
Abstract
Traditional drug design faces significant challenges due to inherent chemical and biological complexities, often resulting in high failure rates in clinical trials. Deep learning advancements, particularly generative models, offer potential solutions to these challenges. One promising algorithm is DrugGPT, a transformer-based model, that generates small molecules for input protein sequences. Although promising, it generates both chemically valid and invalid structures and does not incorporate the features of approved drugs, resulting in time-consuming and inefficient drug discovery. To address these issues, we introduce DrugGen, an enhanced model based on the DrugGPT structure. DrugGen is fine-tuned on approved drug-target interactions and optimized with proximal policy optimization. By giving reward feedback from protein–ligand binding affinity prediction using pre-trained transformers (PLAPT) and a customized invalid structure assessor, DrugGen significantly improves performance. Evaluation across multiple targets demonstrated that DrugGen achieves 100% valid structure generation compared to 95.5% with DrugGPT and produced molecules with higher predicted binding affinities (7.22 [6.30–8.07]) compared to DrugGPT (5.81 [4.97–6.63]) while maintaining diversity and novelty. Docking simulations further validate its ability to generate molecules targeting binding sites effectively. For example, in the case of fatty acid-binding protein 5 (FABP5), DrugGen generated molecules with superior docking scores (FABP5/11, -9.537 and FABP5/5, -8.399) compared to the reference molecule (Palmitic acid, -6.177). Beyond lead compound generation, DrugGen also shows potential for drug repositioning and creating novel pharmacophores for existing targets. By producing high-quality small molecules, DrugGen provides a high-performance medium for advancing pharmaceutical research and drug discovery.
Similar content being viewed by others

Introduction
Traditional drug design often falls short in handling the vast chemical and biological space features involved in ligand-receptor interactions1,2. Usually, a major proportion of suggested drug candidates fail in clinical trials3, making drug discovery a time-consuming and costly process. Recent advances in deep learning (DL), particularly in generative models, offer promising solutions for these obstacles4,5. Deep learning models have been extensively used in molecular design6,7, pharmacokinetics8,9,10,11, pharmacodynamics predictions12, and toxicity assessments10. These models improve the efficiency and accuracy of various tasks in drug development, contributing to different stages of drug discovery and optimization projects13,14. However, due to the insufficiency of available datasets, complexity of drug-target interactions, and complication of manipulating complex chemical structures, generative DL models also seem to be insufficient in proposing optimal answers to drug design problems15. Nevertheless, with the advancement of transformer-based architecture in large language models (LLMs), new horizons have opened up in various biological contexts. ProGen, a model developed to design new proteins with desired functionality and protein–ligand binding affinity prediction using pre-trained transformers (PLAPT), a model for protein–ligand binding affinity prediction, are successful examples of the application of LLMs in bioinformatics16,17. DrugGPT, an LLM based on the generative pre-trained transformer (GPT) architecture18 is another example that has shown potential in generating novel drug-like molecules having interactions with biological targets19.
DrugGPT leverages the transformer architecture to comprehend structural properties and structure–activity relationships. Receiving the amino acid sequence of a given target protein, this model generates simplified molecular input line entry system (SMILES)20 strings of interacting small molecules. By learning from large datasets of known drugs and their targets, DrugGPT can propose new compounds with desired properties by employing autoregressive algorithms for a stable and effective training process21, thus accelerating the lead discovery phase in drug development. However, the effectiveness of generative models in drug discovery relies heavily on the quality and relevance of the training data5. Models trained on comprehensive and accurately curated datasets are more likely to produce viable drug candidates22. Additionally, fine-tuning these models can enhance their performance for predictive applications23.
In this study, we developed “DrugGen”, an LLM based on the DrugGPT architecture, finetuned using a curated dataset of approved drug-target pairs; which is further enhanced using a policy optimization method. By utilizing this approach, DrugGen is optimized to generate drug candidates with optimized properties. Furthermore, we evaluated the model’s performance using custom metrics—validity, diversity, and novelty—to comprehensively assess the quality and properties of the generated compounds. Our results indicated that DrugGen generates chemically sound and valid molecules in comparison with DrugGPT while maintaining diversity and validity of generated structures. Notably, DrugGen excels in generating molecules with higher predicted binding affinities, increasing the likelihood of strong interactions with biological targets. Docking simulations further demonstrated the model’s capability to accurately target binding sites and suggest new pharmacophores. These findings highlight DrugGen’s promising potential to advance pharmaceutical research. Moreover, we proposed evaluation metrics that can serve as objective and practical measures for comparing future models.
Results
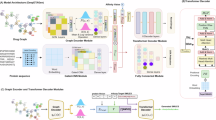
In order to develop an algorithm to generate drug-like structures, we gathered a curated dataset of approved drug-target pairs. We began by selecting a pre-trained model and then enhanced its performance through a two-step process. First, we employed supervised fine-tuning (SFT) on a dataset of approved sequence-SMILES pairs to fine-tune the model. Next, we utilized a reinforcement learning algorithm—proximal policy optimization (PPO)—along with a customized reward system to further optimize its performance. The final model was named DrugGen. The schematic design of the study is illustrated in Fig. 1.

Schematic representation of model development and evaluation. The top section illustrates the dataset creation and the training of DrugGen through supervised fine-tuning (SFT) and proximal policy optimization (PPO) using a customized reward function. The bottom section outlines the assessment process, based on validity, diversity, novelty, and binding affinity for both DrugGen and DrugGPT, along with docking simulations for DrugGen.
DrugGen is effectively fine-tuned on a dataset of approved drug-target
Supervised fine-tuning using the SFT trainer exhibited a steady decrease in training and validation loss over the epochs, indicating effective learning (Fig. 2A and Supplementary file 1). After three epochs of training, the loss of both the training and validation datasets reached a plateau. Therefore, checkpoint number three was selected for the second phase. In the second phase, the model was further optimized using PPO based on the customized reward system. Over 20 epochs of optimization, the model generated 30 unique small molecules for each target in each epoch, ultimately reaching a plateau in the reward diagram (Fig. 2B and Supplementary file 2).

Training process of DrugGen. (A) Learning curve of the model during supervised fine-tuning (SFT) and (B) reward trend during proximal policy optimization (PPO).
DrugGen generates valid, diverse, and novel small molecules
Eight proteins were selected for models assessments which include two targets with a high probability of association with diabetic kidney disease (DKD) from the DisGeNet database, angiotensin-converting enzyme (ACE) and peroxisome proliferator-activated receptor gamma (PPARG) and six proteins without known approved drugs, i.e., galactose mutarotase (GALM), putative fatty acid-binding protein 5-like protein 3 (FB5L3), short-wave-sensitive opsin 1 (OPSB), nicotinamide phosphoribosyltransferase (NAMPT), phosphoglycerate kinase 2 (PGK2), and fatty acid-binding protein 5 (FABP5), that identified as having a high probability of being targeted by approved small molecules through our newly developed druggability scoring algorithm, DrugTar24. For each target, 500 molecules were generated. The validity of generated molecules was 95.45% and 99.90% for DrugGPT and DrugGen, respectively (Chi-Squared, P < 10–38, Supplementary file 3). These molecules had an average diversity of 84.54% [74.24–90.48] for DrugGPT and 60.32% [38.89–92.80] for DrugGen (U = 358,245,213,849, P = 0, Fig. 3A and Supplementary file 4), indicating the generation of more similar molecules in DrugGen. These results suggest that DrugGen still generates a wide range of structurally diverse drug candidates rather than producing similar or redundant molecules. To assess the novelty of generated molecules, 100 unique small molecules were generated for each target. The validity scores for DrugGPT and DrugGen were in agreement with previous results (95.5% and 100%, respectively, Chi-Squared, P < 10–8, Supplementary file 5). After removing invalid structures, the novelty scores for DrugGPT and DrugGen were 66.84% [55.28–73.57] and 41.88% [24–59.66], respectively ([Mann–Whitney, U = 475980, P < 10–80], Fig. 3B and Supplementary file 5), indicating that fewer novel molecules were generated in DrugGen. These values indicate a good balance between diversity and novelty for DrugGen.

Comparison of molecular diversity, novelty, and binding affinity across different targets. (A) Comparison of molecular diversity distribution is shown as the frequency of the “1 – Tanimoto similarity” in percent for each target. (B) Scatter plots comparing novelty, with the “1 – Tanimoto similarity” in percent plotted against the molecular index. (C) Violin plots depicting the distribution of binding affinity for each target.
DrugGen generates small molecules with high affinity for their targets
We used two different measures to assess the binding affinity of the generated molecules to their respective targets: PLAPT, an LLM for predicting binding affinity, and molecular docking.
PLAPT: The same set of small molecules generated in novelty assessment (100 unique small molecules for each target) were used for assessing the quality of generated structures. Except for FABP5, DrugGen consistently produced small molecules with significantly higher binding affinities compared to DrugGPT ([7.22 [6.30–8.07] vs. 5.81 [4.97–6.63], U = 137,934, P < 10–85], Fig. 3C, Table 1, and Supplementary file 5). This finding underscores DrugGen’s superior capability to generate high-quality structures.
Molecular docking: Docking simulations were performed on the targets that had reliable protein data bank files and could be successfully re-docked, i.e., FABP5, NAMPT, and ACE. To further evaluate the model’s ability to generate potential drug candidates, docking was also performed against approved drugs, assessing how well the generated molecules compared to existing therapeutics. GALM protein was included to emphasize the model’s capability to create molecules for unexplored targets with no reference molecules. The results showed that the generated molecules included agents with high binding affinities for the binding site of their respective targets (Table 2 and Supplementary file 6). Except for ACE which has multiple proven binding sites with docked molecules binding to different locations than the reference molecule, all other docked molecules were positioned in the same binding site as the reference in their best-docked poses (Fig. 4). Furthermore, the model has generated molecules with better docking scores than the reference for FABP5 (-9.537 and -8.399 vs. -6.177) and NAMPT (-8.381 vs. -8.300). Notably, for NAMPT, the model suggested a novel pharmacophore that occupies the same active site as the reference molecule (Fig. 5). ID cards of generated small molecules with their related SMILES are presented in Supplementary file 7.

Visualization of ligand binding in active sites across selected targets. (A) FABP5/11 (dark pink) and Palmitic acid (light pink, reference) in the FABP5 active site. (B) GALM13 (light green) and GALM2 (lime) associated with GALM. (C) NAMPT40 (teal) and Daporinad (blue, reference) in the NAMPT active site. (D) ACE29 (red), ACE28 (yellow), ACE17 (peach), ACE14 (orange), and Lisinopril (copper, reference) associated with ACE.

Comparison of pharmacophores of NAMPT inhibitors in the same active site. Daporinad (left) and NAMPT40 (right) with fundamentally different pharmacophores, both placed in the active site of NAMPT.
Discussion
In this study, we developed DrugGen, a large language model designed to generate small molecules based on the desired targets as input. DrugGen is based on a previously developed model known as DrugGPT, achieving improvements by supervised fine-tuning on approved drugs and reinforcement learning. These improvements aim to facilitate the generation of novel small molecules with stronger binding affinities and a higher probability of approval in future clinical trials. The results indicate that DrugGen can produce high-affinity molecules with robust docking scores, highlighting its potential to accelerate the drug discovery process.
DrugGen is primarily based on the DrugGPT, which utilizes a GPT-2 architecture trained on datasets comprising SMILES and SMILES-protein sequence pairs for generation of small molecules. Although DrugGPT shows promise, it became evident that the creation of high-quality small molecules requires more than merely ensuring ligand-target interactions. These molecules may also exhibit essential properties, including favorable chemical characteristics (such as stability and the absence of cytotoxic substructures), pharmacokinetic profiles (acceptable ADME properties—absorption, distribution, metabolism, and excretion), pharmacodynamic attributes (efficacy and potency)25,26,27. Hence, due to the hypothesis that approved drugs have intrinsic properties that make them become approved28, DrugGen was fine-tuned on approved sets of small molecules. This fine-tuning was enhanced through binding affinity feedback from another LLM, PLAPT, resulting in improved quality of generated molecules. Our findings demonstrate that DrugGen produces small molecules with significantly better chemical validity and binding affinity compared to DrugGPT while maintaining chemical diversity.
To assess the capability of DrugGen in generating high-quality molecules, we selected eight targets. The inclusion of six targets without known approved small molecules demonstrates DrugGen’s potential to introduce novel candidates for previously untargeted or unexplored therapeutic areas. Among the assessed targets, generated molecules showed enhanced validity and stronger binding affinities compared to those produced by DrugGPT. This consistency suggests that DrugGen’s reinforcement learning process effectively enhances its ability to generate potent drug candidates. Moreover, docking simulations further confirmed the quality of DrugGen in generating high-quality small molecules. The comparison of docking scores between generated and reference molecules, NAMPT40 vs. Daporinad and FABP5/11 and FABP5/5 vs. Palmitic acid, shows that DrugGen can design molecules with predicted interactions stronger than the known drugs. This observation highlights DrugGen’s capability to innovate beyond the existing drug design approaches. Furthermore, the diversity of generated molecules, reflected in the wide range of docking scores, emphasizes the model’s flexibility in producing varied chemical structures. Additionally, in the case of NAMPT, the model generated one structure with a strong docking score possessing a pharmacophore very different from that of the reference molecules, meaning that core drug structure was dissimilar to the reference molecule. This structure occupied the same binding site as the reference molecule, which is a potentially new pharmacophore for this target. In addition to these improvements, in the process of reinforcement learning, penalties were applied for generating repetitive structures, resulting in a diverse and valid set of molecules whilst retaining the possibility of regenerating approved drugs in the case of drug repurposing29. Thus, DrugGen demonstrates applicability in both de novo drug design and repurposing efforts.
Despite these achievements, our study has some limitations that should be considered in future research. Variability in binding affinity results across assessed targets was observed. For instance, FABP5’s performance improvement was less pronounced compared with others. This might suggest that with certain target classes or protein sequences, unique challenges emerge for our model, requiring additional fine-tuning or alternative strategies for further optimization. In addition, DrugGen cannot target a specific binding site, as can be seen in the case of ACE, which has multiple binding sites30. Ligand prediction using the DrugGen model led to molecules with fairly strong ligand binding to different binding sites; however, this may not be desirable in some cases. The existing reward function relies on an affinity-predictor deep learning model that has inherent accuracy and specificity limitations due to the limitations of the databases and input representation, which could be addressed in future works. Our model is primarily focused on predicting novel cores and structures for targets with limited bioactive molecules, thus it does not generate fully optimized structures. These predicted structures should undergo structural manipulation for structural optimization to better fit the active site of the target. Future improvements will involve incorporating active site interactions into the reward system to enhance structural accuracy. Additionally, integrating this model with others like PocketGen31, which focuses on designing ligand-binding pockets, could be a promising approach that enables the joint optimization of both ligand generation and protein pocket design. This combined approach could enhance drug discovery, leading to more precise, effective, and innovative therapeutics for a wide range of diseases. Finally, the reliance on in silico validation, while useful, needs to be complemented with experimental validation to confirm the practical efficacy and safety of the generated molecules.
In conclusion, DrugGen represents a powerful tool for early-stage drug discovery, with the potential to significantly accelerate the process of identifying novel lead compounds. With further refinement and integration with experimental validation, DrugGen could become an integral part of future drug discovery pipelines, contributing to the development of new therapeutics across a wide range of diseases.
Materials and methods
Dataset preparation
A dataset of small molecules, each approved by at least one regulatory body, was collected to enhance the safety and relevance of the generated molecules. First, 1710 small molecules from the DrugBank database (version: 5.1.10) were retrieved32,33, 117 of which were labeled as withdrawn. After initial assessments of withdrawn drugs by a physician (Ali Motahharynia) and a pharmacist (Mahsa Sheikholeslami) through a literature review, consensus was reached to omit 50 entries due to safety concerns. Consequently, 1660 approved small molecules and their respective targets were selected to retrieve target-related sequences from UniProt database34,35. From the total of 2116 approved drug targets, retrieved from DrugBank database, 27 were not present in the UniProt database. After further assessment, these 27 proteins were manually replaced with equivalently reviewed UniProt IDs by searching the UniProt database, identifying identical proteins via NCBI Gene, and confirming the matches using basic local alignment search tools (BLAST)36. The protein with UniProt ID “Q5JXX5” was deleted from the UniProt database and therefore, omitted from the collected dataset as well. Finally, 1660 small molecules and 2093 related protein targets were selected. Available SMILEs (1634) were retrieved from DrugBank, ChEMBL37, and ZINC20 databases38. Protein sequences were retrieved from the UniProt database.
Data preprocessing
Similar to the structure used by DrugGPT, the small molecules and target sequences were merged into the pair of a string consisting of protein sequence and SMILES in the following format: “ <|startoftext|> + < P > + target protein sequence + < L > + SMILES + <|endoftext|> ." To ensure the compatibility of this input format with the original model, the resulting strings were tokenized using the trained DrugGPT’s byte-pair encoding (BPE) tokenizer (53,083 tokens). The strings were padded to the maximum length of 768, and longer strings were truncated. The “ <|startoftext|> ”, “ <|endoftext|> ”, and “ < PAD > ” were defined as special tokens.
DrugGen development overview
Using the supervised fine-tuning (SFT) trainer module from the transformer reinforcement learning (TRL) library (version: 0.9.4)39, the original DrugGPT model was finetuned on our dataset. Afterward, reinforcement learning was applied to further improve the model. For this purpose, a Tesla V100 GPU with 32 GB of VRAM, 64 GB of RAM, and a 4-core CPU were utilized for both phases, i.e., SFT and reinforcement learning using a PPO trainer.
Supervised fine-tuning
The training dataset consisted of 9398 strings. The base model was trained using the SFT trainer class for five epochs with the following configuration: Learning rate: 5e-4, batch size: 8, warmup steps (linear warmup strategy): 100, and eval steps: 50. AdamW optimizer with a learning rate of 5e-4 and epsilon value of 1e-8 was used for optimizing the model parameters. The model’s performance on the training and validation sets (ratio of 8:2) was evaluated using the cross-entropy loss function during the training phase.
Proximal policy optimization
Hugging Face’s PPO Trainer, which is based on OpenAI’s original method for “Summarize from Feedback”40 was used in this study. PPO is a reinforcement learning algorithm that improves the policy by taking small steps during optimization, avoiding overly large updates that could lead to instability. The key formula used in PPO is:
In this equation, LCLIP(θ) represents the clipped objective function that PPO aims to optimize during training. The expectation \({E}_{t}\) denotes the average over time steps \(t\), capturing the overall performance of the policy. The term \({r}_{t}(\theta )\) is the probability ratio of taking action ɑt under the new policy compared to the old policy, defined as \(r_{t} \left( \theta \right) = \frac{{\pi_{\theta } \left( {\left. {a_{t} } \right|s_{t} } \right)}}{{\pi_{{\theta_{old} }} \left( {\left. {a_{t} } \right|s_{t} } \right)}}\). The advantage estimate \({A}_{t}\) quantifies the relative value of the action taken in relation to the expected value of the policy. The clipping function, \(\text{clip}({\text{r}}_{\text{t}}(\uptheta ),1-\upepsilon ,1+\upepsilon )\), restricts the ratio to a defined range, preventing large updates to the policy that could destabilize training. This formulation allows PPO to balance exploration and stability, enabling effective policy updates while minimizing the risk of performance degradation. There are three main phases in training a model with PPO. First, the language model generates a response based on an input query in a phase called the rollout phase. In our study, the queries were protein sequences, and the generated responses were SMILES strings. Then in the evaluation phase, the generated molecules were assessed with a custom model that predicts binding affinity. Finally, the log probabilities of the tokens in the generated SMILES sequences were calculated based on the query/response pairs. This step is also known as the optimization phase. Additionally, to maintain the generated responses within a reasonable range from the reference language model, a reward signal was introduced in the form of the Kullback–Leibler (KL) divergence between the two outputs. This additional signal ensures that the new responses do not deviate too far from the original model’s outputs. Thus, PPO was applied to train the active language model.
In our study, the rollout section had the following generation parameters: “do_sample”: True, “top_k”: 9, “top_p”: 0.9, “max_length”: 1024, and “num_return_sequences”: 10. In each epoch, generation was continued until 30 unique small molecules were generated for each target. Keeping initial model’s structure in mind, the dataset was filtered based on the length of each protein sequence. After creating the prompts according to the specified format, i.e., “ <|startoftext|> + < P > + target protein sequence + < L > ”, prompts with a tensor size greater than 768 were omitted, resulting in 2053 proteins (98.09% of the initial dataset).
The PPO trainer configuration included: “mini_batch_size”: 8, “batch_size”: 240, and “learning_rate”: 1.41e-5. Score scaling and normalization were handled with the PPO trainer’s built-in functions.
Reward function
PLAPT: PLAPT, a cutting-edge model designed to predict binding affinities with remarkable accuracy was used as a reward function. PLAPT leverages transfer learning from pre-trained transformers, ProtBERT and ChemBERTa, to process one-dimensional protein and ligand sequences, utilizing a branching neural network architecture for the integration of features and estimation of binding affinities. The superior performance of PLAPT has been validated across multiple datasets, where it achieved state-of-the-art results16. The affinities of the generated structures with their respective targets were evaluated using PLAPT’s neg_log10_affinity_M output.
Customized invalid structure assessor: We developed a customized algorithm using RDKit library (version: 2023.9.5)41 to assess invalid structure, where specific checks were performed to identify potential issues such as atom count, valence errors, and parsing errors. Invalid structures, including those with fewer than two atoms, incorrect valence states, or parsing failures were flagged and penalized accordingly. To promote the generation of valid molecules, a reward value of 0 was assigned to any invalid SMILES structures. These reward systems provide a rigorous scoring system for model development.
To further shift the model toward generating novel molecules, a multiplicative penalty was applied to the reward score when a generated SMILES string matched a molecule already present in the approved SMILES dataset. Specifically, the reward was multiplied by 0.7 for such occurrences, to retain a balance between generating new structures as well as repurposing approved drugs.
DrugGen assessment
To evaluate the performance of DrugGen, several metrics were employed to measure its efficacy in generating viable and high-affinity drug candidates. For this purpose, eight targets consisting of two DKD targets with the highest score in DisGeNet database (version 3.12.1)42, i.e., “ACE” and “PPARG” and six targets without any known approved small molecules for them were selected. The selection of these six targets was according to our recent study “DrugTar Improves Druggability Prediction by Integrating Large Language Models and Gene Ontologies”24. According to this study, 6 out of the 10 most probable proteins for future targets were selected. The selected targets are “GALM”, “FB5L3”, “OPSB”, “NAMPT”, “PGK2”, and “FABP5”. The generative quality of DrugGPT and DrugGen in terms of validity, diversity, novelty, and binding affinity was assessed. Additionally, we performed in silico validation of the molecules generated by DrugGen using a rigorous docking method.
Validity assessment
The validity of the generated molecules was evaluated using the previously mentioned customized invalid structure assessor. The percentage of valid to total generation was reported as models’ capability to construct valid structures.
Diversity assessment
To assess the diversity of the generated molecules, 500 ligands were generated for each target by DrugGPT and DrugGen. The diversity of the generated molecules was quantitatively assessed using the Tanimoto similarity index43. The diversity evaluation process involved the following steps: First, each generated molecule was converted to its corresponding molecular fingerprint using Morgan fingerprints (size = 2048 bits, radius = 2)44. For each molecule, pairwise Tanimoto similarities were calculated between all possible pairs of fingerprints, and the average value was calculated. Thus, the diversity of the generated set was determined as the “1—average of Tanimoto similarity” within a generated batch. The distribution of diversity for each target was plotted. The invalid structures were not involved in diversity assessments. Statistical analyses were performed using Mann–Whitney U test.
Novelty assessment
For each target, a set of 100 unique molecules was generated by DrugGPT and DrugGen. The novelty of the generated molecules was evaluated by comparing them to a dataset of approved drugs. After converting the molecules into Morgan fingerprints, the similarity of each generated molecule to the approved drugs was calculated using Tanimoto similarity index, retaining only the maximum similarity value. The novelty was reported as the “1—max_Tanimoto similarity”. The invalid structures were not included in the novelty assessments. Statistical analyses were performed using Mann–Whitney U test.
PLAPT binding affinity assessment
The same set of molecules generated during the novelty assessment was used to evaluate the binding affinities of the compounds produced by DrugGPT and DrugGen. The invalid structures were involved in the binding affinity assessments. Statistical analysis was conducted using the Mann–Whitney U test, and corrections for multiple comparisons were applied using the Bonferroni method.
Molecular docking
Molecular docking was conducted for selected targets with available protein data bank (PDB) structures, specifically ACE, NAMPT, GALM, and FABP5. A set of 100 newly generated molecules, following duplicate removal, were docked into the crystal structures of ACE (PDB ID: 1o86), NAMPT (PDB ID: 2gvj), GALM (PDB ID: 1snz), and FABP5 (PDB ID: 1b56). Overall, blind docking45 was employed for all 122 generated molecules and their references to thoroughly search the entire protein surface for the most favorable active site (Supplementary file 6 and 7). The reference ligands used were Lisinopril for ACE and Palmitic acid for FABP5, both of which were bound in the active site. For NAMPT, Daporinad, a molecule currently in phase 2 clinical trials, served as the highest available reference. In the case of GALM, no reference ligand was found. The retrieved PDB files were prepared using the protein preparation wizard46 available in the Schrödinger suite, ensuring the addition of missing hydrogens, assignment of appropriate charge states at physiological pH, and reconstruction of incomplete side chains and rings. LigPrep47 with the OPLS4 force field48 was employed to generate all possible stereoisomers and ionization states at pH 7.4 ± 0.5. The prepared structures were used for docking.
Docking simulations were performed using the GLIDE program49. Ligands were docked using the extra precision (XP) protocol. Ligands were allowed full flexibility during the docking process, while the protein was held rigid. The information of the grid boxes is summarized in Table 3.
The GLIDE XP scoring function was used to evaluate docking poses. Negative values of the GLIDE score (XP GScore) were reported for readability. The robustness of the docking procedures was validated by redocking the reference ligands into their respective binding sites. The computed root-mean-squared deviation (RMSD) values were 0.7233Å, 0.2961Å, and 2.0119Å for ACE, NAMPT, and FABP5, respectively, confirming the reliability of the docking protocol.
Data availability
All data generated or analyzed during this study are included in the manuscript and supporting files. The sequence-SMILES dataset of approved drug-target pairs used in this study is publicly available at “alimotahharynia/approved_drug_target” on Hugging Face (https://huggingface.co/datasets/alimotahharynia/approved_drug_target).
Code availability
The checkpoints, code for generating small molecules, and customized invalid structure assessor are publicly available at https://huggingface.co/alimotahharynia/DrugGen and https://github.com/mahsasheikh/DrugGen. Explore the interactive user interface of DrugGen at https://huggingface.co/spaces/alimotahharynia/GPT-2-Drug-Generator
References
Bai, L. et al. AI-enabled organoids: construction, analysis, and application. Bioactive Materials 31, 525–548 (2024).
Coley, C. W. Defining and exploring chemical spaces. Trends Chem. 3, 133–145 (2021).
Sun, D., Gao, W., Hu, H. & Zhou, S. Why 90% of clinical drug development fails and how to improve it?. Acta Pharm. Sin. B 12, 3049–3062 (2022).
Tong, X. et al. Generative models for de novo drug design. J. Med. Chem. 64, 14011–14027 (2021).
Zeng, X. et al. Deep generative molecular design reshapes drug discovery. Cell Rep. Med. https://doi.org/10.1016/j.xcrm.2022.100794 (2022).
Meyers, J., Fabian, B. & Brown, N. D. novo molecular design and generative models. Drug Discovery Today 26, 2707–2715 (2021).
Méndez-Lucio, O., Baillif, B., Clevert, D.-A., Rouquié, D. & Wichard, J. D. novo generation of hit-like molecules from gene expression signatures using artificial intelligence. Nat. Commun. 11, 10 (2020).
Janssen, A. et al. A Generative and causal pharmacokinetic model for factor VIII in hemophilia a: a machine learning framework for continuous model refinement. Clin. Pharmacol. Ther. 115, 881–889 (2024).
Ota, R. & Yamashita, F. Application of machine learning techniques to the analysis and prediction of drug pharmacokinetics. J. Control. Release 352, 961–969 (2022).
Horne, R. I. et al. Using generative modeling to endow with potency initially inert compounds with good bioavailability and low toxicity. J. Chem. Inf. Model. 64, 590–596 (2024).
Ghayoor, A. & Kohan, H. G. Revolutionizing pharmacokinetics: the dawn of AI-powered analysis. J. Pharm. Pharm. Sci. 27, 12671 (2024).
Menke, J. & Koch, O. Using domain-specific fingerprints generated through neural networks to enhance ligand-based virtual screening. J. Chem. Inf. Model. 61, 664–675 (2021).
Qureshi, R. et al. AI in drug discovery and its clinical relevance. Heliyon 9 (2023).
Zhuang, D. & Ibrahim, A. K. Deep learning for drug discovery: a study of identifying high efficacy drug compounds using a cascade transfer learning approach. Appl. Sci. 11, 7772 (2021).
Gangwal, A. & Lavecchia, A. Unlocking the potential of generative AI in drug discovery. Drug Discovery Today, 103992 (2024).
Rose, T., Monti, N., Anand, N. & Shen, T. PLAPT: protein-ligand binding affinity prediction using pretrained transformers. bioRxiv 2024(2002), 575577 (2008).
Madani, A. et al. Progen: Language modeling for protein generation. Preprint https:\\arXiv.org.\2004.03497 (2020).
Brown, T. B. Language models are few-shot learners. Preprint https:\\arXiv.org.\2005.14165 (2020).
Li, Y. et al. DrugGPT: A GPT-based strategy for designing potential ligands targeting specific proteins. bioRxiv 2023(2006), 543848 (2023).
Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inform. Comput. Sci. 28, 31–36 (1988).
Michailidis, G. & d’Alché-Buc, F. Autoregressive models for gene regulatory network inference: Sparsity, stability and causality issues. Math. Biosci. 246, 326–334 (2013).
Kim, T. K., Paul, H. Y., Hager, G. D. & Lin, C. T. Refining dataset curation methods for deep learning-based automated tuberculosis screening. J. Thorac. Dis. 12, 5078 (2020).
Stokes, J. M. et al. A deep learning approach to antibiotic discovery. Cell 180(688–702), e613 (2020).
Borhani, N., Izadi, I., Motahharynia, A., Sheikholeslami, M. & Gheisari, Y. DrugTar improves druggability prediction by integrating large language models and gene ontologies. biorxiv https://doi.org/10.1101/2024.09.21.614218 (2024).
Roskoski, R. Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2024 update. Pharmacol Res. 200, 107059. https://doi.org/10.1016/j.phrs.2024.107059 (2024).
Loftsson, T. in Essential Pharmacokinetics (ed Thorsteinn Loftsson) 85–104 (Academic Press, 2015).
Di, L. & Kerns, E. H. in Drug-Like Properties (Second Edition) (eds Li Di & Edward H. Kerns) 1–3 (Academic Press, 2016).
Li, B. et al. DrugMetric: quantitative drug-likeness scoring based on chemical space distance. Briefings Bioinform. https://doi.org/10.1093/bib/bbae321 (2024).
Kulkarni, V. S., Alagarsamy, V., Solomon, V. R., Jose, P. A. & Murugesan, S. Drug repurposing: an effective tool in modern drug discovery. Russ J Bioorg Chem 49, 157–166. https://doi.org/10.1134/s1068162023020139 (2023).
Cozier, G. E., Lubbe, L., Sturrock, E. D. & Acharya, K. R. Angiotensin-converting enzyme open for business: structural insights into the subdomain dynamics. Febs j 288, 2238–2256. https://doi.org/10.1111/febs.15601 (2021).
Zhang, Z., Shen, W. X., Liu, Q. & Zitnik, M. Efficient generation of protein pockets with PocketGen. Nat. Machine Intell. 6, 1382–1395. https://doi.org/10.1038/s42256-024-00920-9 (2024).
Wishart, D. S. et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 34, D668–D672 (2006).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074-d1082. https://doi.org/10.1093/nar/gkx1037 (2018).
UniProt: the universal protein knowledgebase in 2023. Nucleic acids research 51, D523-D531 (2023).
Consortium & T. U,. UniProt: the Universal Protein Knowledgebase in 2025. Nucleic Acids Res. 53, D609–D617. https://doi.org/10.1093/nar/gkae1010 (2024).
Sf, A. Basic local alignment search tool. J Mol Biol 215, 403–410 (1990).
Zdrazil, B. et al. The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 52, D1180–D1192 (2024).
Irwin, J. J. et al. ZINC20—a free ultralarge-scale chemical database for ligand discovery. J. Chem. Inf. Model. 60, 6065–6073 (2020).
Transformer Reinforcement Learning, <https://huggingface.co/docs/trl/en/index> (
Summarize from Feedback, <https://github.com/openai/summarize-from-feedback> (
RDKit, <https://www.rdkit.org/> (
Piñero, J. et al. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic acids research, gkw943 (2016).
Bajusz, D., Rácz, A. & Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations?. J. Cheminformatics 7, 1–13 (2015).
Morgan, H. L. The generation of a unique machine description for chemical structures-a technique developed at chemical abstracts service. J. Chem. Doc. 5, 107–113 (1965).
Hassan, N. M., Alhossary, A. A., Mu, Y. & Kwoh, C.-K. Protein-ligand blind docking using QuickVina-W with inter-process spatio-temporal integration. Sci. Rep. 7, 15451 (2017).
Madhavi Sastry, G., Adzhigirey, M., Day, T., Annabhimoju, R. & Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Design 27, 221–234 (2013).
Schrödinger Release 2024–2: LigPrep (Schrödinger, LLC, New York, NY, 2024).
Lu, C. et al. OPLS4: Improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput. 17, 4291–4300 (2021).
Friesner, R. A. et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 (2004).
Acknowledgements
We sincerely thank Dr. Mehdi Rahmani for his invaluable assistance with technical and software issues related to training our model on the cluster servers.
Author information
Authors and Affiliations
Contributions
Conceptualization: M.S, Y.G, M.I, A.M. Dataset preparation: M.S, A.M. Model development: M.S, N.M, M.I, A.M. Statistical analysis: M.S, N.M, A.M. In silico validation: M.S, A.F. Data interpretation: All authors. Drafting original manuscript: M.S, N.M. Revising the manuscript: Y.G, A.F, M.I, A.M. All the authors have read and approved the final version for publication and agreed to be responsible for the integrity of the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sheikholeslami, M., Mazrouei, N., Gheisari, Y. et al. DrugGen enhances drug discovery with large language models and reinforcement learning. Sci Rep 15, 13445 (2025). https://doi.org/10.1038/s41598-025-98629-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-98629-1
