
Abstract
During the acute phase (1–3 days) of cerebral infarction, neutrophils and macrophages accumulate at the infarction site, inducing inflammation and cerebral oedema. However, the role of neutrophils in oedema formation after ischaemic stroke remains unclear. This study examined neutrophil involvement in cerebral oedema using a transient middle cerebral artery occlusion (tMCAO) rat model, primary cultured neutrophils, and astrocytes. Brain specimens were stained with myeloperoxidase (MPO) and lymphocyte antigen 6 complexes, locus G (Ly6G), and the number of MPO+/Ly6G+ cells was counted. Neutrophil infiltration began in the leptomeninges at 3 h, reaching the ischaemic cortex by 6 h and the striatum by 24 h, peaking at 24–48 h before declining. Neutrophils attached to endothelial walls and infiltrated the brain parenchyma, correlating with oedema severity. Infiltrating neutrophils strongly expressed IL-1α and TNF in the ischaemic brain. Co-culturing LPS-activated neutrophils with astrocytes increased Aqp4 mRNA and protein expression, which was inhibited by IL-1RI and TNF antagonists. These findings suggest that activated neutrophils exacerbate cerebral oedema by inducing astrocytic AQP4 expression via IL-1α and TNF in peri-infarct and ischaemic core tissues.
Similar content being viewed by others


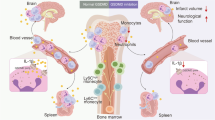
Introduction
Stroke is an acute cerebrovascular disease characterised by high morbidity, disability, and mortality and imposes a huge burden on the physical and financial well-being of families and society1. Cerebral ischaemia or subsequent reperfusion causes cerebral capillary dysfunction, resulting in cerebral oedema2. Cerebral oedema is defined as brain swelling due to excess fluid accumulation in the brain parenchyma, which can be severe and directly cause death. Important treatment options for cerebral oedema include pharmacological treatments with hyperosmotic solutions (e.g., d-mannitol or hypertonic saline) and intravenous steroids, as well as conventional surgical approaches such as decompressive craniectomy for cerebral oedema, which often leads to brain herniation.
Neutrophils, the most abundant cell type among leukocytes, are regarded as the first peripheral responders to acute inflammation and as crucial immune components in ischaemic stroke exacerbation3,4. The size of the ischaemic infarction and level of neurological deficits positively correlate with an increase in the number and activity of neutrophils, which in turn leads to an increased risk of death5. Neutrophils worsen the outcome of ischaemic stroke by serving as a scaffold for thrombosis6, impairing vascular remodelling7, aggravating blood-brain barrier (BBB) damage8, increasing microvessel occlusion, and promoting neuronal death9,10. The role of neutrophils in post-stroke inflammation has been clarified; however, their role in cerebral oedema formation, the most influential prognostic factor, remains unclear.
The expression of aquaporin-4 (AQP4), the most abundant water channel protein in the brain, increases after infarction and plays an important role in post-ischaemic oedema and its clearance11,12. In the brain, AQP4 is densely abundant in the subarachnoid space, epithelial cells of the choroid plexus, and perivascular astrocytic endfeet13.
After brain injury or disease, these astrocytes change to a phenotype called ‘reactive’, affecting surrounding cells14,15. Liddelow et al. reported that interleukin-1 alpha (IL-1α), tumour necrosis factor (TNF), and C1q secreted by activated microglia are closely involved in phenotypic changes toward reactive astrocytes16. We recently demonstrated that IL-1α and TNF secreted by activated microglia in the ischaemic core induce astrocytic AQP4 expression and exacerbate cerebral oedema17. Furthermore, an IL-1 receptor antagonist (IL-1RA), an anti-inflammatory cytokine and a type of myokine, suppresses astrocytic AQP4 expression and brain oedema in transient middle cerebral artery occlusion (tMCAO) model rats18.
Previously, we observed a negative correlation between the magnitude of weight loss and brain oedema at both 1 and 7 days post-tMCAO17,18. This finding led us to hypothesise that weight loss rate could serve as a useful indicator of brain oedema. In this study, we sought to investigate whether neutrophils are involved in the formation of cerebral oedema following cerebral infarction. To test this hypothesis, we investigated the effects of neutrophils in the ischaemic lesions on the expression of AQP4 and the development of brain oedema using a rat model of tMCAO and primary cultures of rat neutrophils and astrocytes.
Methods
Animal and surgical procedures
Animal care and experiments conformed to the Guidelines for Animal Experiments of Osaka University and were approved by the Animal Research Committee of Osaka University (approval no. 01-024-002). The experiments were performed following the ARRIVE guidelines (https://arriveguidelines.org) (Animal Research: Reporting in Vivo Experiments). Seven-week-old male Wistar rats (n = 204, weight: 192.0 ± 238.4 g; Kiwa Laboratory Animals Co., Wakayama, Japan) were subjected to tMCAO for 90 min, as previously described19. Rats were anaesthetised with 1.5–2% isoflurane via a face mask, and the right middle cerebral artery was occluded using a 4 − 0 nylon monofilament suture advanced from the internal carotid artery. The rats were allowed to recover from anaesthesia during occlusion, re-anaesthetised 90 min later, and reperfused by withdrawing the thread. For sham surgery, the animals were anaesthetised, a midline incision was made in the neck, and the right carotid artery was isolated. Rats subjected to tMCAO were weighed before surgery and those with less than 10% weight loss on the day after tMCAO were excluded from the analysis17. A heating pad was used to maintain body temperature during surgery. Neurological dysfunction was assessed using the Longa score 30 min after tMCAO. Rats with a score of 2 were considered to have successfully established the model. The neurological findings were scored on a five-point scale: 0 points, no discernible neurological dysfunction; 1 point, mild neurological dysfunction, characterised by an inability to extend the contralateral forepaw fully; 2 points, moderate neurological dysfunction, manifested by circling behaviour toward the contralateral side; 3 points, severe neurological dysfunction, characterised by falling toward the contralateral side; and 4 points, complete inability to ambulate independently and loss of consciousness20. In vitro experiments were performed using neonatal Wistar rats (n = 80; Kiwa Laboratory Animals Co.) aged 0–2 days. The rats were kept at room temperature (22 ± 1 °C) with free access to water and food (light period: 7:00 am to 7:00 pm; dark period: 7:00 pm to 7:00 am) until the experiments.
Isolation of circulating neutrophils
Neutrophils were isolated from rat blood using Histopaque gradients as previously described10. Briefly, 3 mL Histopaque-1077 (Sigma-Aldrich, St. Louis, MO, USA) was layered on 3 mL Histopaque-1119 (Sigma-Aldrich) in a 15 ml tube, and 4 mL of heparinised rat blood was carefully placed on the top of the Histopaque mixture, forming a three-step gradient. The tube was centrifuged at 400 × g for 30 min using a swinging rotor. The first ring, containing mononuclear cells, was slowly aspirated, and the second ring, containing neutrophils, was transferred to another 15 mL tube. Phosphate-buffered saline (PBS)-BG, comprising PBS with 0.1% bovine serum albumin (BSA) (Sigma-Aldrich) and 10% glucose, was added to the tube and centrifuged at 1500 × g for 10 min. The pellet was resuspended in 3 mL of PBS-BG, placed on 3 mL of Histopaque-1119, and centrifuged at 1500 × g for 10 min at 4 °C. The ring-containing neutrophils were suspended in RPMI (Fujifilm Wako Pure Chemicals, Osaka, Japan) containing 10% foetal bovine serum (FBS). For some experiments, neutrophils were cultured for 6 h in RPMI medium supplemented with 10 ng/mL lipopolysaccharide (LPS; Escherichia coli serotype 055:B5; Sigma-Aldrich). The cells were seeded onto 0.4-µm pore size (high pore density) inserts (Corning) in 6- or 24-well plates or coated coverslips (Matsunami Glass, Osaka, Japan) in a serum-free E2 medium comprising PSA-supplemented DMEM-based medium with 10 mM HEPES (pH 7.4), 4.5 mg/mL glucose, 10 µg/mL insulin, 38.7 nM sodium selenite, 5.5 µg/mL transferrin (GIBCO, Grand Island, NY, USA), and 1 mg/mL BSA.
Primary culture of astrocytes
The cerebrum was extirpated from neonatal rats, and the meninges and hippocampi were removed. The processed cerebrums were dissociated into individual cells using a nylon bag with 160-µm pores. Dissociated cells are a mixture of glial cells, including astrocytes. The suspensions were seeded in 75 cm2 flasks (Corning, NY, USA) coated with poly-L-lysine hydrobromide (PLL) (Sigma-Aldrich) in two flasks per animal and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Fujifilm Wako Pure Chemicals) containing 10% FBS and 1% penicillin-streptomycin-amphotericin B suspension (PSA) (Fujifilm Wako Pure Chemicals). The medium was changed every other day, and the culture was maintained for 13 days. Glial cells cultured for 13 days were shaken using a BioShaker (Titech Co., Saitama, Japan) at 200 rpm at 37 °C for 1 h. The astrocytes were purified using the anti-GLAST (ACSA-1) MicroBead Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) and seeded in PLL-coated 6- or 24-well plates (Corning) or PLL-coated coverslips (Matsunami Glass, Osaka, Japan). The next day, cells were cultured in E2 medium supplemented with either recombinant rat IL-1 receptor type I (IL-1RI) antagonist (100 µM; Calbiochem, La Jolla, CA, USA) or TNF-α antagonist (100 µM; Calbiochem) for 2 h and then co-cultured with neutrophils.
Expression of recombinant rat AQP4
HEK-293T cells were kindly provided by Dr. Hidekazu Tanaka, College of Life Sciences, Ritsumeikan University, Shiga, Japan, and maintained in 10% FBS-DMEM. HEK-293T cells (2.5 × 104 cells/cm2) were seeded in 4- or 6-well plates, or PLL-coated coverslips, one day before transfection. The next day, the expression plasmids containing gene encoding rat Aqp4 (VB190626-1034msf; VectorBuilder Inc., Chicago, IL, USA) were introduced into HEK-293T cells using a Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) cell transfection kit.
Immunohistochemistry
At 3 and 6 h, and 1, 2, 3, 4, 5, and 7 days after tMCAO, the rats were euthanised using CO2, perfused with cold 2% heparin-containing PBS, and subsequently perfused with 4% paraformaldehyde phosphate buffer solution (Fujifilm Wako Pure Chemicals). The brains were dissociated, frozen, and sliced into 10-µm-thick coronal sections using a cryostat. For immunocytochemical staining, cultured cells were seeded on PLL-coated coverslips and fixed with 4% paraformaldehyde. Subsequently, tissues and cells were incubated with the primary antibodies against AQP4 (1:50; rabbit polyclonal, AB3594; Chemicon, Temecula, CA, USA), glial fibrillary acidic protein (GFAP) (1:250; goat polyclonal, ab53554; Abcam, Cambridge, UK), IL-1α (1:50; mouse monoclonal, ab239517; Abcam), Ly6G (1:100; rat monoclonal, 11-9668-82; Invitrogen), myeloperoxidase (MPO) (1:100; rabbit monoclonal, ab208670; Abcam), or TNF (1:50; Rabbit polyclonal, NBP1-19532; Novus Biologicals, Centennial, CO, USA) in Tris-buffered saline containing 0.1% Tween-20 (TBST) and 0.1% BSA. After washing with TBST, the tissues and cells were incubated with DyLight 488-, DyLight 549-, and DyLight 649-labelled secondary antibodies (1:1000; Invitrogen), and Hoechst 33,342 (1:1000; Invitrogen) was used for nuclear staining. Finally, the immune-stained specimens were observed using an all-in-one fluorescence microscope (BZ-X710; Keyence, Osaka, Japan) with a CFI Plan Fluor 20× lens (Nikon, Tokyo, Japan) or a confocal laser scanning microscope (AX-R Ti2; Nikon) with a Plan Apo λD 60× Oil OFN25 DIC N2 lens (Nikon in the Nikon Imaging Center, Osaka University). The images were quantified using the Adobe Photoshop software (Adobe Systems Inc., San Jose, CA, USA). The number of Ly6G+/MPO+ cells in 1.0 mm2 (1.0 × 1.0 mm) was counted. Additionally, images stained with Hoechst 33,342 were used to measure the area of the left and right (or ischaemic) hemispheres, and the extent of brain oedema was calculated using the following equation: right hemisphere area / left hemisphere area21. In some experiments, grayscale images were converted to black and white, and the stained areas were selected. The area and mean fluorescence intensity averages were calculated by superimposing the stained areas and raw images.
Triphenyltetrazolium chloride stain
Rats subjected to tMCAO were euthanised using CO2 gas on days 0.25, 1, 2, 3, 5, and 7. Brains were dissected, and 2-mm-thick coronal sections were prepared at the caudo-putaminal level. Brain slices were stained with 0.1% 2, 3, 5-triphenyl-2 H-tetrazolium chloride (Fujifilm Wako Pure Chemicals), dissolved in phosphate-buffered saline (PBS) for 5 min and divided into the contralateral cortex, peri-infarct tissue, and ischaemic core22. These tissues were homogenised using 5-mm stainless steel beads (Titech Inc.) in reagents for mRNA or protein.
Quantitative real-time polymerase chain reaction (qPCR)
Brain tissue RNA was extracted using the mirVana miRNA Isolation Kit (Thermo Fisher Scientific, Waltham, MA, USA), whereas cultured cells were extracted using ISOGEN (Nippon Gene, Tokyo, Japan). Extracted RNA was used for complementary DNA synthesis using ReverTra Ace qPCR RT Master Mix with gDNA Remover (Toyobo, Osaka, Japan). Subsequently, the cells were treated with TB Green Premix Ex Taq II (Takara Bio Inc., Shiga, Japan), followed by qPCR using a CFX Connect Real-Time System (Bio-Rad Laboratories, Hercules, CA, USA). All gene-specific mRNA expression values were normalised to the expression value of glyceraldehyde-3-phosphate dehydrogenase mRNA. The primer sequences for each gene are listed in Supplementary Table S1.
Western blot analysis
Cultured cells were lysed using the Laemmli sample solution. The lysate was electrophoresed on a 10–20% acrylamide gel (SuperSep Ace, Fujifilm Wako Pure Chemicals) and transferred onto a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA), which was then cut at 37 kDa (Supplementary Fig. S1. Membranes containing 25–37 kDa or 37–50 kDa were immunoblotted with primary antibodies against AQP4 (1:100; rabbit polyclonal, AQP-004; Alomone, Jerusalem, Israel) or β-actin (1:500; mouse monoclonal, A5441; Sigma-Aldrich). Following incubation with a horseradish peroxidase-conjugated secondary antibody (1:5000; GE Healthcare, Marlborough, MA, USA), immunoreactions were visualised using Western BLoT Chemiluminescence HRP Substrate (Takara Bio Inc.) and photographed using a ChemiDoc Touch Imaging System (Bio-Rad Laboratories). Immunoreactive bands were analysed by densitometry using Adobe Photoshop software. The densitometry data were standardised against the immunoreactivity of β-actin.
Statistical analysis
Values are expressed as the mean ± standard deviation. Data were analysed using GraphPad Prism 9 (GraphPad Software, La Jolla, CA, USA). Data normality was examined using the Shapiro–Wilk normality test. The equality of variance between two normally distributed datasets was examined using an F-test. Data were analysed using a two-tailed Student’s t-test (unpaired). Multiple comparison tests were performed using one-way or two-way analysis of variance, followed by post hoc tests (Dunnett’s or Tukey’s test). Statistical significance was set at P < 0.05.
Results
Spatial-temporal profiles of neutrophil trafficking-related factors in the ischaemic brain after tMCAO
Initially, we evaluated the expression timeline of factors associated with neutrophil trafficking to determine the mechanism of neutrophil infiltration into the infarct region. Endothelial cells are activated by local production of inflammatory cytokines, such as IL-1α (Il1a), IL-1β (Il1b), and TNF (Tnf), and the induced expression of adhesion molecules E-selectin (Sele), P-selectin (Selp), VCAM-1 (Vcam1), ICAM-1 (Icam1), and ICAM-2 (Icam2)23,24. Neutrophils bind to these adhesion molecules, slowing their movement and causing them to roll along the endothelium. Neutrophils bind to CXCL1 (Cxcl1), CXCL2 (Cxcl2), CXCL3 (Cxcl3), CXCL5 (alias Cxcl6 in rats), CXCL7 (Cxcl7), and CXCL8 (no mouse and rat) on the endothelial luminal surface and infiltrate the infarcted lesion25. We also examined the expression of CXCR2 (Cxcr2), the receptor for these chemokines25. The qPCR results showed that the expression of inflammatory cytokines peaked at 6 h after tMCAO in the peri-infarct tissue and ischaemic core (Fig. 1A-C). Sele and Selp expression were highly remarked in the peri-infarct tissue and ischaemic core from 6 h to 2 days after tMCAO (Fig. 1D and E). The expression of Icam1 increased in the peri-infarct tissue and ischaemic core on the day after tMCAO and again on day 7 (Fig. 1G). Vcam1 and Icam2 expression gradually increased and peaked on day 7 (Fig. 1F and H). Furthermore, the expression of neutrophil-attracting chemokines increased at 6 h after tMCAO, peaked at 24 h, and then gradually decreased (Fig. 1I-K and M). However, only the expression of Cxcl6 peaked on day 7 (Fig. 1L). Cxcr2 increased at 6 h, peaked at days 1 and 2, and then gradually decreased (Fig. 1N).

Spatial-temporal profiles of neutrophil trafficking-related factors in the ischaemic brain after transient middle cerebral artery occlusion (tMCAO). Quantitative real-time polymerase chain reaction analysis of Il1a (A), Il1b (B), Tnf (C), Sele (E-selectin) (D), Selp (P-selectin) (E), Vcam1 (F), Icam1 (G), Icam2 (H), Cxcl1 (I), Cxcl2 (J), Cxcl3 (K), Cxcl6 (Cxcl5) (L), Cxcl7 (M), and Cxcr2 (N) mRNA expression in the ischaemic core, peri-infarct tissue (peri), and contralateral cortex (cont) of tMCAO rats and sham rats (S) (n = 6). Data are expressed as mean ± standard deviation. *, **, and *** indicate P < 0.05, 0.01, and 0.001, respectively, as compared to the sham (S) group.
Spatial-temporal profiles of neutrophils in ischaemic brain after tMCAO
We evaluated the spatiotemporal profiles of neutrophil marker genes and proteins to investigate the distribution of neutrophil infiltration in the ischaemic brain. First, TTC-stained brains were divided into the contralateral cortex, peri-infarct tissue, and ischaemic core and examined for the expression of Ly6c mRNA, a neutrophil marker. The qPCR results showed that Ly6c mRNA was clearly increased only in the ischaemic core on days 1 and 2, then gradually decreased (Fig. 2A). Next, we focused on the ischaemic core with high Ly6c mRNA expression and investigated the spatial and temporal distribution of neutrophil infiltration. Brain specimens were stained with neutrophil-specific markers, MPO and Ly6G, and the numbers of MPO+/Ly6G+ cells in the ischaemic cortex, striatum, and contralateral cortex (shown in Fig. 2B) were counted. The infiltration of MPO+/Ly6G+ cells began in the leptomeninges at 3 h after tMCAO and then spread to the ischaemic cortex at 6 h and the striatum at 24 h (Fig. 2D and E). Subsequently, we counted Ly6G+/MPO+ cells within a 1.0 mm2 area (1.0 × 1.0 mm) (Supplementary Fig. S2). Their numbers peaked at 24 and 48 h, then gradually decreased (Fig. 2C). MPO+/Ly6G+ cells were observed in the perivascular and vascular lumens, flattened, and attached to the luminal sides of the endothelial walls (Fig. 2E). The counts of MPO+/Ly6G+ cells in ischaemic brains indicated that the first MPO+/Ly6G+ cell entry occurred through the leptomeninges at ~ 3 h, and then they were observed in the ischaemic cortex at 6 h and in the striatum 24 h after tMCAO.

Spatial-temporal profiles of neutrophils in the ischaemic brain after transient middle cerebral artery occlusion (tMCAO). (A) Quantitative real-time polymerase chain reaction analysis of Ly6c mRNA expression in the ischaemic core, peri-infarct tissue (peri), and contralateral cortex (cont) of tMCAO rats and sham rats (S) (n = 6). (B and C) Images of Ly6G (green) and MPO (red) fluorescence immunostaining in the ischaemic brain at 2 days after tMCAO, showing the cont, ischaemic striatum (St), and ischaemic cortex (Ct) (B). The number of MPO+/Ly6G+ cells in the Cont, St, and Ct (C) (n = 3). (D and E) Images of Ly6G (green) and MPO (red) fluorescent immunostaining in the leptomeninges (D) and ischaemic cortex and striatum (E) from 1 h to 24 h after tMCAO. Hoechst 33,258 fluorescence (blue) indicates the nuclei (n = 3). White and yellow arrows indicate MPO+/Ly6G+ and MPO+/Ly6G− cells, respectively.
Correlation between neutrophil infiltration and severity of illness after tMCAO
Subsequently, we investigated the correlation between the number of neutrophil infiltrations in the brain parenchyma and disease severity after tMCAO. We previously observed a negative correlation between the magnitude of weight loss and brain oedema 1 or 7 days after tMCAO17,18. We, therefore, used the magnitude of weight loss after tMCAO as a prognostic indicator. Two days after tMCAO, TTC-stained brains were measured for Ly6c and Aif1 (Iba1) mRNA expression, a marker for microglia and macrophages, in the contralateral cortex, peri-infarct tissue, and ischaemic core. We examined the correlation between Ly6c and Aif1 (Iba1) mRNA expression and the magnitude of weight loss 2 days after tMCAO. The expression of Ly6c mRNA in the peri-infarct tissue (Fig. 3A, P < 0.05) and ischaemic core (Fig. 3B, P < 0.05) was negatively correlated with the magnitude of weight loss but not with Iba1 (Fig. 3C and D). The number of MPO+/Ly6G+ cells in the ischaemic peri-infarct tissue, striatum, and cortex was also measured 2 days after tMCAO (Supplementary Fig. S3A), and its correlation with the magnitude of weight loss was examined. There was a negative correlation between the magnitude of weight loss two days after tMCAO and MPO+/Ly6G+ cells in the peri-infarct tissue (Fig. 3E, P < 0.05), ischaemic striatum (Fig. 3F, P < 0.05), and ischaemic cortex (Fig. 3G, P < 0.05). Hoechst-stained images from the same individuals were used to determine the area ratio between the right and left hemispheres (Supplementary Fig. S3B). The area ratio was negatively correlated with the rate of weight loss (Supplementary Fig. S3C, P < 0.01) and positively correlated with MPO+/Ly6G+ cells in the ischaemic striatum (Fig. 3I, P < 0.05) and ischaemic cortex (Fig. 3J, P < 0.05). However, this was not observed in the peri-infarct tissue (Fig. 3H). These results indicate a correlation between the post-tMCAO severity of illness and the number of neutrophils infiltrating the brain parenchyma.

Correlation between neutrophil infiltration and severity of illness after transient middle cerebral artery occlusion (tMCAO). Correlation between the magnitude of weight loss and Ly6c mRNA expression in the peri-infarct tissue (A) or ischaemic core (B) and Aif1 (Iba1) mRNA expression in the peri-infarct tissue (C) or ischaemic core (D) 2 days after tMCAO (n = 10). (E–G) Correlation between the magnitude of weight loss and the number of MPO+/Ly6G+ cells in the peri-infarct tissue (E), ischaemic striatum (F), or ischaemic cortex (G) at 2 days after tMCAO (n = 10). (H–J) Correlation between the magnitude of weight loss and the area ratio of the right and left hemispheres in the peri-infarct tissue (H), ischaemic striatum (I), or ischaemic cortex (J) at 2 days after tMCAO (n = 10).
Correlation between neutrophil trafficking-related factors and severity of illness after tMCAO
We evaluated the correlation between neutrophil trafficking-related factors and illness severity to investigate the mechanism by which more neutrophils infiltrate severely ill rats. Expression of Sele (Supplementary Fig. S4A, P < 0.01), Cxcl2 (Supplementary Fig. S4G, P < 0.05), and Cxcl3 (Supplementary Fig. S4H, P < 0.05) in the peri-infarct tissue and Sele (Fig. 4A, P < 0.05), Cxcl2 (Fig. 4G, P < 0.05) and Cxcl3 (Fig. 4H, P < 0.05) in the ischaemic core were negatively correlated with the magnitude of weight loss at 2 days after tMCAO. There were no significant correlations with other neutrophil trafficking-related factors, such as Selp (Supplementary Fig. S4B and Fig. 4B), Vcam1 (Supplementary Fig. S4C and Fig. 4C), Icam1 (Supplementary Fig. S4D and Fig. 4D), Icam2 (Supplementary Fig. S4E and Fig. 4E), Cxcl1 (Supplementary Fig. S4F and Fig. 4F), Cxcl6 (Supplementary Fig. S4I and Fig. 4I), or Ppbp (Supplementary Fig. S4J and Fig. 4J). These results indicate that E-selectin was expressed in endothelial cells, and CXCL2 and CXCL3 produced by the ischaemic brain were highly expressed in severely ill rats after tMCAO. Increased E-selectin levels may loosen neutrophils rolling on the endothelium and cooperate with CXCL2 and CXCL3 to enhance neutrophil recruitment to the infarct region.

Correlation between neutrophil trafficking-related factors and severity of illness after transient middle cerebral artery occlusion (tMCAO). Correlation between the magnitude of weight loss and Sele (E-selectin) (A), Selp (P-selectin) (B), Vcam1 (C), Icam1 (D), Icam2 (E), Cxcl1 (F), Cxcl2 (G), Cxcl3 (H), Cxcl6 (Cxcl5) (I), or Ppbp (Cxcl7) mRNA expression (J) in the ischaemic core at 2 days after tMCAO (n = 10).
Correlation between neutrophils and cerebral oedema-related genes after tMCAO
Subsequently, we investigated the involvement of neutrophils in the development of cerebral oedema. Since the expression of AQP4 in astrocytes is closely related to the formation of brain oedema, we investigated the correlation between the expression levels of Aqp4 and Ly6c 2 days after tMCAO. The expression level of Aqp4 mRNA in the peri-infarct tissue (Fig. 5A, P < 0.001) was positively correlated with Ly6c mRNA, but not in the ischaemic core (Supplementary Fig. S5). Therefore, we examined the correlation between Ly6c and cytokines that induce AQP4 expression in astrocytes in peri-infarct tissue. The results showed that both Il1a and Tnf mRNA levels positively correlated with Ly6c mRNA levels (Fig. 5B, P < 0.01 and 5 C, P < 0.05). As shown in Fig. 5D, Ly6G+ cells strongly expressed IL-1α and TNF in the ischaemic brain. In addition, Ly6G+ and AQP4+/GFAP+ cells were closely located in the peri-infarct tissue (Fig. 5E). Neutrophils may be involved in the elevated AQP4 expression in astrocytes.

Correlation between neutrophils and cerebral oedema-related genes after transient middle cerebral artery occlusion (tMCAO). Correlation between Ly6c and Aqp4 (A), Il1a (B), and Tnf (C) mRNA expression in peri-infarct tissue 2 days after tMCAO (n = 10). (D) Images of Ly6G (green), IL-1α (red), and TNF (pink) fluorescent immunostaining in the ischaemic brain at 2 days after tMCAO. Hoechst 33,258 fluorescence (blue) indicates the nuclei (n = 3). White arrows indicate Ly6G+/IL-1α+/TNF+ cells. (E) Images of Ly6G (green), AQP4 (red), and GFAP (pink) fluorescent immunostaining in the ischaemic brain at 2 days after tMCAO. Hoechst 33,258 fluorescence (blue) indicates the nuclei (n = 3). White arrows indicate Ly6G+ cells.
Activated neutrophils increased AQP4 expression in cultured neonatal rat astrocytes
Activated neutrophils and astrocytes were co-cultured to investigate whether activated neutrophils affect astrocytic AQP4 expression. LPS-activated neutrophils were seeded on the cell culture inserts with polyethene terephthalate track-etched membranes (0.4-µm high pore density), and the cultured astrocytes were seeded on companion plates. After activated neutrophils and astrocytes were co-cultured for 18 h, AQP4 expression in astrocytes was evaluated. Activated neutrophils increased the expression of Aqp4 mRNA (Fig. 6A) and protein (Fig. 6B). When the astrocytes were pre-incubated with IL-1RI and TNF antagonists for 2 h, activated neutrophil-induced AQP4 expression was inhibited (Fig. 6A and B). Immunofluorescence staining showed that astrocytes co-cultured with activated neutrophils displayed activated short, thick processes and enlarged somata (Fig. 6D; LPS alone). These morphological changes were suppressed by IL-1RI and TNF antagonists (Fig. 6D). Furthermore, the AQP4-positive area was significantly increased relative to the GFAP-positive area in activated neutrophils (Fig. 6C; LPS alone). Additionally, IL-1RI and TNF antagonists suppressed the effects of the activated neutrophils (Fig. 6C). These data suggest that activated neutrophils induce AQP4 expression in cultured astrocytes.

Effect of activated neutrophils on primary cultured astrocytes. (A) Quantitative real-time polymerase chain reaction analysis of Aqp4 mRNA expression in astrocytes co-cultured with lipopolysaccharide (LPS; 10 ng/mL)-activated neutrophils after treatment with antagonists (IL-1RI; 100 µM or/and TNF; 100 µM) (n = 4). (B) Western blot analysis showing the protein levels of AQP4 in astrocytes co-cultured with LPS-activated neutrophils after treatment with antagonists (IL-1RI or/and TNF) (n = 8). (C and D ) Images of fluorescence immunostaining of AQP4 (green) and GFAP (red) in astrocytes (D). Hoechst 33,258 fluorescence (blue) indicates nuclei. Quantitative analysis of AQP4-positive area in GFAP-positive area in astrocytes co-cultured with LPS-activated neutrophils after treatment with antagonists (IL-1RI or/and TNF) (C) (n = 8). White arrows indicate AQP4+/GFAP+ cells. Data are expressed as mean ± standard deviation. *, **, and *** indicate P < 0.05, 0.01, and 0.001, respectively.
Discussion
Neutrophils are among the first immune cells to infiltrate brain lesions after an ischaemic stroke. Neutrophil infiltration within the infarct lesion is followed by a rapid response. The present study described the time course of neutrophil infiltration, neutrophil-attracting chemokines, and neutrophil-related inflammatory alterations and examined the correlation between these factors and the severity of illness. Furthermore, we showed that one mechanism involved the activation of neutrophils in cerebral oedema formation after ischaemic stroke.
Endothelial cells are activated by the local production of inflammatory cytokines such as IL-1 and TNF, which induces the expression of adhesion molecules and chemokines23,24. The expression of IL-1 and TNF peaked at 6 h after tMCAO, while that of most adhesion molecules and chemokines began to increase at 6 h and peaked at 24 h (Fig. 1). Neutrophils bind to selectins and integrin ligands, slowing their movement and causing them to roll along the endothelium. Neutrophil adhesion occurs when CXCR2 binds to neutrophil-attracting chemokines on the luminal surface of the endothelium. CXCR2 expression peaked between days 1 and 2 (Fig. 1N). In this study, neutrophils began in the leptomeninges 3 h after tMCAO and then spread to the ischaemic cortex at 6 h and the striatum at 24 h (Fig. 2D and E). The spatiotemporal profile data revealed a timeline of increased adhesion molecules and chemokines before neutrophil infiltration.
Severely ill rats showed a higher number of neutrophils in infarcted lesions (Fig. 3E-J). Increased E-selectin, CXCL2, and CXCL3 expression may be involved in high neutrophil infiltration in severely ill rats. Neutrophils, the most abundant cell type among leukocytes, are traditionally considered the first line of innate immune defence against microbes26. In addition to playing a role in killing bacteria, the activation of neutrophils causes the release of nuclear and granular contents to form extensive web-like structures of neutrophil extracellular traps (NETs)27. NETs neutralise pathogens and play a defensive role in the immune response; however, recent studies have reported that the formation of NETs exacerbates ischaemic brain damage28,29,30, and pharmaceutical modulation of NETs induces neuroprotection after stroke31. Even without NET formation, neutrophils may form a mechanical obstruction in brain capillaries, contributing to no flow in ischaemic stroke and leading to an exacerbation of stroke outcomes32. Furthermore, neutrophils play a crucial role in post-ischaemic inflammation by generating reactive oxygen and nitrogen species33,34 and disrupting the integrity of the BBB via the release of matrix metalloproteinases35,36. Therefore, it may be important to inhibit neutrophil infiltration into the cerebral infarct lesions. These results are consistent with those reported by Lux et al. in that a higher neutrophil-lymphocyte ratio and lower lymphocyte-monocyte ratio at 24 h were significantly associated with poorer functional outcomes in patients37. However, there are reports that N2 neutrophils and neutrophil-derived cathelicidin play a protective role in the pathogenesis of stroke38,39.
Furthermore, we revealed a novel function of neutrophils in the pathogenesis of cerebral infarction. There was a positive correlation between neutrophil and AQP4 expression in the peri-infarct tissue 2 days after tMCAO (Fig. 5A). Furthermore, there was also a positive correlation between neutrophils and IL-1α or TNF, which induced the expression of AQP4 (Fig. 5B and C). Neutrophils infiltrating the infarct lesion expressed IL-1α and TNF (Fig. 5D). They were closely present with astrocytes in the peri-infarct tissue (Fig. 5E). Liddelow et al. reported that IL-1α and TNF secreted by activated microglia are closely involved in the phenotypical changes toward reactive astrocytes16. Not only activated microglia but also IL-1α and TNF produced by neutrophils may play a major role in phenotypical changes in astrocytes. One limitation of this study is that in vitro experiments cannot fully replicate the complex inflammatory environment that occurs following a stroke, where interactions among neurons, microglia, and macrophages, neutrophils, and astrocytes play a critical role. However, our findings demonstrate that IL-1α and TNF from activated neutrophils are key mediators of brain oedema formation. Therefore, inhibiting neutrophil infiltration and activation may offer a potential strategy for alleviating brain oedema. We hope this research contributes to the development of targeted therapies for ischaemic stroke, focusing on neutrophils as a therapeutic target.
Conclusion
Neutrophil infiltration began within 3 h post-stroke and peaked at 24–48 h in the ischaemic cortex and striatum. The level of neutrophil infiltration was correlated with the severity after ischaemic stroke. Additionally, activated neutrophils, when co-cultured with astrocytes, increased the expression of AQP4, a protein involved in brain oedema. This effect was blocked by IL-1RI and TNF antagonists, suggesting that neutrophil-induced AQP4 expression via IL-1α and TNF exacerbates cerebral oedema in ischaemic brain regions.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
GBD 2019 Stroke Collaborators. Global, regional, and National burden of stroke and its risk factors, 1990–2019: A systematic analysis for the global burden of disease study 2019. Lancet Neurol. 20, 795–820. https://doi.org/10.1016/S1474-4422(21)00252-0 (2021).
Simard, J. M., Kent, T. A., Chen, M., Tarasov, K. V. & Gerzanich, V. Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol. 6, 258–268. https://doi.org/10.1016/S1474-4422(07)70055-8 (2007).
Dhanesha, N. et al. PKM2 promotes neutrophil activation and cerebral thromboinflammation: therapeutic implications for ischemic stroke. Blood 139, 1234–1245. https://doi.org/10.1182/blood.2021012322 (2022).
Segal, A. W. How neutrophils kill microbes. Annu. Rev. Immunol. 23, 197–223. https://doi.org/10.1146/annurev.immunol.23.021704.115653 (2005).
Gökhan, S. et al. Neutrophil lymphocyte ratios in stroke subtypes and transient ischemic attack. Eur. Rev. Med. Pharmacol. Sci. 17, 653–657 (2013).
Laridan, E. et al. Neutrophil extracellular traps in ischemic stroke thrombi. Ann. Neurol. 82, 223–232. https://doi.org/10.1002/ana.24993 (2017).
Kang, L. et al. Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat. Commun. 11, 2488. https://doi.org/10.1038/s41467-020-16191-y (2020).
Rosell, A. et al. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 39, 1121–1126. https://doi.org/10.1161/STROKEAHA.107.500868 (2008).
Huang, X. et al. Microglial IL-1RA ameliorates brain injury after ischemic stroke by inhibiting astrocytic CXCL1-mediated neutrophil recruitment and microvessel occlusion. Glia 71, 1607–1625. https://doi.org/10.1002/glia.24359 (2023).
Kim, S. W., Lee, H., Lee, H. K., Kim, I. D. & Lee, J. K. Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain. Acta Neuropathol. Commun. 7, 94. https://doi.org/10.1186/s40478-019-0747-x (2019).
Vella, J., Zammit, C., Di Giovanni, G., Muscat, R. & Valentino, M. The central role of aquaporins in the pathophysiology of ischemic stroke. Front. Cell. Neurosci. 9, 108. https://doi.org/10.3389/fncel.2015.00108 (2015).
Badaut, J. et al. Temporal and regional evolution of aquaporin-4 expression and magnetic resonance imaging in a rat pup model of neonatal stroke. Pediatr. Res. 62, 248–254. https://doi.org/10.1203/PDR.0b013e3180db291b (2007).
Rash, J. E., Yasumura, T., Hudson, C. S., Agre, P. & Nielsen, S. Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc. Natl Acad. Sci. U.S.A. 95, 11981–11986. https://doi.org/10.1073/pnas.95.20.11981) (1998).
Amantea, D. et al. Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front. Neurosci. 9, 147. https://doi.org/10.3389/fnins.2015.00147 (2015).
Czyżewski, W. et al. Astroglial cells: Emerging therapeutic targets in the management of traumatic brain injury. Cells 13, 148. https://doi.org/10.3390/cells13020148 (2024).
Liddelow, S. A. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. https://doi.org/10.1038/nature21029 (2017).
Murata, Y. et al. Activated microglia-derived macrophage-like cells exacerbate brain edema after ischemic stroke correlate with astrocytic expression of aquaporin-4 and interleukin-1 alpha release. Neurochem Int. 140, 104848. https://doi.org/10.1016/j.neuint.2020.104848 (2020).
Gono, R. et al. Molecular mechanism of cerebral edema improvement via IL-1RA released from the stroke-unaffected hindlimb by treadmill exercise after cerebral infarction in rats. J. Cereb. Blood Flow Metab. 43, 812–827. https://doi.org/10.1177/0271678X231151569 (2023).
Matsumoto, H. et al. Antibodies to CD11b, CD68, and lectin label neutrophils rather than microglia in traumatic and ischemic brain lesions. J. Neurosci. Res. 85, 994–1009. https://doi.org/10.1002/jnr.21198 (2007).
Longa, E. Z., Weinstein, P. R., Carlson, S. & Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20, 84–91. https://doi.org/10.1161/01.str.20.1.84 (1989).
Nishioka, R. et al. Treadmill exercise ameliorates ischemia-induced brain edema while suppressing Na⁺/H⁺ exchanger 1 expression. Exp. Neurol. 277, 150–161. https://doi.org/10.1016/j.expneurol.2015.12.016 (2016).
Sugimoto, K. et al. Activated microglia in a rat stroke model express NG2 proteoglycan in peri-infarct tissue through the involvement of TGF-β1. Glia 62, 185–198. https://doi.org/10.1002/glia.22598 (2014).
Markovic, S. et al. In vitro effects of cyclosporin A on the expression of adhesion molecules on human umbilical vein endothelial cells. Clin. Chim. Acta. 316, 25–31. https://doi.org/10.1016/s0009-8981(01)00732-x (2002). (PubMed: 11750271).
Kagnoff, M. F. & Eckmann, L. Epithelial cells as sensors for microbial infection. J. Clin. Invest. 100, 6–10. https://doi.org/10.1172/JCI119522 (1997).
Griffith, J. W., Sokol, C. L. & Luster, A. D. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu. Rev. Immunol. 32, 659–702. https://doi.org/10.1146/annurev-immunol-032713-120145 (2014).
Borregaard, N. Neutrophils from marrow to microbes. Immunity 33, 657–670. https://doi.org/10.1016/j.immuni.2010.11.011 (2010).
Sorvillo, N., Cherpokova, D., Martinod, K. & Wagner, D. D. Extracellular DNA NET-works with dire consequences for health. Circ. Res. 125, 470–488. https://doi.org/10.1161/CIRCRESAHA.119.314581 (2019).
Yipp, B. G. & Kubes, P. NETosis: How vital is it? Blood 122, 2784–2794. https://doi.org/10.1182/blood-2013-04-457671 (2013).
Li, C. et al. Neutrophil extracellular traps exacerbate ischemic brain damage. Mol. Neurobiol. 59, 643–656. https://doi.org/10.1007/s12035-021-02635-z (2022).
Denorme, F. et al. Neutrophil extracellular traps regulate ischemic stroke brain injury. J. Clin. Invest. 132, e154225. https://doi.org/10.1172/JCI154225 (2022).
Peña-Martínez, C. et al. Neutrophil extracellular trap targeting protects against ischemic damage after fibrin-rich thrombotic stroke despite non-reperfusion. Front. Immunol. 13, 790002. https://doi.org/10.3389/fimmu.2022.790002 (2022).
El Amki, M. et al. Neutrophils obstructing brain capillaries are a major cause of no-reflow in ischemic stroke. Cell Rep. 33, 108260. https://doi.org/10.1016/j.celrep.2020.108260 (2020).
Forster, C., Clark, H. B., Ross, M. E. & Iadecola, C. Inducible nitric oxide synthase expression in human cerebral infarcts. Acta Neuropathol. 97, 215–220. https://doi.org/10.1007/s004010050977 (1999).
Garcia-Bonilla, L. et al. Inducible nitric oxide synthase in neutrophils and endothelium contributes to ischemic brain injury in mice. J. Immunol. 193, 2531–2537. https://doi.org/10.4049/jimmunol.1400918 (2014).
Anrather, J. & Iadecola, C. Inflammation and stroke: An overview. Neurotherapeutics 13, 661–670. https://doi.org/10.1007/s13311-016-0483-x (2016).
Ludewig, P. et al. Carcinoembryonic antigen-related cell adhesion molecule 1 inhibits MMP-9-mediated blood-brain-barrier breakdown in a mouse model for ischemic stroke. Circ. Res. 113, 1013–1022. https://doi.org/10.1161/CIRCRESAHA.113.301207 (2013).
Lux, D. et al. The association of neutrophil-lymphocyte ratio and lymphocyte-monocyte ratio with 3-month clinical outcome after mechanical thrombectomy following stroke. J. Neuroinflammation. https://doi.org/10.1186/s12974-020-01739-y (2020).
Cuartero, M. I. et al. N2 neutrophils, novel players in brain inflammation after stroke: modulation by the PPARγ agonist rosiglitazone. Stroke 44, 3498–3508. https://doi.org/10.1161/STROKEAHA.113.002470 (2013).
Xie, W. et al. Neutrophil-derived cathelicidin promotes cerebral angiogenesis after ischemic stroke. J. Cereb. Blood Flow Metab. 43, 1503–1518. https://doi.org/10.1177/0271678X231175190 (2023).
Acknowledgements
We are grateful to the Nikon Imaging Center at Osaka University for their assistance with confocal microscopy and image acquisition and Hidekazu Tanaka, College of Life Sciences, Ritsumeikan University for kindly gifting HEK-293T cells. We would like to thank Editage (www.editage.com) for the English language editing.
Funding
This work was supported in part by the Japan Society for the Promotion of Science [Grants 22K09256 and 24H00656].
Author information
Authors and Affiliations
Contributions
KS designed the project, obtained funding, and was a major contributor to writing the manuscript. CY and MO performed the qPCR. MS provided technical support. RK performed the formal analyses. HM obtained funding, reviewed the manuscript and made critical revisions related to its content. All the authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All animal care and experimental procedures were performed in accordance with the Guidelines for Animal Experiments of Osaka University, with approval from the Animal Research Committee of Osaka University (Approval Number: 01-024-002).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sugimoto, K., Yang, C., Ono, M. et al. Neutrophils induce astrocytic AQP4 expression via IL-1α and TNF, contributing to cerebral oedema in ischaemic stroke rats. Sci Rep 15, 13923 (2025). https://doi.org/10.1038/s41598-025-98758-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-98758-7
