
Abstract
The activation of microglia and the resulting neuroinflammation play crucial regulatory roles in the pathogenesis and progression of neurological diseases, although the specific mechanisms remain incompletely understood. Cytidine monophosphate kinase 2 (CMPK2) is a key mitochondrial nucleotide kinase involved in cellular energy metabolism and nucleotide synthesis. Recent studies suggest that CMPK2 plays a role in microglial-mediated neuroinflammation; however, its specific impact on microglial activation remains unclear. In this study, we hypothesize that CMPK2 promotes microglial-mediated neuroinflammation by activating the cGAS-STING signaling pathway. To investigate this mechanism, we employed lipopolysaccharide (LPS)-treated microglial cells to investigate the detailed mechanisms by which CMPK2 regulates neuroinflammation. Our experimental results indicate that in the BV2 and mouse primary microglial neuroinflammation model, both CMPK2 protein and transcript levels were significantly elevated, accompanied by microglial activation phenotypes such as increased cell size, shortened processes, transformation to round or rod-like shapes, and elevated CD40 expression. Concurrently, there was an increase in pro-inflammatory cytokine levels and a decrease in anti-inflammatory cytokine levels. Further investigation revealed that in the microglial, the expression of cGAS and STING was elevated, along with an increase in oxidative products and inflammatory responses. CMA stimulation further intensified these changes, while cGAS knockdown mitigated them. Finally, we demonstrated that cGAS knockdown inhibited the oxidative stress, cell activation-related changes, and neuroinflammatory responses induced by CMPK2 overexpression in the BV2 neuroinflammation model. Molecular docking experiments showed that CMPK2 stably binds to cGAS at the protein level. These findings suggest that the cGAS-STING pathway mediates CMPK2-induced microglial activation. In summary, our study demonstrates that LPS-induced CMPK2 overactivity promotes microglial activation and neuroinflammatory through the cGAS-STING pathway.
Similar content being viewed by others

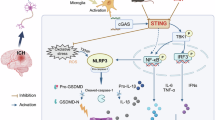

Introduction
Microglia are resident macrophage-like immune cells in the brain, often referred to as the “first line of defense” of the central nervous system, playing a pivotal role in maintaining neural homeostasis and responding to pathological stimuli1,2,3. Under physiological conditions, microglia primarily exist in a resting state, secreting anti-inflammatory cytokines and neurotrophic factors to promote neural repair, tissue regeneration, and the maintenance of the central nervous system’s homeostasis. However, under pathological conditions, microglia are rapidly activated and undergo phenotypic transitions to distinct functional states, among which M1-activated microglia exhibit neurotoxic effects4, whereas resting microglia are more involved in tissue repair. This phenotypic transition mechanism makes microglia indispensable in regulating neuronal functions3.
In recent years, studies have shown that microglial activation is closely associated with the onset and progression of neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease, as well as stroke5,6,7,8. In particular, during stroke, neuroinflammation has been widely recognized as a critical factor in its initiation and progression. When brain tissue experiences ischemic or hemorrhagic injury, microglia are rapidly activated, triggering neuroinflammatory responses that can persist for several days or even longer. Activated microglia respond swiftly to ischemic or hemorrhagic damage by releasing pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and nitric oxide (NO), directly influencing the pathological progression of stroke9,10,11,12. Furthermore, activated microglia exhibit macrophage-like functions, including the phagocytosis of cellular debris and foreign microorganisms, as well as the secretion of cytokines and matrix metalloproteinases (MMPs). These substances not only compromise the integrity of the blood-brain barrier but also exacerbate inflammatory responses and tissue damage, thereby accelerating the progression of stroke and other related diseases13,14,15.Therefore, an in-depth investigation into the molecular mechanisms underlying microglial-mediated neuroinflammation is essential for uncovering the fundamental pathological processes of central nervous system diseases.
CMPK2 (cytidine monophosphate kinase 2) is a key mitochondrial nucleotide kinase involved in cellular energy metabolism and nucleotide synthesis16,17,18. The study of CMPK2 in inflammation-related diseases has garnered increasing attention. Previous studies have demonstrated that CMPK2 plays a crucial role in inflammatory activation, particularly in response to viral infections. Inhibiting CMPK2 has been shown to significantly reduce synovial inflammation and cartilage damage, effectively slowing the progression of inflammation19,20,21. During stroke, ischemia and reperfusion injury lead to metabolic dysregulation and inflammatory responses in brain tissue, with CMPK2 playing a critical role in this process. First, CMPK2 promotes mitochondrial DNA synthesis and repair, maintaining mitochondrial function and cellular energy metabolism, which may be crucial for post-ischemic brain tissue recovery. Additionally, upregulation of CMPK2 expression can initiate mtDNA synthesis in LPS-induced immune cells, triggering inflammasome-mediated inflammatory responses22. AAV-mediated microglial/macrophage CMPK2 gene knockout or the use of dehydroabietic acid to inhibit CMPK2 reduces neuroinflammation and improves ischemic injury, suggesting its potential as a therapeutic target for neurological diseases23. CMPK2 not only plays a role in neuroinflammation but also shows potential in neuroprotection. Some studies suggest that regulating CMPK2 expression and activity can reduce neuronal damage. For example, techniques based on gene silencing or pharmacological interventions to lower CMPK2 activity may improve central neuron survival16. Despite significant progress in the study of CMPK2 in neuroinflammation, the specific mechanisms involved require further exploration.
The cGAS-STING signaling pathway, as a novel and crucial immune signaling mechanism, is closely linked to neuroinflammatory responses24,25. The mechanism of this pathway has been well characterized. Upon binding to intracellular or extracellular DNA, cGAS catalyzes the synthesis of cGAMP from ATP and GTP. Acting as a second messenger, cGAMP binds to STING, triggering a conformational change that activates STING and promotes its translocation from the endoplasmic reticulum to the Golgi apparatus. This activation subsequently induces the expression of interferons (IFNs) and pro-inflammatory cytokines such as TNF-α and IL-6, thereby amplifying the host immune response against pathogens26. Thus, the cGAS-STING pathway may play a significant role in microglial-mediated neuroinflammation. Studies have shown that in herpes simplex encephalitis (HSE), microglia can be activated through the cGAS-STING pathway, leading to IFN production and initiating antiviral defense mechanisms. Mice lacking cGAS are more susceptible to HSV-1 infection27. In chronic neurodegenerative conditions, the activation of the cGAS-STING pathway and elevated cytokine levels often coexist28,29.Therefore, the cGAS-STING pathway may play a crucial role in microglial-mediated neuroinflammation, but its involvement in CMPK2-mediated neuroinflammation remains unclear. It is known that cytosolic mtDNA is a classical activator of cGAS20,30, and abnormal activation of CMPK2 is associated with mtDNA release. However, whether it induces microglial-mediated neuroinflammation through the activation of the cGAS-STING pathway has not been reported.
Therefore, This study aims to comprehensively investigate the specific molecular mechanisms by which CMPK2 activates microglial-mediated neuroinflammation through the cGAS-STING pathway, thereby elucidating its central role in microglial-mediated neuroinflammation within the central nervous system. Specifically, by regulating CMPK2 and its downstream signaling pathways, it may be possible to mitigate pathological inflammatory responses caused by aberrant microglial activation, thereby reducing neuronal damage, promoting functional recovery, and providing foundational theoretical guidance for the treatment of neurological diseases.
Methods
Cell culture and treatment
BV2 microglial cell were purchased from Fuheng Biotechnology Co., Ltd. (Shanghai, China) and utilized to establish a neuroinflammation cell model. The cells were cultured in DMEM/DF12 medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin, maintained in a humidified incubator at 37 °C with 5% CO2. microglial-mediated neuroinflammation was induced using 1 µg/ml LPS (Sigma-Aldrich, Cat. No. L5293). Additionally, BV2 activation of the cGAS-STING signaling pathway was induced using 100 µg/ml CMA, A small molecule drug capable of highly and specifically activating the cGAS-STING signaling pathway. (Sigma-Aldrich, Cat. No. 17927)31.
Mouse primary microglial cultures
We extracted primary microglial cells from 1-3-day-old C57BL/6 mice. First, the whole brain tissue was isolated and the meninges were carefully removed. The brain tissue was then minced and digested with trypsin at 37 °C in a 5% CO₂ incubator. The digested tissue was passed through a 70 μm nylon mesh to remove large debris. Subsequently, a single-cell suspension was prepared by repeated pipetting, followed by density gradient centrifugation to remove non-cellular components, resulting in a mixed glial cell suspension. The suspension was plated into culture flasks containing DMEM/F12 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. The cells were cultured at 37 °C in a 5% CO₂ incubator for 7–10 days, with regular medium changes to maintain cell viability. Once the mixed glial cell culture reached confluence, microglial cells were isolated using the shake-off method. Specifically, the flasks were shaken at 200 rpm at 37 °C for 2 h. The culture supernatant containing microglial cells was collected, centrifuged, and resuspended to obtain highly purified microglial cells. These microglial cells were then used for subsequent analyses and experiments32,33. This study was approved by the Ethical Committee for Animal Experiments at The First Affiliated Hospital of Nanchang University and conducted in accordance with the “Guidelines for Ethical Review of Welfare of Laboratory Animals in China” (2018/09) and the “Guide for the Care and Use of Laboratory Animals.” All procedures were performed in compliance with the ARRIVE guidelines.
Transfection
To overexpress/knockdown CMPK2 and knockdown cGAS in BV2 cells, we obtained the CMPK2 (NM_207315) Human Tagged ORF clone (RC218794), CMPK2 Human siRNA Oligo Duplex (SR315070), and Human cGAS shRNA Plasmid Kit (TL305813) from OriGene Technologies (Rockville). Concurrently, the pCMV6-Entry Mammalian Expression Vector (Rockville) was acquired from OriGene Technologies(Rockville) as a control plasmid. One day before transfection, BV2 cells were seeded in 6-well plates at a density of 1 × 10^5 cells per well. The following day, cells were transfected. The old medium was replaced with medium containing the overexpression plasmid, and transfection reagents were added according to the Lipofectamine 2000 kit protocol (Invitrogen). After thorough mixing, the medium was incubated with the cells, and transfection efficiency was assessed by Western blot34.
Western blotting
Protein expression in BV2 cells was detected by Western blot. First, total protein from cells and tissues was extracted using RIPA lysis buffer (P0013B; Beyotime) and protease and phosphatase inhibitors (B14001 and B15001; BioTools). Protein concentration was determined by the BCA protein assay (P0009; Beyotime). Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; Boster) and transferred onto PVDF membranes (Solarbio). Membranes were then blocked with blocking solution containing 5% milk and subsequently probed with the following antibodies: mouse anti-β-actin (8H10D10; Cell Signaling Technology), rabbit anti-CMPK2 (ab139720; Abcam), mouse anti-cGAS (36468; Cell Signaling Technology), and rabbit anti-STING (ab239074; Abcam). Detection was performed using horseradish peroxidase-conjugated secondary antibodies (goat anti-mouse/rabbit IgG, 1:1000)12.
Enzyme-linked immunosorbent assay (ELISA)
BV2 cells were seeded in a 24-well plate and incubated overnight. Cells were treated with LPS, CMA, CMPK2, si-CMPK2 and si-cGAS for 24 h, and then, according to the manufacturer’s instructions, the concentrations of pro-inflammatory cytokines lL-6(Pl326; Beyotime), TNF-α (PT512; Beyotime), lL-1β (Pl301; Beyotime), and TGF-β (PT878; Beyotime) in the culture supernatant were measured using ELlSA assay kits12.
RNA extraction and RT-qPCR analysis
Total RNA was extracted from samples using the EZ-press RNA Purification Kit (EZBioscience). RNA concentration and purity were determined using a NanoDrop ND-2000 spectrophotometer (Thermo Fisher Scientific, Inc.). Complementary DNA (cDNA) was synthesized from 1 µg of total RNA using the PrimeScript™ RT reagent Kit with gDNA Eraser (Takara). The resulting cDNA product was diluted 1:10 in ddH2O. RT-qPCR was performed using SYBR® Premix Ex Taq™ II and the CFX96 Real-Time PCR System (Bio-Rad). The expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as a normalization control. Relative gene expression levels were calculated and normalized using the ΔΔCt method12.
Immunofluorescence staining
BV2 cells were cultured in 12-well plates and blocked with buffer containing 0.03% Triton X-100 and 5% goat serum albumin (C0265; Beyotime). The cells were then incubated overnight at 4 °C with antibodies against CMPK2 (1:100), cGAS (1:50), and STING (1:200). Subsequently, the cells were incubated for 2 h at room temperature with Cy3-conjugated goat anti-rabbit IgG (GB21303; 1:200; Servicebio) or Alexa Fluor 488-conjugated goat anti-mouse IgG (GB25301; 1:200; Servicebio). Immunostained images were captured using a confocal microscope (Leica)12,35.
Detection of cell surface CD40
CD40 is a marker of microglial activation and plays a crucial role in inflammatory responses36. The surface expression of CD40 on BV2 cells was detected using a fluorescein isothiocyanate (FITC)-conjugated CD40 antibody (BioLegend; 124607), Meanwhile, FITC Rat lgG2a, lsotype Ctrl Antibody (BioLegend; 400505) were used. BV2 cells were seeded at a density of 2 × 10^5 cells per well in 12-well plates and incubated overnight. Cells were then treated with LPS, CMA. According to the manufacturer’s instructions, 1 µL of FITC-conjugated CD40 antibody was added to each well and incubated with the BV2 cells for 15 min. The cells were then washed twice with PBS containing fetal bovine serum and analyzed for fluorescence using an LSRII flow cytometer (BD Biosciences). Data were processed using FlowJo software (version 10.8.1; https://www.flowjo.com)12,37.
ROS detection
The levels of reactive oxygen species (ROS) in BV2 cells under various treatment conditions were detected using DCFH-DA (Sigma-Aldrich). BV2 cells were seeded at a density of 2 × 10^5 cells per well in 12-well plates and incubated overnight, followed by treatment with LPS, CMA. DCFH-DA was diluted to a final concentration of 10 µM according to the manufacturer’s instructions and incubated with the BV2 cells for 30 min. The cells were then harvested, washed twice with PBS, and fluorescence was analyzed using an LSRII flow cytometer (BD Biosciences). Data were analyzed using FlowJo software (version 10.8.1; https://www.flowjo.com)38.
Molecular docking
This study employed the HDOCK online platform (http://h-dockphys.hust.edu.cn/) for molecular docking analysis. HDOCK was used to explore various conformations of protein docking, the binding activity under these conformations, and interactions between amino acid residues within 5 Å. The 3D structure of CMPK2 was sourced from the Alphafold protein database, with non-redundant protein structures being selected. The structure of CMPK2 was also retrieved from the Protein Data Bank. PyMOL software (version 2.3.0; https://pymol.org) was used to isolate the original ligands and protein structures, perform dehydration, and remove organic molecules. The “prepare” module in Discovery Studio software was utilized for protein preparation, including hydrogenation and protonation. Ligplus software was used to analyze the 2D interactions between the proteins. The “analysis interface” module in Discovery Studio was employed to investigate the protein-protein interaction interface. PyMOL was used to visualize the amino acid residues involved in the interactions between the two proteins13.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software (version 10.0; https://www.graphpad.com). Data are presented as mean ± SD. The statistical significance between two independent groups was determined using Student’s t-test, while one-way ANOVA was used to assess differences among multiple groups, and the Bonferroni post-test was used. A p-value of < 0.05 was considered statistically significant, with differences between groups indicated by asterisks (*)35.
Results
CMPK2 upregulation in LPS-stimulated BV2 Cells
In this study, we investigated the expression of CMPK2 in BV2 cells using RT-qPCR and compared CMPK2 levels between resting and activated microglia. For this purpose, BV2 cells were stimulated with different concentrations of LPS (0, 0.1, 0.2, 0.5, and 1 µg/ml) for 24 h. We observed that CMPK2 expression was significantly induced in a dose-dependent manner (Fig. 1A). Additionally, BV2 cells were stimulated with 1 µg/ml LPS for various durations (0, 1, 6, 12, and 24 h), and CMPK2 expression levels were found to be upregulated in a time-dependent manner, with a more pronounced increase at 24 h (Fig. 1B). These results indicate that CMPK2 expression levels are significantly higher in LPS-stimulated BV2 cells compared to the control group.

CMPK2 Upregulation in LPS-Stimulated BV2 Cells. (A) Analysis of CMPK2 mRNA expression under different concentrations of LPS treatment. (B) Analysis of CMPK2 mRNA expression at different time points following LPS treatment. The presented data represent the mean ± standard deviation (SD) derived from three independent experiments. Statistical significance of the fold change was determined, with ****P < 0.0001 denoting the significance levels.
LPS Induces microglial-mediated neuroinflammation by activating CMPK2
Aberrant expression of CMPK2 is potentially associated with neurodegenerative diseases. Elevated intracellular CMPK2 can induce mitochondrial dysfunction, leading to the leakage of mitochondrial DNA (mtDNA) into the cytoplasm, which is linked to neuroinflammatory processes23. To further elucidate the role of CMPK2 in the microglial-mediated neuroinflammation, we observed that both the transcript and protein levels of CMPK2 were upregulated upon LPS stimulation, and knockdown of CMPK2 (LPS + si-CMPK2 group) significantly reduced this upregulation (Fig. 2A, B). To investigate the potential of CMPK2 knockdown in mitigating microglial.

LPS Induces microglial-mediated neuroinflammation by Activating CMPK2. (A) Western blot analysis of CMPK2 protein levels under different treatments. (B) Analysis of CMPK2 mRNA expression under different treatments. (C) Immunofluorescence analysis of CMPK2 expression in mouse primary microglial under different treatments. (D) Analysis of morphological differences between treated and control cells. (E) Analysis of the microglial activation marker CD40 expression.F,G. Analysis of mRNA and protein expression levels of IL-6, TNF-α, IL-1β, and TGF-β. The presented data represent the mean ± standard deviation (SD) derived from three independent experiments. Statistical significance of the fold change was determined, with *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 denoting the significance levels.
activation and cytokine release, we utilized immunofluorescence staining, which revealed that CMPK2 knockdown significantly reduced CMPK2 expression in mouse primary microglial (Fig. 2C). Further analysis of the impact of CMPK2 knockdown on LPS-stimulated BV2 cell activation and neuroinflammation showed morphological changes and differences in surface marker expression. LPS stimulation increased cell activation, characterized by enlarged cell bodies, shortened processes, and transformation to a round or rod-like shape. CMPK2 knockdown restored cells to a smaller, more ramified resting state (Fig. 2D). Additionally, LPS-induced upregulation of the microglial activation marker CD40 was observed, which was downregulated following CMPK2 knockdown (Fig. 2E). We also evaluated the expression levels of IL-6, TNF-α, IL-1β, and TGF-β after LPS stimulation and CMPK2 inhibition. RT-qPCR and ELISA analysis showed that CMPK2 knockdown inhibited LPS-induced increases in IL-6, TNF-α, and IL-1β, while counteracting the downregulation of the anti-inflammatory cytokine TGF-β (Fig. 2F–G). Overall, our findings suggest that CMPK2 knockdown effectively inhibits LPS-induced microglial activation.
LPS Enhances microglial-mediated neuroinflammation via the cGAS/STING Pathway
The cGAS-STING pathway plays a crucial role in the activation and regulation of inflammation by recognizing cytosolic DNA and activating associated signaling pathways to mediate anti-infection and anti-tumor immune responses, as well as participating in the regulation of various physiological function39. Understanding the structure and molecular biology of the cGAS-STING pathway can aid in developing new therapeutic strategies for inflammation-related diseases34. In our study, we first confirmed that the expression of key markers cGAS and STING in the cGAS-STING signaling pathway was upregulated in BV2 cells stimulated with LPS. The simultaneous use of the cGAS-STING pathway inducer CMA further enhanced this expression (Fig. 3A and B), accompanied by an increase in cellular lipid oxidation products ROS (Fig. 3C). Lastly, RT-qPCR and ELISA analysis indicated that induction of cGAS-STING significantly exacerbated the LPS-induced increase in IL-6, TNF-α, and IL-1β, while counteracting the downregulation of the anti-inflammatory cytokine TGF-β (Fig. 3D-E).In summary, these results suggest that the cGAS-STING pathway is involved in regulating microglial-mediated neuroinflammation.

LPS Enhances microglial-mediated neuroinflammation via the cGAS/STING Pathway. (A) Western blot analysis of cGAS and STING protein levels under different treatments. (B) Analysis of cGAS and STING mRNA expression under different treatments. (C) Flow cytometry analysis of oxidative stress (ROS) levels under different treatments.D,E. Analysis of mRNA and protein expression levels of IL-6, TNF-α, IL-1β, and TGF-β. The presented data represent the mean ± standard deviation (SD) derived from three independent experiments. Statistical significance of the fold change was determined, with *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 denoting the significance levels.
Inhibition of the cGAS/STING pathway alleviates microglial-mediated neuroinflammation
Subsequently, to further elucidate the regulatory value of targeting the cGAS-STING pathway in neuroinflammation, we constructed RNA interference plasmids to transfect BV2 microglial cells and interfere with the expression of the cGAS protein. We first confirmed that si-cGAS effectively reduced the intracellular expression levels of cGAS protein, along with a concomitant decrease in STING protein levels (Fig. 4A and C), thereby effectively inhibiting the activity of the intracellular cGAS-STING pathway. Furthermore, we explored the specific impact of cGAS-STING pathway inhibition on microglial-mediated neuroinflammation. Initially, we found that silencing cGAS significantly reduced cellular oxidative stress levels (Fig. 4D). Finally, analysis of inflammatory cytokines revealed that following cGAS knockdown, the expression levels of pro-inflammatory cytokines IL-6, TNF-α, and IL-1β decreased, whereas the expression level of the anti-inflammatory cytokine TGF-β was restored (Fig. 4E, F). In summary, our findings indicate that the cGAS-STING pathway is involved in the activation of microglial, and inhibiting the cGAS-STING pathway can effectively mitigate microglial-mediated neuroinflammation.

Inhibition of the cGAS/STING Pathway Alleviates microglial-mediated neuroinflammation. (A) Western blot analysis of cGAS and STING protein levels under different treatments. (B) Analysis of cGAS and STING mRNA expression under different treatments. (C) Immunofluorescence analysis of cGAS and STING expression in mouse primary microglial under different treatments. (D) Flow cytometry analysis of oxidative stress (ROS) levels under different treatments. E, F. Analysis of mRNA and protein expression levels of IL-6, TNF-α, IL-1β, and TGF-β. The presented data represent the mean ± standard deviation (SD) derived from three independent experiments. Statistical significance of the fold change was determined, with *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 denoting the significance levels.
CMPK2 induces microglial-mediated neuroinflammation via the cGAS/STING Pathway
Previous studies have demonstrated that both CMPK2 and the cGAS/STING pathway independently influence microglial-mediated neuroinflammation. However, the mechanism by which CMPK2 exacerbates inflammation and whether it interacts with the cGAS/STING pathway requires further investigation. To address this question, we first demonstrated that transfection with CMPK2 overexpression plasmid could effectively increase CMPK2 protein expression in BV2 cells (Fig. 5A). Subsequently, we found that overexpression of CMPK2 induced cGAS-STING pathway activation, while knockdown of cGAS inhibited the molecular expression of the cGAS-STING pathway (Fig. 5B and C). Morphological observations indicated that cGAS knockdown inhibited CMPK2 overexpression-induced microglial activation, characterized by increased cell size, shortened processes, and transformation into a round or rod-like shape (Fig. 5D). This was accompanied by a downregulation of the microglial activation marker CD40 (Fig. 5E). These results suggest that the cGAS-STING pathway plays a crucial role in CMPK2-mediated cell activation and neuroinflammation, implying potential interactions that regulate cell function. Molecular docking visualization based on PYMOL further supports this interaction, showing 15 amino acid pairs forming hydrogen bonds, with the tightest bond between the N atom of lysine at position 368 of the CMPK2 peptide and the O atom of aspartic acid at position 95 of the cGAS peptide. There were no disulfide bonds but the presence of salt bridges and other connection structures (Fig. 5F). Finally, RT-qPCR and ELISA results showed that cGAS knockdown significantly mitigated the CMPK2 overexpression-induced increases in IL-6, TNF-α, and IL-1β, and significantly upregulated the anti-inflammatory cytokine TGF-β (Fig. 5G-H). In summary, these findings indicate that CMPK2 can regulate microglial-mediated neuroinflammation through the cGAS-STING pathway.

CMPK2 Induces microglial-mediated neuroinflammation via the cGAS/STING Pathway.A. Western blot analysis of CMPK2 under control plasmid and CMPK2 overexpression plasmid groups.B.Western blot analysis of CMPK2, cGAS, and STING protein levels under different treatments.C. Analysis of CMPK2, cGAS, and STING mRNA expression under different treatments.D. Analysis of morphological differences between treated and control cells.E. Analysis of microglial activation marker CD40 expression.F. Molecular docking analysis of CMPK2 and cGAS interaction.G,H. Analysis of mRNA and protein expression levels of IL-6, TNF-α, IL-1β, and TGF-β. The presented data represent the mean ± standard deviation (SD) derived from three independent experiments. Statistical significance of the fold change was determined, with *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 denoting the significance levels.
Discussion
microglial-mediated neuroinflammation is involved in the progression of many neurological disorders, contributing to the exacerbation of diseases such as stroke through the release of a large amount of inflammatory mediators34,38,40,41. Under physiological conditions, microglia exhibit a ramified morphology, performing critical functions such as environmental monitoring, synaptic pruning, and clearance of apoptotic neurons to maintain central nervous system (CNS) homeostasis42 In the event of brain injury, microglia are the first responders, transitioning into an activated state43,44. Microglial activation significantly increases the number of apoptotic neurons. Compounds such as salvianolic acid B and borneol ester have been shown to reduce BV2 microglial activation, inhibit NF-κB activity, and suppress the production of pro-inflammatory mediators, thereby reducing infarct volume and improving sensory, motor, and cognitive functions45. However, the specific factors driving microglial activation remain incompletely understood. In this study, we established a neuroinflammation cell model by stimulating BV2 cells with LPS, and we found that CMPK2 expression was elevated in a time- and dose-dependent manner. Elevated CMPK2 expression was associated with BV2 microglial activation, characterized by increased cell size, shortened processes, and transformation into round or rod-like shapes, along with upregulation of the microglial activation marker CD40. At the inflammatory level, we detected increases in pro-inflammatory cytokines IL-6, TNF-α, and IL-1β, and a significant decrease in the anti-inflammatory cytokine TGF-β. This indicates that LPS can significantly activate microglia and induce neuroinflammation, with CMPK2 potentially playing a key role in this process.
CMPK2 primarily regulates mitochondrial function and energy metabolism, maintaining cell survival and function, and plays a modulatory role in immune responses and inflammation processes46,47. Studies have shown that CMPK2 expression levels are positively correlated with infarct volume and NIHSS scores, indicating the potential regulatory value of CMPK2 in neurological diseases48,49. Downregulation of CMPK2 activity using cannabidiol has been shown to inhibit inflammasome activation50. Additionally, knockdown of CMPK2 in a mouse tMCAO model can inhibit inflammation associated with caspase-1 activation and IL-1β release, and in vitro it suppresses LPS-induced microglial activation. Although these data suggest that the CMPK2 gene may play an indirect and complex role in neuroinflammation, the regulatory value of CMPK2 in microglial-mediated neuroinflammation remains to be elucidated. In our study, we first demonstrated that CMPK2 protein levels were abnormally elevated in LPS-stimulated BV2 cells. This abnormal CMPK2 activity was potentially associated with microglial activation and increased pro-inflammatory cytokines IL-6, TNF-α, and IL-1β, along with decreased anti-inflammatory cytokine TGF-β. To clarify this regulatory effect, we utilized RNA interference to disrupt CMPK2 gene translation. We further found that CMPK2 knockdown inhibited LPS-induced microglial activation and subsequent neuroinflammatory responses. Thus, our findings preliminarily demonstrate the important role of CMPK2 in microglial-mediated neuroinflammation during PD pathogenesis.
To further elucidate the specific molecular mechanisms by which CMPK2 regulates microglial-mediated neuroinflammation, we conducted an in-depth investigation of the cGAS-STING pathway. The cGAS-STING pathway is a critical and potent regulatory mechanism in autoimmune and neuroinflammatory responses and serves as a key mechanism for the central nervous system to detect pathogenic DNA. In most neurodegenerative diseases, elevated levels of neuroinflammation and pro-inflammatory cytokines are observed, and inhibiting the cGAS-STING pathway offers a potential therapeutic intervention51,52,53. During the progression of neurodegenerative diseases, the cGAS-STING pathway appears to play a similarly important regulatory role. For instance, in LRRK2 knockout cells, an increased mitochondrial fission caused by dynamin-related protein 1 (Drp1) and heightened oxidative stress leads to the activation of the cGAS-STING pathway. Modulating this pathway can alleviate the oxidative stress response induced by LRRK2 knockout54.To determine the involvement and potential regulatory value of the cGAS-STING pathway in LPS-stimulated BV2 microglial, we first confirmed the elevated expression levels of key effector molecules within the cGAS-STING pathway in our neuroinflammation cell model. These elevated effector molecules were potentially linked to microglial activation, oxidative stress response, and neuroinflammation. This pro-microglial activation and neuroinflammatory effect were further intensified when the cells were stimulated with the cGAS-STING pathway inducer CMA. To further confirm that the cGAS-STING pathway is a crucial intermediary in LPS-induced neuroinflammatory responses, we knocked down the cGAS gene. Successful inhibition of the cGAS-STING pathway resulted in a significant reduction in LPS-induced microglial activation, oxidative stress response, and the release of inflammatory cytokines. This evidence strongly supports the crucial role of the cGAS-STING pathway in microglial-mediated neuroinflammation.
Abnormal expression of CMPK2 has been confirmed as a key trigger for microglial-mediated neuroinflammation, with the cGAS-STING pathway serving as an indispensable intermediate mechanism in this neuroinflammatory process. To investigate whether the cGAS-STING pathway functions as the molecular mechanism through which CMPK2 mediates neuroinflammatory responses, we first conducted CMPK2 overexpression experiments. The results showed that overexpression of CMPK2 activated BV2 microglial cells, leading to morphological changes characterized by enlarged cell bodies, shortened processes, and transformation into round or rod-like shapes. Additionally, the expression of the activation marker CD40 was significantly upregulated, accompanied by increases in pro-inflammatory cytokines IL-6, TNF-α, and IL-1β, as well as a decrease in the anti-inflammatory cytokine TGF-β.To further verify the role of the cGAS-STING pathway in CMPK2-mediated neuroinflammatory responses, we knocked down cGAS to block the activation of the cGAS-STING pathway while overexpressing CMPK2. The results indicated that microglial-mediated neuroinflammation induced by CMPK2 overexpression were significantly alleviated. Finally, molecular docking visualization based on PYMOL revealed a strong interaction between CMPK2 and cGAS, further confirming that CMPK2 can activate microglia through the cGAS-STING pathway, thereby triggering microglial-mediated neuroinflammation.
However, it is undeniable that this study has certain limitations. Firstly, during the LPS-induced abnormal elevation of CMPK2 protein levels, we did not conduct an in-depth investigation into the structural changes of the CMPK2 gene caused by LPS. Consequently, we were unable to determine whether the abnormal elevation of CMPK2 protein was due to gene locus mutations or transcriptional modifications. Additionally, this study only explored the regulation of the cGAS-STING pathway by CMPK2 in activating BV2 microglial cells and inducing neuroinflammatory responses through in vitro cell experiments. We did not validate these findings in vivo using animal models. Future research should address these issues through sequencing and animal experiments.
Conclusion
In summary, during the pathological process of microglial-mediated neuroinflammation, LPS induces an abnormal elevation in CMPK2 activity, subsequently activating the cGAS-STING pathway. This activation leads to the activation of microglia and the initiation of neuroinflammatory responses. These findings provide valuable theoretical insights for understanding the mechanisms underlying neurological diseases and for developing novel clinical therapeutic strategies.
Data availability
The data supporting the findings of this study are derived from the experiments conducted by our research team. They can be provided by the corresponding authors upon reasonable request.
References
Lan, X., Han, X., Li, Q., Yang, Q. W. & Wang, J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat. Rev. Neurol. 13 (7), 420–433 (2017).
Scholz, R., Brösamle, D., Yuan, X., Beyer, M. & Neher, J. J. Epigenetic control of microglial immune responses. Immunol. Rev. 323 (1), 209–226 (2024).
Subramaniam, S. R. & Federoff, H. J. Targeting microglial activation States as a therapeutic avenue in Parkinson’s disease. Front. Aging Neurosci. 9, 176 (2017).
Tang, Y. & Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 53 (2), 1181–1194 (2016).
Mee-Inta, O., Zhao, Z. W. & Kuo, Y. M. Phys. Exerc. Inhibits Inflamm. Microglial Activation Cells 8(7) (2019).
Yan, Y. Q. et al. Parkin regulates microglial NLRP3 and represses neurodegeneration in Parkinson’s disease. Aging Cell. 22 (6), e13834 (2023).
Zhong, L. et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nat. Commun. 10 (1), 1365 (2019).
Wang, H. et al. TREM2, microglial and ischemic stroke. J. Neuroimmunol. 381, 578108 (2023).
Yang, X. et al. Small extracellular vesicles-derived from 3d cultured human nasal mucosal mesenchymal stem cells during differentiation to dopaminergic progenitors promote neural damage repair via miR-494-3p after manganese exposed mice. Ecotoxicol. Environ. Saf. 280, 116569 (2024).
Abdu, H., Tadese, F. & Seyoum, G. Clinical profiles, comorbidities, and treatment outcomes of stroke in the medical ward of Dessie comprehensive specialized hospital, Northeast Ethiopia; a retrospective study. BMC Neurol. 22 (1), 399 (2022).
Guruswamy, R. & ElAli, A. Complex roles of microglial cells in ischemic stroke pathobiology: New insights and future directions. Int. J. Mol. Sci. 18(3) (2017).
Zheng, Z. et al. LRRK2 regulates ferroptosis through the system Xc-GSH-GPX4 pathway in the neuroinflammatory mechanism of Parkinson’s disease. J. Cell. Physiol. 239 (5), e31250 (2024).
Yao, L. et al. LRRK2 Gly2019Ser mutation promotes ER stress via interacting with THBS1/TGF-β1 in Parkinson’s disease. Adv. Sci. (Weinh). 10 (30), e2303711 (2023).
Iadecola, C. & Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 17 (7), 796–808 (2011).
Zheng, Z. et al. Mechanisms of Autoimmune Cell in DA Neuron Apoptosis of Parkinson’s Disease: Recent Advancement.Oxid Med Cell Longev(2022) 7965433. (2022).
Zhao, M. et al. Loss of function of CMPK2 causes mitochondria deficiency and brain calcification. Cell. Discov. 8 (1), 128 (2022).
Xian, H. et al. Metformin Inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity 54 (7), 1463–1477e11 (2021).
Chen, Y. et al. Electroacupuncture inhibits NLRP3 activation by regulating CMPK2 after spinal cord injury. Front. Immunol. 13, 788556 (2022).
Tang, Z. et al. Drugs targeting CMPK2 inhibit pyroptosis to alleviate severe pneumonia caused by multiple respiratory viruses. J. Med. Virol. 96 (5), e29643 (2024).
Riley, J. S. & Tait, S. W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 21 (4), e49799 (2020).
Jin, L. et al. The FTO-CMPK2 pathway in Fibroblast-like synoviocytes modulates rheumatoid arthritis synovial inflammation and cartilage homeostasis via MtDNA regulation. Int. J. Biol. Sci. 20 (5), 1617–1633 (2024).
Newman, L. E. & Shadel, G. S. Mitochondrial DNA release in innate immune signaling. Annu. Rev. Biochem. 92, 299–332 (2023).
Guan, X. et al. Microglial CMPK2 promotes neuroinflammation and brain injury after ischemic stroke. Cell. Rep. Med. 5 (5), 101522 (2024).
Beavan, M. et al. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol. 72 (2), 201–208 (2015).
Hong, C. et al. Foijer, cGAS-STING drives the IL-6-dependent survival of chromosomally instable cancers. Nature 607 (7918), 366–373 (2022).
Decout, A., Katz, J. D., Venkatraman, S. & Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 21 (9), 548–569 (2021).
Reinert, L. S. et al. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 7, 13348 (2016).
Hofer, M. J. & Campbell, I. L. Type I interferon in neurological disease-the devil from within. Cytokine Growth Factor. Rev. 24 (3), 257–267 (2013).
Baruch, K. et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346 (6205), 89–93 (2014).
Han, F. et al. Retraction notice to: Silencing of LncRNA LINC00857 enhances BIRC5-Dependent Radio-Sensitivity of lung adenocarcinoma cells by recruiting NF-κB1. Mol. Ther. Nucleic Acids. 28, 538 (2022).
Stansley, B., Post, J. & Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflammation. 9, 115 (2012).
Ryu, K. Y. et al. Dasatinib regulates LPS-induced microglial and astrocytic neuroinflammatory responses by inhibiting AKT/STAT3 signaling. J. Neuroinflammation. 16 (1), 190 (2019).
Zhu, Y. et al. PTP1B inhibitor alleviates deleterious microglial activation and neuronal injury after ischemic stroke by modulating the ER stress-autophagy axis via PERK signaling in microglia. Aging (Albany NY). 13 (3), 3405–3427 (2021).
Yao, L. et al. MicroRNA-124 regulates the expression of MEKK3 in the inflammatory pathogenesis of Parkinson’s disease. J. Neuroinflammation. 15 (1), 13 (2018).
Huang, W. et al. Triggering receptor expressed on myeloid cells 2 protects dopaminergic neurons by promoting autophagy in the inflammatory pathogenesis of Parkinson’s disease. Front. Neurosci. 15, 745815 (2021).
Plastira, I. et al. Sattler, 1-Oleyl-lysophosphatidic acid (LPA) promotes polarization of BV-2 and primary murine microglia towards an M1-like phenotype. J. Neuroinflammation. 13 (1), 205 (2016).
Ismail, E. N., Jantan, I., Vidyadaran, S., Jamal, J. A. & Azmi, N. Phyllanthus Amarus prevents LPS-mediated BV2 microglial activation via MyD88 and NF-κB signaling pathways. BMC Complement. Med. Ther. 20 (1), 202 (2020).
Yao, L. et al. MicroRNA-124 regulates the expression of p62/p38 and promotes autophagy in the inflammatory pathogenesis of Parkinson’s disease. Faseb J. 33 (7), 8648–8665 (2019).
Oduro, P. K. et al. The cGAS-STING signaling in cardiovascular and metabolic diseases: Future novel target option for pharmacotherapy. Acta Pharm. Sin B. 12 (1), 50–75 (2022).
Emmrich, J. V., Ejaz, S., Neher, J. J., Williamson, D. J. & Baron, J. C. Regional distribution of selective neuronal loss and microglial activation across the MCA territory after transient focal ischemia: quantitative versus semiquantitative systematic immunohistochemical assessment. J. Cereb. Blood Flow. Metab. 35 (1), 20–27 (2015).
Liao, S. et al. A novel compound DBZ ameliorates neuroinflammation in LPS-stimulated microglia and ischemic stroke rats: Role of Akt(Ser473)/GSK3β(Ser9)-mediated Nrf2 activation. Redox Biol. 36, 101644 (2020).
Fetler, L. & Amigorena, S. Neuroscience. Brain under surveillance: The microglia patrol. Science 309 (5733), 392–393 (2005).
Kim, J. Y., Kim, N. & Yenari, M. A. Mechanisms and potential therapeutic applications of microglial activation after brain injury. CNS Neurosci. Ther. 21 (4), 309–319 (2015).
Hu, X. et al. Microglial and macrophage polarization—new prospects for brain repair. Nat. Rev. Neurol. 11 (1), 56–64 (2015).
Lalancette-Hébert, M. et al. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J. Neurosci. 32 (30), 10383–10395 (2012).
Li, D. et al. Upregulation of microglial ZEB1 ameliorates brain damage after acute ischemic stroke. Cell. Rep. 22 (13), 3574–3586 (2018).
Luo, Y. et al. CMPK2 accelerates liver ischemia/reperfusion injury via the NLRP3 signaling pathway. Exp. Ther. Med. 22 (6), 1358 (2021).
Liu, Z., Cai, Y. & He, J. High serum levels of 8-OHdG are an independent predictor of post-stroke depression in Chinese stroke survivors. Neuropsychiatr Dis. Treat. 14, 587–596 (2018).
Lorente, L. et al. García-Marín, DNA and RNA oxidative damage are associated to mortality in patients with cerebral infarction. Med. Intensiva (Engl Ed). 45 (1), 35–41 (2021).
Qi, X. et al. CBD promotes oral ulcer healing via inhibiting CMPK2-Mediated inflammasome. J. Dent. Res. 101 (2), 206–215 (2022).
Haag, S. M. et al. Targeting STING with covalent small-molecule inhibitors. Nature 559 (7713), 269–273 (2018).
Hansen, A. L. et al. Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc. Natl. Acad. Sci. U S A. 115 (33), E7768–e7775 (2018).
Hansen, A. L., Mukai, K., Schopfer, F. J., Taguchi, T. & Holm, C. K. STING palmitoylation as a therapeutic target. Cell. Mol. Immunol. 16 (3), 236–241 (2019).
Weindel, C. G. et al. LRRK2 maintains mitochondrial homeostasis and regulates innate immune responses to Mycobacterium tuberculosis. Elife 9 (2020).
Acknowledgements
All sections relevant to the research content of this paper are comprehensively covered in the manuscript and have been approved for publication.
Funding
This study was supported by the Key Project of National Natural Science Foundation of Jiangxi Province (20202ACBL206005).
Author information
Authors and Affiliations
Contributions
Feng Gao and Zijian Zheng: Conceptualization, Methodology, Experimentation, Data Analysis, Writing - Original Draft, Xinjie Liu: Investigation, Data Curation, Writing - Review & Editing. Jianwei Li: Investigation, Data Curation, Writing - Review & Editing. All authors contributed to the literature search, read, and approved the final manuscript. All authors confirm their accountability for the research presented in this manuscript, and no further changes to the authorship will be made.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent to participate and publication
All the authors actively participated in the experiments, contributed to the writing of the article, and thoroughly reviewed the manuscript. They have provided their authorization for its publication.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gao, F., Zheng, Z., Liu, X. et al. CMPK2 promotes microglial activation through the cGAS-STING pathway in the neuroinflammatory mechanism. Sci Rep 15, 11807 (2025). https://doi.org/10.1038/s41598-025-97232-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-97232-8
