratory syncytial Virus - United States, 2017–2023. MMWR Morb Mortal. Wkly. Rep. 72, 355–361. https://doi.org/10.15585/mmwr.mm7214a1 (2023).
Bardsley, M. et al. Epidemiology of respiratory syncytial virus in children younger than 5 years in England during the COVID-19 pandemic, measured by laboratory, clinical, and syndromic surveillance: a retrospective observational study. Lancet Infect. Dis. 23, 56–66. https://doi.org/10.1016/S1473-3099(22)00525-4 (2023).
Olsen, S., Azziz-Baumgartner, E. & Budd, A. Decreased influenza activity during the COVID-19 Pandemic — United States, Australia, Chile, and South Africa, 2020. Morbidity Mortal. Wkly. Rep. (MMWR). 69, 1305–1309. https://doi.org/10.15585/mmwr.mm6937a6 (2020).
Kim, D. H., Nguyen, T. M. & Kim, J. H. Infectious respiratory diseases decreased during the COVID-19 pandemic in South Korea. Int. J. Environ. Res. Public. Health. 18 https://doi.org/10.3390/ijerph18116008 (2021).
Sinha, P., Reifler, K., Rossi, M. & Sagar, M. C. Disease 2019 mitigation strategies were associated with decreases in other respiratory virus infections. Open. Forum Infect. Dis. 8, ofab105. https://doi.org/10.1093/ofid/ofab105 (2021).
Htun, Y. M. et al. Initial presenting symptoms, comorbidities and severity of COVID-19 patients during the second wave of epidemic in Myanmar. Trop. Med. Health. 49, 62. https://doi.org/10.1186/s41182-021-00353-9 (2021).
Htun, Y. M. et al. Impact of containment measures on community mobility, daily confirmed cases, and mortality in the third wave of COVID-19 epidemic in Myanmar. Trop. Med. Health. 50, 23. https://doi.org/10.1186/s41182-022-00413-8 (2022).
Htun, Y. M. et al. Trajectory of confirmed cases and deaths: fourth wave of COVID-19 epidemic in Myanmar. Virol. J. 20, 3. https://doi.org/10.1186/s12985-023-01960-0 (2023).
Scally, G., Jacobson, B. & Abbasi, K. The UK’s public health response to covid-19. BMJ 369, m (1932). https://doi.org/10.1136/bmj.m1932 (2020).
Reicherz, F. et al. Waning immunity against respiratory syncytial virus during the coronavirus disease 2019 pandemic. J. Infect. Dis. 226, 2064–2068. https://doi.org/10.1093/infdis/jiac192 (2022).
Sarah Hamid, A. W. et al. Hall. Seasonality of respiratory syncytial Virus - United States, 2017–2023. Weekly 72, 355–361. https://doi.org/10.15585/mmwr.mm7214a1 (2023).
Lee, C. Y. et al. Delayed respiratory syncytial virus outbreak in 2020 in Taiwan was correlated with two novel RSV-A genotype ON1 variants. Influenza Other Respir Viruses. 16, 511–520. https://doi.org/10.1111/irv.12951 (2022).
Dolores, A. et al. RSV reemergence in Argentina since the SARS-CoV-2 pandemic. J. Clin. Virol. 149, 105126. https://doi.org/10.1016/j.jcv.2022.105126 (2022).
Mercer, T. R. & Salit, M. Testing at scale during the COVID-19 pandemic. Nat. Rev. Genet. 22, 415–426. https://doi.org/10.1038/s41576-021-00360-w (2021).
Zhang, R. F. et al. Human respiratory syncytial virus in children with acute respiratory tract infections in China. J. Clin. Microbiol. 48, 4193–4199. https://doi.org/10.1128/JCM.00179-10 (2010).
Virant, M. J., Lustrek, M., Kogoj, R., Petrovec, M. & Ursic, T. Changes in HRSV Epidemiology but Not Circulating Variants in Hospitalized Children due to the Emergence of SARS-CoV-2. Viruses 15, (2023). https://doi.org/10.3390/v15061218.
Pretorius, M. A. et al. Replacement and positive evolution of subtype A and B respiratory syncytial virus G-protein genotypes from 1997–2012 in South Africa. J. Infect. Dis. 208 (Suppl 3), 227–237. https://doi.org/10.1093/infdis/jit477 (2013).
Redlberger-Fritz, M., Springer, D. N., Aberle, S. W., Camp, J. V. & Aberle, J. H. Respiratory syncytial virus surge in 2022 caused by lineages already present before the COVID‐19 pandemic. J. Med. Virol. 95 https://doi.org/10.1002/jmv.28830 (2023).
Adams, G. et al. Viral lineages in the 2022 RSV surge in the united States. N Engl. J. Med. 388, 1335–1337. https://doi.org/10.1056/NEJMc2216153 (2023).
Yu, J. M., Fu, Y. H., Peng, X. L., Zheng, Y. P. & He, J. S. Genetic diversity and molecular evolution of human respiratory syncytial virus A and B. Sci. Rep. 11, 12941. https://doi.org/10.1038/s41598-021-92435-1 (2021).
Martinelli, M. et al. Phylogeny and population dynamics of respiratory syncytial virus (Rsv) A and B. Virus Res. 189, 293–302. https://doi.org/10.1016/j.virusres.2014.06.006 (2014).
Yoshihara, K. et al. Molecular evolution of respiratory syncytial virus subgroup A genotype NA1 and ON1 attachment glycoprotein (G) gene in central Vietnam. Infect. Genet. Evol. 45, 437–446. https://doi.org/10.1016/j.meegid.2016.10.010 (2016).
Acknowledgements
We thank members of the virology Section of the National Health Laboratory, Myanmar, for providing the study samples and Dr. Kyi Kyi Wai for collecting samples. We also thank, Dr. Nay chi Win, Dr. Lasham Di Ja, Dr. Aung Htet Kaung, Ms. April Ko Ko, Ms. Khine Wutyi Lwin, Ms. Akemi Watanabe, Ms. Saori Nedachi, Ms. Toshiko Yabe, Ms. Kaoru Takizawa, and Ms. Risa Mi-zuno; IDRC members in Myanmar and Japan for managing this study. We would like to express our gratitude to Dr. Stephanie Goya at the University of Washington, USA, Dr. Mariana Viegas at the Ricardo Gutiérrez Children’s Hospital, Argentina, and the HRSV Genotyping Consensus Consortium (RGCC) for their invaluable support in genetic sequencing.
Funding
This research was funded by the Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) from Japan Agency for Medical Research and Development (AMED) (15fm0108009 h0001 - 19fm0108009 h0005, 20wm0125005 h0001 - 24wm0125005 h0005). The funders had no role in the study design, data collection and analysis, publication intention, or manuscript preparation.
Author information
Authors and Affiliations
Contributions
Conceptualization, Y.K., H.H.T., H.W., and R.S.; methodology, Y.K., M.M.A., S.S., S.M.K.W., H.H.T., H.W., and R.S.; software, J.L.; validation, I.C., S.Y., and T.O.; formal analysis, J.L., I.C., W.W.P., S.Y., W.H., W.K., Y.S., and T.O.; investigation, Y.K., M.M.A, S.M.K.W., H.W., and R.S.; resources, H.W., and R.S.; data curation, T.O.; writing—original draft preparation, J.L.; writing—review and editing, W.K., T.B.P., T.O., T.T., and R.S.; visualization, J.L.; supervision, Y.K., M.M.A, S.S., H.H.T., H.W., and R.S.; project ad-ministration, Y.K., H.H.T., H.W., and R.S.; funding acquisition, H.W., and R.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Institutional review board statement
This study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Review Committee of Niigata University (No. 2015–2533 and 2020 − 0155) and the Review Committee of the Department of Medical Research, Ministry of Health, Myanmar (No. 016516 and 2023 - 16).
Informed Consent
Informed consent was obtained from all subjects involved in this study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, J., Chon, I., Phyu, W.W. et al. Molecular epidemiological surveillance of respiratory syncytial virus infection in Myanmar from 2019 to 2023. Sci Rep 15, 13126 (2025). https://doi.org/10.1038/s41598-025-97103-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-97103-2

{kind=link}
{kind=link}