
Abstract
Homologous recombination deficiency (HRD) has been recognized as a key biomarker for poly-ADP ribose polymerase inhibitors (PARPi) and platinum-based chemotherapy in breast cancer (BC). HRD prediction typically relies on molecular biology assays, which have a high turnaround time, and cost. In contrast, tissue sections stained with hematoxylin and eosin (H&E) are ubiquitously available. However, current HRD prediction methods that utilize pathological images are usually based on attention-based multiple instance learning, which is ineffective for modeling the global context of whole slide images (WSIs). To address this challenge, we propose a Sufficient and Representative Transformer (SuRe-Transformer) for WSI-based prediction of HRD. Experimental results demonstrate the superior performance of SuRe-Transformer in predicting HRD status compared to state-of-the-art methods, achieving an AUROC of 0.887 ± 0.034. Furthermore, SuRe-Transformer demonstrates generalizability across multiple external patient cohorts and achieves state-of-the-art performance in predicting several gene mutation biomarkers from BC WSIs.
Similar content being viewed by others


Introduction
Breast cancer (BC) has the highest incidence rate among women globally and continues to be a leading cause of cancer-related mortality in women1; advancing the treatment of BC is of paramount importance. Homologous recombination repair (HRR) is a high fidelity repair pathway of double-strand DNA breaks. Homologous recombination deficiency (HRD) has been recognized as a key biomarker for poly-ADP ribose polymerase inhibitors (PARPi) in BC. Several methods have been developed to detect HRD, including testing for mutations in HRR-related genes2, analyzing genomic instability3 and mutational signatures4, and conducting functional assays5. However, standard diagnostic tests for detecting HRD via molecular profiling, pose economic challenges, particularly in low-resource settings6. Consequently, there is an urgent need to develop affordable and available methods for HRD prediction.
Hematoxylin and eosin (H&E) stained pathological images are ubiquitously available in clinical practice. Recent work by Lazard et al.7 indicates that morphological features, including high tumor cell density, conspicuous nucleoli, tissue necrosis, distinctive laminated fibrosis, and tumor infiltration, serve as the key indicators for differentiating HRD and homologous recombination proficient (HRP). Therefore, we decide to predict HRD status from WSIs stained with H&E. WSIs typically exhibit extremely high resolution and are often stored in a multi-resolution pyramid structure8. Consequently, it is generally considered ineffective to directly apply deep learning methods to the entire WSI for training9. To address this issue, we select several patches from WSI and aggregate these patches as WSI-based feature representations for HRD status prediction. While several approaches10,11 randomly select patches from WSIs, and others utilize color-based methods12 or patch-level classifier predictions13, these methods often fail to ensure the representativeness of the selected patches. Therefore, a good patch selection method, especially one that ensures the representativeness of the selected patches, is necessary for accurate HRD status prediction.
In the evolving landscape of bioinformatics, machine learning approaches are progressively demonstrating transformative potential14,15,16,17,18,19,20,21. This trend is particularly evident in computational pathology, where recent advances in deep learning22 have demonstrated accurate, reliable, and reproducible performance across various pathological diagnosis tasks, including the capacity to identify patterns not typically recognized by human experts. Schirris et al.23 introduce DeepSMILE, which uses a two-stage multiple instance learning (MIL) that combines self-supervised learning (SSL) for convolutional neural network (CNN) pre-training to extract features and variability-aware multiple instance learning (VarMIL) for feature aggregation to predict HRD status. To mitigate a bias among computational pathology workflows, Lazard et al.7 introduce a strategic sampling approach. They utilize Momentum Contrast (MoCo) for feature extraction and aggregate these features based on MIL to predict HRD status from tissue slides and identify related morphological phenotypes associated with HRD. Bergstrom et al.24 propose DeepHRD, which is based on ResNet18 and MIL, to predict the HRD status from BC WSIs.
Currently, although progress has been made in MIL-based HRD prediction methods using pathological images, these methods are ineffective in capturing the global context of WSIs. However, a new type of model architecture has emerged to replace the attention-based MIL method: Transformers. Originally developed for natural language processing (NLP), Transformers25 are robust tools for sequence processing, effectively harnessing the potential of large-scale data. Following the success of Transformer in NLP, Transformer has recently achieved remarkable success in various visual recognition tasks, primarily due to its ability to effectively capture long-range dependencies in sequences through attention mechanisms26. Dosovitskiy et al.27 proposed ViT, which directly applies a Transformer architecture to non-overlapping image patches for image classification. However, vanilla Transformers are constrained by computational complexity and can only handle shorter sequences of fewer than 1000 tokens28. To address these challenges, several Transformer-based methods for WSI analysis have adopted linear attention mechanisms29,30, including the Nyströmformer28 and Performer31, which replace the traditional softmax attention to reduce memory usage. Additionally, several studies have adopted sparse attention32 and deformable attention33 to reduce computational costs and memory requirements. However, the above Transformer-based methods often encounter overfitting issues when dealing with limited pathological images due to the nature of self-attention.
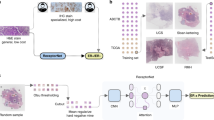
In the present study, we propose a novel Transformer-based framework, the Sufficient and Representative Transformer (SuRe-Transformer), for predicting HRD status from BC WSIs, as illustrated in Fig. 1. Altogether, we demonstrate that SuRe-Transformer addresses several key challenges in applying the Transformer to WSI through the following methods: (1) Instead of randomly selecting patches from WSI, we employ cluster-size-weighted sampling to ensure the representativeness of the selected patches. Additionally, we provide a mathematical proof for the effectiveness of this patch selection method. (2) We propose a radial decay self-attention (RDSA) to extend the input sequence length in Transformer, ensuring that a sufficient number of patches are considered. (3) We adapt an unsupervised feature extractor, pretrained using DINO on the largest BC WSI dataset we curated, to extract patch embeddings, enabling better feature representation for clustering and Transformer. Experiments conducted on BC from The Cancer Genome Atlas (TCGA) dataset demonstrate the superior performance of SuRe-Transformer in predicting HRD status compared to state-of-the-art methods. Furthermore, predictions from SuRe-Transformer generalize effectively to an external BC cohort. Additionally, our proposed framework can capture the general morphological features related to HRD, as demonstrated by an HRD classifier trained on TCGA BC that is able to predict HRD in TCGA ovarian and pancreas cancers. Moreover, our proposed framework is a general WSI representation learning framework that can predict various gene mutation biomarkers, including TP53, GATA3, MAP3K1, ERBB2, BRCA1, and PIK3CA, from BC WSIs.

a Overview of the 6 cohorts of BC, which are used in the study for pre-training feature extractors using self-supervised learning. Created with BioRender.com. b The preprocessing workflow for WSIs consists of several steps: WSI digitization, tissue segmentation, and tessellation, patch embedding based on self-supervised learning, and patch selection through clustering and cluster-size-weighted sampling. c The illustration of the SuRe-Transformer and network details of the SuRe-Transformer encoder.
Results
The experiment in this study is divided into four parts. First, we use TCGA BC dataset to compare SuRe-Transformer with state-of-the-art methods in evaluating the HRD status. Second, we conduct three external tests to compare SuRe-Transformer with DeepSMILE, as shown in Fig. 2a. Third, we evaluate the prediction performance of SuRe-Transformer in predicting six gene mutation biomarkers on TCGA BC dataset, as illustrated in Fig. 2b. Finally, we provide a visualization analysis to illustrate the results of patch clustering and conduct an ablation study to assess the contribution of each component of SuRe-Transformer.

a The area under the receiver operating characteristic curve provides an overall measure of SuRe-Transformer accuracy in predicting homologous recombination deficiency, evaluated using held-out test sets comprising different cancer types within a single data center (TCGA-PAAD, TCGA-OV) as well as data from distinct centers for the same cancer type (ChosenMed-BRCA). b The area under the receiver operating characteristic curve provides an overall assessment of SuRe-Transformer accuracy in predicting gene mutation biomarkers-including TP53, PIK3CA, MAP3K1, GATA3, ERBB2, and BRCA1-from breast cancer whole slide images. Created with BioRender.com.
Dataset description
We acquire Formalin-Fixed, Paraffin-Embedded (FFPE) WSIs for BC from TCGA. The dataset, collected from multiple medical centers across the United States, is available for download at https://portal.gdc.cancer.gov/. A description of the patient cohort is shown in detail in supplemental material (Supplementary Tables 1–7). We retrieve genomic DNA Damage Response (DDR) related labels, including HRD Score, from Knijnenburg et al.34. The HRD score is calculated based on several criteria: the number of subchromosomal regions with allelic imbalance extending to the telomere35, the number of chromosomal breakpoints between adjacent regions of at least 10 megabases36, and the number of regions with a loss of heterozygosity event of intermediate size37. The methods for labeling HRD status are inspired by binary (mHRD) and ternary (tHRD) strategies proposed by Schirris et al.23. The binary approach involves partitioning the HRD score at the median to distinguish HRP from HRD. That is,
where HRDm is the HRD score of the patient m and q(α) indicates the α-quantile of the HRD scores. Another approach utilizes ternary strategy, which divides the HRD scores into three portions. Patients with HRD scores in the top third are assigned HRD, those with HRD scores in the bottom third are assigned HRP, and patients with HRD scores in the middle third are discarded. That is:
Compared methods and evaluation metrics
1) Compared methods. For a direct comparison of the performance of SuRe-Transformer with other published methods, we select a weakly supervised method based on Transformer38 and two MIL methods: DMIL39 and DeepSMILE23. DeepSMILE uses a two-stage MIL, utilizing SSL to pre-train CNN for feature extraction and VarMIL for feature aggregation to achieve HRD status prediction. Only the untrained version of the DeepSMILE code is released to the public. Benchmark testing of these three methods, in comparison with SuRe-Transformer, is conducted on BC pathological images to predict HRD status. All source codes for these methods are downloaded and retrained to ensure a fair and accurate comparison.
2) Evaluation metrics. To evaluate the performance of our framework, we adopt the area under the receiver operating characteristic curve (AUROC, AUC) and F1 score as the primary evaluation metrics, consistent with the previous methods23. A higher F1 score indicates that the model exhibits high precision and recall in distinguishing between HRD and HRP. This means that the model not only accurately identifies HRD but also minimizes the misclassification of HRP. Additionally, two more curves are adopted for performance analysis, i.e., receiver operating characteristic (ROC) curves with the AUC values and precision recall curves (PRC) with the AUC values.
Implementation detail
We conduct all experiments utilizing a 5-fold cross-validation, where both validation and test sets are split off from the full dataset at the patient level. The dataset is randomly partitioned and is stratified by class, into a training set (80% of cases), and an internal validation (20% of cases). The partitioning was performed at the patient level and therefore all slides from the same patient are always placed into the same set. DINO uses Vision Transformer (ViT) as the backbone and employs the Adam optimizer with a learning rate of 0.0001. For K-means clustering, we take K = 50 following Yan et al.40,41. Considering computational efficiency, we divide the WSI into four radius ranges (Rad = 0 ~ r, r ~ 2r, 2r ~ 3r, . . . , (m − 1)r ~ mr) in RDSA of SuRe-Transformer, with m = 4. For training SuRe-Transformer, we perform data augmentation. Each WSI is clustered into 50 clusters, and subsequently, patches are then sampled from each cluster based on cluster-size-weighted sampling, resulting in the creation of M patches. Specifically, M takes values of 600, 800, 1000, 1200, 1400, and 1600. Repeating this random selection process S times yields a total of S groups from each WSI, effectively increasing the overall sample size by S times. Here, S = 30. The SuRe-Transformer is trained with AdamW42 optimizer with a weight decay of 2 × 10−5 and a learning rate of 1 × 10−4. All models are trained for 25 epochs with a batch size of 1. All experiments are performed on a 4-node cluster using the PyTorch framework and scikit-learn. Each node is equipped with a 32 cores CPU, four MI50 GPUs (AMD Vega20, 3840 stream processors, 16GB HBM2 memory), and 128GB of memory.
Overall prediction performance
Given the limited number of studies on HRD status prediction based on WSIs and the variability in labeling methods across different studies, achieving a perfect comparison is challenging. In this paper, we compare the performance of the proposed framework, SuRe-Transformer, with DeepSMILE, DMIL and Transformer-based methods for mHRD and tHRD prediction. To comprehensively demonstrate the predictive performance of our proposed framework, we evaluate the AUROC and F1 scores utilizing a 5-fold cross-validation approach and present the results as mean ± standard deviation. The performance of our proposed framework is shown in Table 1. Our proposed framework with RDSA trained on the patches selected using cluster-size-weighted sampling from WSI achieves the best performance, reaching 0.810 ± 0.047 AUROC and 0.737 ± 0.041 F1 for mHRD, and 0.887 ± 0.034 AUROC and 0.820 ± 0.034 F1 for tHRD. Compared to existing methods, SuRe-Transformer achieves statistically significant improvements across several metrics: it outperforms both DeepSMILE and Wagner et al. in AUROC and F1-score for tHRD (p < 0.05), while it exhibits a significant F1-score improvement over DeepSMILE for mHRD (p < 0.05). The increase in AUROC on mHRD, though numerically higher, is not statistically significant (p > 0.05). Among WSI-based HRD prediction methods, DeepSMILE demonstrates relatively lower performance, primarily due to its reliance on a feature extractor pre-trained on natural images, limiting its ability to effectively capture complex inter-patch relationships characteristic of histopathological images. In contrast, DMIL and Wagner et al. perform better than DeepSMILE as they utilize feature extractors specifically pre-trained on histopathological images. Our proposed method achieves superior performance due to the use of a feature extractor pre-trained on the largest available breast pathology image dataset, effectively capturing relevant histopathological features and inter-patch relationships. Additionally, we visualize the ROC curves and PRC of the above methods in Figs. 3 and 4 for mHRD and tHRD. To evaluate the generalizability of patch embedding based on DINO in our proposed framework, we conduct extensive comparative experiments across all methods. The results indicate that DeepSMILE and DMIL, using DINO for feature extraction, achieve significantly better performance compared to methods that do not incorporate DINO.

Top row, from left to right: ROC curves for a DeepSMILE, b Wanger et al., c DMIL and d SuRe-Transformer. Bottom row, from left to right: PRC for e DeepSMILE, f Wanger et al., g DMIL and h SuRe-Transformer. In the ROC curves, the black solid line represents the mean value of 5-fold cross-validation and the shaded area around the curves represents the standard deviation of 5-fold cross-validation. In the PRC, AUC (area under the curve) represents the area under the PRC.

Top row, from left to right: ROC curves for a DeepSMILE, b Wanger et al., c DMIL and d SuRe-Transformer. Bottom row, from left to right: PRC for e DeepSMILE, f Wanger et al., g DMIL and h SuRe-Transformer. In the ROC curves, the black solid line represents the mean value of 5-fold cross-validation and the shaded area around the curves represents the standard deviation of 5-fold cross-validation. In the PRC, AUC (area under the curve) represents the area under the PRC.
In addition, we evaluate the effectiveness of RDSA and patch selection based on K-means clustering method with cluster-size-weighted sampling within our proposed framework, using features extracted by DINO1K. Specifically, when the K-means is not used, patches are randomly selected from each WSI for model training and prediction. As shown in Table 1, the framework utilizing RDSA performs better than the one without it, reaching 0.796 ± 0.030 AUROC and 0.720 ± 0.060 F1 score for mHRD, and 0.869 ± 0.014 AUROC and 0.803 ± 0.035 F1 score for tHRD. We also observe that the framework with cluster-size-weighted sampling increases AUROC by 0.014 up to 0.883 ± 0.028 and increases F1 score by 0.009 up to 0.812 ± 0.032 for tHRD. For mHRD, it demonstrates a comparable performance enhancement, with the F1 score improving by 0.014 to 0.734 ± 0.049. Nevertheless, the improvement in AUROC for mHRD is relatively marginal, exhibiting merely a 0.004 increment to 0.800 ± 0.035. The results illustrate that all components of our proposed framework are essential for the performance of SuRe-Transformer.
Next, we aim to investigate whether morphological features associated with HRD in one cancer type could be used to predict HRD status in other cancer types. We conduct a comparative analysis of the performance of SuRe-Transformer and DeepSMILE by training both models on TCGA-BRCA and subsequently evaluating their effectiveness on TCGA-OV and TCGA-PAAD. The performance of the models is assessed based on AUROC and F1 score for both mHRD and tHRD, as summarized in Table 2. The results show that SuRe-Transformer, trained on TCGA-BRCA, outperforms DeepSMILE in predicting HRD status for TCGA-OV and TCGA-PAAD. Specifically, SuRe-Transformer achieves higher AUROC and F1 scores across all evaluations. For TCGA-OV, SuRe-Transformer achieves an AUROC of 0.713 and an F1 score of 0.664 for mHRD, compared to DeepSMILE’s AUROC of 0.640 and F1 score of 0.471. Similarly, for TCGA-PAAD, SuRe-Transformer achieves an AUROC of 0.599 and an F1 score of 0.577 for mHRD, while DeepSMILE achieves an AUROC of 0.552 and an F1 score of 0.419. Additionally, for tHRD, SuRe-Transformer demonstrates comparable performance improvements over DeepSMILE. These findings indicate that SuRe-Transformer, trained on TCGA-BRCA, can predict HRD status from histological features in other cancer types, suggesting a common “HRD morphology" shared across different cancers. The TCGA-PAAD and TCGA-OV used in this study are available for download at https://portal.gdc.cancer.gov/.
Finally, to assess the generalizability and robustness of our proposed framework, we conduct external validation experiments using a total of 192 BC patients (WSI + HRD status) collected from the Beijing ChosenMed Clinical Laboratory Co. Ltd as an independent test cohort. The HRD score is calculated using the scarHRD (https://github.com/sztup/scarHRD), following the methodology outlined in previous studies43,44. scarHRD uses whole genome sequencing single nucleotide polymorphism (SNP) array data to obtain the patient-wise HRD scores. Based on the 194 WSIs, we conduct an external validation (train on TCGA, test on ChosenMed) and show the results in Table 2. SuRe-Transformer outperforms DeepSMILE in predicting HRD status. Specifically, SuRe-Transformer achieves an AUROC of 0.712 and an F1 score of 0.656 for mHRD, and an AUROC of 0.750 and an F1 score of 0.692 for tHRD. These results highlight the superior performance of SuRe-Transformer in predicting HRD status, underscoring its generalizability and robustness across different sites. However, the predictive performance of SuRe-Transformer on the external validation set drops significantly, however, it is still slightly better than that of DeepSMILE. This is in line with our expectations, because from the naked eye, there is a large difference in color and appearance between the pathological images of ChosenMed and TCGA.
Prediction of breast cancer gene mutation biomarkers
Gene mutation biomarkers are crucial for accurate diagnosis and precise treatment of cancer. In the treatment of BC, TP53 mutations and ERBB2 overexpression45,46 are associated with poor prognosis and treatment response, while PIK3CA and BRCA1 mutations are linked to the applicability of targeted therapy drugs47,48. GATA3 mutations49 are relevant to the applicability of immunotherapy, and MAP3K1 mutations50 are associated with the regulation of cellular signaling pathways. The number of patients with various gene mutations in TCGA BC is shown in Supplementary Table 8. Notably, TP53, PIK3CA, GATA3, and MAP3K1 are the most frequently mutated genes in TCGA-BRCA51. We evaluate the prediction performance of SuRe-Transformer, which predicts six gene mutation biomarkers on TCGA-BRCA. The results are shown in Table 3. SuRe-Transformer can predict five gene mutation biomarkers (TP53, GATA3, MAP3K1, ERBB2, BRCA1) with an AUROC above 0.844. However, the performance for predicting PIK3CA gene mutation is not as robust. Additionally, we compute the gene variation results on six gene mutations of TCGA-BRCA, as shown in Supplementary Fig. 1. We find that PIK3CA gene mutations, which exhibit poor prediction performance, have higher mutation frequencies, with missense mutations accounting for a significant proportion. The model tends to overfit on missense mutations, leading to poor learning outcomes for rarer mutations, thus affecting the overall predictive performance.
Additionally, SuRe-Transformer demonstrates superior prediction performance across multiple gene biomarkers compared to the method proposed by Qu et al.52, underscoring its robustness and effectiveness. To evaluate the model, we acquire a total of 967 breast cancer patients’ data (WSI + gene mutation status) from TCGA. As shown in Table 4, compared to the method proposed by Qu et al., SuRe-Transformer achieves higher AUROC for all cancer gene biomarkers, including TP53, PIK3CA, MAP3K1, GATA3, ERBB2, and BRCA1. These results indicate that SuRe-Transformer improves prediction performance and provides a more reliable tool for identifying key gene mutations in BC.
The ROC and PRC curves
To comprehensively demonstrate the performance of our proposed framework on TCGA BC dataset, we visualize the ROC curves and PRC of the proposed architecture (Transformer + DINO1K, Transformer + DINO1K + Sparse, Transformer + DINO1K + Sparse + K-means) for tHRD and mHRD classification in Supplementary Fig. 2 and Supplementary Fig. 3, respectively. It can be shown that the framework (Transformer + DINO1K + Sparse + K-means), which utilizes RDSA and is trained on patches selected using cluster-size-weighted sampling from WSI, achieves the best performance in both ROC curves and PRC. Notably, the PRC for tHRD is relatively flatter compared to that for mHRD, likely because the tHRD discards patients with an HRD score close to the median, leading to a more distinct separation between HRD and HRP.
The visualization of patch clustering result
We follow the experimental settings outlined in previous work from Yan et al.40, utilizing K-means clustering with 50 clusters. In this study, we implement the “DINO + K-means” approach to cluster patches within each WSI. The visualization of patch clustering results for BC using K-means (K = 50) is shown in Supplementary Fig. 4. The clustering results indicate that spatially adjacent patches exhibit similar feature representations. Furthermore, discernible spatial heterogeneity of the tumor is observed. Employing clustering-based methodologies facilitates the selection of a diverse array of phenotypic patches, thereby enabling a nuanced characterization of tumor spatial heterogeneity.
Ablation study
We conduct several ablation studies to assess the benefits of different components in our proposed architecture. All experiments are conducted using a 5-fold cross-validation approach on BC dataset from TCGA. First, we evaluate the impact of the number of patches randomly selected from WSI. We fix the rest of the framework (Transformer + DINO1K), and only change the number of the patches randomly selected from WSI. The experimental results are shown in Supplementary Fig. 5 (random sample). When the input patch sequence length of the Transformer is 1000, it reflects the rich information contained in the WSI. Based on the experimental results, we observe that when the number of randomly selected patches is less than 1000, the performance of the model improves with the increase in the number of selected patches. However, when the number of randomly selected patches exceeds 1000, the performance of the model tends to decrease as the number of patches increases. This phenomenon can be attributed to the longer input sequence, which exacerbates overfitting.
Second, we evaluate the impact of RDSA in ViT. The experimental results are shown in Supplementary Fig. 5 (random sample vs. random sample with RDSA). RDSA effectively increases the input sequence length in ViT. The model achieves optimal predictive performance when the input sequence length is 1200. Based on the experimental results, we observe that when the number of randomly selected patches is less than 1000, the performance of the model with RDSA in ViT is slightly higher than that of the model without RDSA. However, when the number of randomly selected patches exceeds 1000, differences arise in the performance of the model with RDSA compared to that without it. The model with RDSA shows improved performance, whereas the model without RDSA exhibits a decline in performance. This also indicates that RDSA mechanism we introduce effectively alleviates the overfitting issue caused by excessively long input sequences.
Third, we evaluate the impact of the hyper-parameter k in RDSA, with the results reported in Table 5. We fix the rest of the framework (Transformer + DINO1K + Sparse), and only change the value of the hyper-parameter k within the range (Rad = 0 ~ r, r ~ 2r, 2r ~ 3r, . . . , (m − 1)r ~ mr) in RDSA to observe its effect on model performance, focusing on metrics such as accuracy, F1 score, and AUROC. From the results reported in the Table 5, we observe that the hyper-parameter k with a value of 4, within the range (Rad = 0 ~ r, r ~ 2r, 2r ~ 3r, . . . , (m − 1)r ~ mr) in RDSA, achieves the best performance. Our framework operates within four ranges, resulting in a total of k × 4 = 16. This finding is consistent with the experimental results reported by Yan et al.40, which also identifies a value of 16 as optimal. When k is too small, it inadequately represents the input sequence, resulting in suboptimal performance. When k is larger than 4, the performance of the proposed framework does not improve with the increase of k. When k is larger than 8, the performance of the proposed framework is lower than that of not using RDSA. These findings highlight the importance of selecting an optimal k value to balance computational efficiency and model performance in RDSA.
Next, we evaluate the impact of the cluster-size-weighted sampling. The experimental results are shown in Supplementary Fig. 5 (random sample with RDSA vs cluster-size-weighted sampling with RDSA). Unlike random patch selection from WSI, cluster-size-weighted sampling ensures that the selected patches are representative. Additionally, we fix the rest of the framework (Transformer + DINO1K + Sparse + K-means) and only modify the patch selection method: 1). Random patch selection, 2). Patch selection using cluster-size-weighted sampling within each WSI cluster, and 3). Selection of a fixed number of patches from each WSI cluster. The experimental results are shown in Supplementary Fig. 6 (random sample with RDSA vs. kmeans clustering(pro) with RDSA vs. kmeans clustering(equ) with RDSA). The patches selected by cluster-size-weighted sampling exhibit the best performance among the three methods, with a smaller standard deviation. As demonstrated in the systematic studies presented in Supplementary, both random patch selection and cluster-size-weighted sampling from the WSI share the same mathematical expectation, which can approximate the mathematical expectation of all patches in the WSI. However, selecting a fixed number of patches from each WSI cluster, which exhibits the worst performance among the three methods, does not approximate the expectation of all patches in the WSI. The relevant mathematical derivations are provided in Supplementary. Additionally, the patches selected by cluster size-weighted sampling lead to better model performance and a smaller standard deviation compared to randomly selected patches. This is primarily due to the fact that cluster-size-weighted sampling ensures a dispersed selection of patches from the WSI, while random patch selection allows for concentration. The coverage of different patch selection methods on WSIs is presented in Supplementary Fig. 7. As the number of samples increases, cluster size-weighted sampling can cover nearly all patches on the WSI.
Additionally, we evaluate the impact of the clustering embedding module in SuRe-Transformer, with the results reported in Table 6. In our experiments, we fix the rest of the framework (Transformer + DINO1K + Sparse + K-means), and only change the type of position embedding in SuRe-Transformer. The results show that the clustering embedding module significantly enhances performance compared to other position embedding strategies. This improvement can be attributed to the module’s ability to accurately capture and represent the relative relationships between different patches within the WSIs.
Finally, we conduct a comparative analysis of various SSL-based patch embedding methods on pathological images within our proposed framework. Building on recent insights from Wölflein et al.53, we include two SSL methods in the comparison: DINO and CTransPath. Introduced by Wang et al.54, CTransPath is a universal feature extractor for pathological images that combines convolutional layers with the Swin Transformer to effectively capture both local and global contextual information. It employs semantically-relevant contrastive learning (SRCL), a novel SSL technique based on MoCo v355, to enhance feature extraction. For a comprehensive comparison, we also include UNI56, a universal self-supervised framework that leverages the DINOv2 (ref. 57) backbone network and enhances data diversity through pre-training on the “Mass-100K” pathological images dataset, designed for applications such as tissue and disease subtype classification. Additionally, we introduce four variants of DINO: DINO32, DINO20, DINO1K, and DINO8K. The DINO32, DINO20, and DINO1K variants are trained on eight A800 blocks over a span of 36 days, utilizing datasets representing 32 common cancer types, 20 prevalent cancer types, and BC pathological images from TCGA, respectively. Specifically, DINO8K is trained on 7723 WSIs of BC collected from publicly available datasets, including TCGA-BRCA and CPTAC-BRCA, as well as a private dataset. In our analysis, we maintain the remaining components of the framework (Transformer + Sparse + K-means) and evaluate six patch embedding methods: CTransPath (768D)54, DINO32 (384D), DINO20 (384D), DINO1K (384D), DINO8K (384D), and UNI (1024D)56. The experimental results, presented in Table 7, indicate that DINO8K achieves the best performance, with an AUROC of 0.887. The comparatively poor performance of UNI can likely be attributed to its exclusion of public pathological image datasets such as TCGA, CPTAC, and PAIP during pre-training. In contrast, DINO and CTransPath utilize datasets like TCGA, which may lead to performance discrepancies, particularly in AUROC results, as shown in Table 7. Additionally, we visualize the attention heatmap of DINO across 6 attention heads in Supplementary Fig. 8. We observe an interesting phenomenon: while foundational models exhibit strong performance across various cancer types, their effectiveness is fundamentally influenced by the volume and specificity of the data used for training. When the focus shifts to a single cancer type, such as BC, it becomes evident that a larger, specialized pre-trained model tailored specifically for BC may yield superior outcomes.
We compare the performance of the proposed framework, SuRe-Transformer, with Transformer-based methods for mHRD and tHRD prediction, including TransMIL, Mamba, and the method proposed by Wagner et al. The performance of our proposed framework is shown in Table 8. SuRe-Transformer achieves the best performance, reaching 0.810 ± 0.047 AUROC and 0.737 ± 0.041 F1 for mHRD, and 0.887 ± 0.034 AUROC and 0.820 ± 0.034 F1 for tHRD.
Discussion
The deployment of HRD biomarker testing in precision oncology for BC presents a promising avenue to enhance treatment efficacy and potentially extend patient survival. However, its adoption has been hindered by significant challenges, primarily due to the complexity and high costs associated with HRD testing, which requires sophisticated diagnostic equipment and specialized expertise. In contrast, tissue sections stained with H&E are ubiquitously available, presenting a viable alternative for assessing HRD status. Therefore, we decide to predict HRD status from WSIs stained with H&E to make HRD testing more accessible and feasible.
In this work, we propose a novel Transformer-based method for the HRD status prediction from WSI, considering the comprehensive correlation among different patches within the WSI. As shown in Fig. 5, there are tens of thousands of patches cropped from a WSI, resulting in extremely long input sequences. The vanilla Transformer exhibits high computational complexity, which restricts the length of the input sequence. Furthermore, the training data for HRD status prediction is limited, making the model susceptible to overfitting. Therefore, using all patches P from each WSI as input is ineffective for predicting HRD status. Instead, we select a small number of informative patches \({P}^{{\prime} }\) and then aggregate them for HRD status prediction. The main contributions are as follows (see Fig. 5):
-
To ensure that the selected patches \({P}^{{\prime} }=\{{P}_{k}^{{\prime} }| k=1,\ldots ,S\}\) are informative to preserve the correlation with biomarker labels, we introduce SuRe-Transformer to capture an informative set of patches from WSI. (i) Departing from randomly selecting patches from WSI, we employ cluster-size-weighted sampling to ensure the representativeness of the selected patches. Additionally, we theoretically demonstrate the performance of the framework when using different patch selection methods. (ii) We propose a RDSA mechanism to extend the length of the input sequence in ViT to ensure that sufficient patches are considered. (iii) We pretrain DINO using the largest BC WSI dataset, which consists of 7723 BC WSIs collected from publicly available and private datasets. The resulting model, DINO8K, serves as an unsupervised feature extractor to obtain better feature representation from image patches within WSIs for clustering and Transformer. (iv) Since the absolute and relative positions between patches after patch sampling are unclear, we propose a clustering embedding module to replace traditional 1D and 2D position embeddings. This module rearranges patches within the same cluster and combines all clusters into SuRe-Transformer’s input sequence, ensuring that spatially or semantically related patches are processed in close proximity.
-
Our method achieves better prediction performance. Experiments conducted on BC from TCGA dataset demonstrate the superior performance of SuRe-Transformer in predicting HRD status, compared to the state-of-the-art methods. Additionally, SuRe-Transformer exhibits strong generalizability, effectively predicting HRD status not only in external BC cohorts but also across different cancer types, including ovarian and pancreatic cancers.
-
Our framework demonstrates strong generalizability. SuRe-Transformer achieves state-of-the-art performance in predicting key gene mutation biomarkers from BC WSIs. Notably, our proposed framework achieves an AUROC above 0.84 for predicting five key gene mutation biomarkers (TP53, GATA3, MAP3K1, ERBB2, BRCA1).

Here, \({P}^{{\prime} }=\{{P}_{k}^{{\prime} }| k=1,\ldots ,S\}\) represents the selected patches for analysis in SuRe-Transformer, and S denotes the number of repeated samplings from each WSI.
From a clinical perspective, our method achieves high predictive performance for HRD status based on histopathological slides and demonstrates generalizability across diverse cancer types and patient cohorts, providing a cost-effective and scalable solution. Additionally, it shows promising utility in predicting gene mutations, indicating potential as a versatile biomarker tool, especially in resource-limited settings. Building upon these features, our proposed approach may facilitate the clinical translation of deep learning-based methods.
Although our research has demonstrated promising results, it is essential to acknowledge that our experimental setup has several limitations. First, while SuRe-Transformer demonstrates better generalizability to external patient cohorts compared to DeepSMILE, its performance declines on external test datasets, particularly when compared to internal test datasets. Future work will explore the integration of domain adaptation techniques to address this issue. Second, SuRe-Transformer demonstrates limited effectiveness in handling class imbalance, particularly for underrepresented mutation types. Differences in mutation frequency distributions among various BC gene mutation biomarkers contribute to this imbalance. Although data augmentation techniques were employed to mitigate the issue, the model still struggles with detecting rare gene mutations, especially those with low variant allele frequencies. These limitations may affect its generalizability to larger or more heterogeneous datasets and should be addressed in future work. Third, our current approach has primarily been evaluated using standardized, high-quality datasets such as TCGA, without specific experimentation on lower-quality imaging data. Future studies should investigate the method’s applicability and robustness with lower-quality imaging data to enhance its real-world practicality and effectiveness. Fourth, our proposed method is currently not fully end-to-end, requiring separate preprocessing and feature extraction steps that could introduce variability. Future research will aim to integrate these steps to streamline and enhance the overall workflow. Fifth, the predictive performance of our method exhibits considerable fluctuations. To improve prediction stability and reliability, we intend to introduce a voting mechanism or ensemble approach in future investigations. Finally, the diversity of HRD labeling strategies in existing studies hinders equitable comparisons across different approaches. To provide a reference, we investigate the performance comparisons of existing HRD classification methods based on BC, as presented in Table 9. The HRD status prediction methods utilize various label acquisition methods for HRD status based on the TCGA BC dataset, including HRD score, mutation signatures, and gene signatures. On TCGA BC dataset, SuRe-Transformer achieves similar performance on mHRD classification as Valieris et al.58, although a perfect comparison is not available due to the different labeling strategies. Additionally, we observe that the predictive performance of tHRD classification is significantly better than the best reported to date. Specifically, SuRe-Transformer achieves an AUROC of 0.89, while DeepSMILE achieved an AUROC of 0.81.
In conclusion, we proposed a novel Transformer-based framework, SuRe-Transformer, for predicting HRD status on WSI from BC with an AUROC of 0.89 ± 0.04 for tHRD and 0.81 ± 0.05 for mHRD. Our model generalizes effectively to an external BC cohort. By publishing the trained models, we hope our proposed framework will provide the researchers and clinicians with new way in HRD prediction using routine histology images.
Methods
Our proposed Transformer-based framework for predicting HRD status comprises four main components: patch embedding based on self-supervised learning, patch selection based on cluster-size-weighted sampling, the architecture of SuRe-Transformer, and radial decay self-attention. Each component will be described in detail in the subsequent subsections.
Patch embedding based on self-supervised learning
In the pre-processing pipeline for WSIs, we use RGB thresholding and Canny edge detection to segment tissue regions, effectively isolating these regions from the background and generating segmentation masks that contain enough tissue cells. Each WSI is tessellated into patches of 256 × 256 pixels (without overlap) at 20 × magnification with a resolution of 0.5 microns per pixel. We view each WSI (known as bag) as a collection of many smaller regions or patches (known as instances). Background patches are removed and the remaining tissue patches are subsequently transformed into feature embeddings. Given the difficulty in obtaining patch-level biomarker annotations, supervised learning methods are not feasible for patch embedding. However, recent advances in SSL offer promising solutions by enabling the training of robust feature extractors without relying on labeled data. Notably, several SSL models trained on large-scale pathology datasets have emerged, providing superior representations for various downstream tasks.
DINO59 is a type of SSL method commonly referred to as knowledge distillation, primarily aimed at training smaller networks to replicate the outputs of larger networks for model compression. A comparison of the feature representation learning abilities of different SSL methods is summarized in Table 7. DINO comprises two networks: the student network and the teacher network. The student network is defined by a set of parameters θs, as shown in Supplementary Fig. 9. The teacher network has the same network structure as the student network but a different set of parameters θt. The teacher network provides the training target for the student network, whose parameters are updated by using an exponential moving average (EMA) on the student weights, following the rule:
where λ follows a cosine scheduled from 0.996 to 1 during training. Given a fixed teacher network gθt, we learn to match the distributions by minimizing the cross-entropy loss with respect to the parameters of the student network gθs,
where \(H(a,b)=-a\log b\). After training converges, the patch embedding output gθt is used as the pathological image representation. Therefore, we utilize DINO, which has been pre-trained on 6 cohort of BC pathological images, as shown in Fig. 1a, to convert each image patch of size 256 × 256 × 3 into a discriminative feature vector of size 384. We save each patch in a hierarchical data format hdf5, which contains an array of extracted features along with their corresponding patch coordinates.
Patch selection based on cluster-size-weighted sampling
Transformer-based architectures are computationally expensive due to the self-attention mechanism, the computation of which is typically quadratic to the length of the input sequence. This poses a considerable challenge for analyzing histopathological images, such as WSIs, which are of extremely high resolution, thereby limiting the number of input patches in existing Transformer-based WSI analysis approaches. Traditional Transformers are only capable of handling short sequences of less than 100028. To address this limitation, we select several patches from WSI and aggregate these patches into WSI-based feature representations. However, random patch selection is insufficient for capturing the representative information from the WSI.
Ensuring the representativeness of the selected patches is essential for accurate HRD status prediction. Therefore, we propose a patch selection method based on cluster-size-weighted sampling, as shown in Fig. 1b, where all patches cropped from the WSI are clustered into K clusters using the K-Means algorithm, a standard unsupervised clustering technique. Subsequently, patches are sampled from each cluster based on the proportion of patches in each cluster relative to the total number of patches in the WSI, ensuring that the required number of patches, denoted as M, is obtained from each WSI. Detailed information can be found in Section “SuRe-Transformer architecture”. Specifically, each patch is transformed into a descriptive, one-dimensional feature representation with a length of 384 based on the DINO8K. The K-means algorithm is then applied to cluster these feature vectors into K clusters. Patches are sampled from each cluster in accordance with the aforementioned selection proportion, ensuring that the selected patches effectively represent the diversity within each cluster for subsequent analysis.
SuRe-Transformer architecture
There are two main obstacles that hinder the application of Transformers for HRD status prediction. First, the inherent computational complexity of the vanilla Transformer imposes limitations on the length of the input sequence. Second, the limited availability of datasets for HRD status prediction increases the risk of model overfitting. Therefore, it is crucial to explore alternative Transformer architectures tailored to address these challenges.
The SuRe-Transformer builds upon the ViT. We define the ith patch obtained from gigapixel WSIs as an instance xi, and consider the set of all patches in the WSI as a bag (B = {xi∣i = 1, . . . , N}), where N represents the number of patches in the WSI. The input of SuRe-Transformer architecture is a dataset P consisting of a sequence of M patches, with the dataset label derived from the corresponding WSI label:
where K is the number of clusters per WSI, \({C}_{k}=\frac{{N}_{k}}{N}\times M\) denotes the number of selected patches from the kth cluster, M satisfies the following relationship \(M=\mathop{\sum }\nolimits_{k=1}^{K}{C}_{k}\), and Nk is the number of patches in the kth cluster. The patch embedding E(⋅) is extracted based on the DINO8K.
Since the Transformer does not account for the position embedding38, position embeddings are added to the patch embeddings to capture the order of the input sequence. However, given that the input of the SuRe-Transformer is sparsely distributed, traditional 1D and 2D position embeddings fail to effectively capture the relative relationship between the patches within the WSI. To address this issue, we propose a clustering embedding module in SuRe-Transformer, making it more suitable for the HRD status prediction. As illustrated in Supplementary Fig. 10, the vanilla Transformer encoder utilizes [*, Patch embedding] plus position embedding. In contrast, the SuRe-Transformer encoder introduces a clustering embedding module, which arranges the patches of the same cluster together and combines all clusters to generate the input sequence of SuRe-Transformer, ensuring that spatially or semantically related patches are processed in closer proximity. This allows the SuRe-Transformer to better understand the contextual information, as each patch is assigned its embedding information based on its position in the sequence.
As illustrated in Fig. 1c, each layer of the SuRe-Transformer encoder consists of a multi-head radial decay self-attention (MHRDSA) module, which is detailed in the subsequent subsection, and a multi-layer perceptron (MLP) block. We follow the ViT, adding the CLS token at the beginning of the input sequence P. The input token embeddings can be represented as:
The Transformer encoder module consists of L-stacked layers, where LN denotes the layer normalization, and MLP comprises two fully connected layers with a non-linear GELU activation function25. The SuRe-Transformer can be expressed as follows:
The CLS token from the output of the L Transformer layers is used as the WSI representation and is passed into the MLP head for predicting the HRD status for BC. The prediction of the HRD status can be formulated as follows:
Radial decay self-attention
The self-attention (SA) in the Transformer computes the query-key product as follows:
where Q, K, and V represent the queries, keys, and values, respectively, and dk denotes the dimensions of Q and K. The multi-head self-attention (MHSA) applies SA in every head and concatenates the heads in a weighted manner:
where headi = Att(Qi, Ki, Vi), i = 1. . . h, and Wo are learnable parameters. Specifically, we choose a Transformer network architecture consisting of six layers, each with six heads. The MHSA is an extension of the SA mechanism in Transformer that enables the model to learn in different representation spaces by parallelizing multiple independent SA operations, allowing it to more effectively capture complex relationships within the input sequence. Most ViT-based architectures, such as the original ViT, perform poorly on small-scale biomarker datasets due to a lack of inductive bias. In the task of HRD status prediction, it is unnecessary for each patch in the input sequence to establish relationships with all other patches. Instead, it suffices for each patch to interact with only a subset of key patches.
We introduce a RDSA mechanism, inspired by Generative Pre-trained Transformer 3 (GPT-3)60. One fundamental idea behind GPT-3 is sparse self-attention, where attention is restricted to tokens within a relative distance of no more than k. That is, attention is selectively applied to tokens at relative distances of k, 2k, 3k, and beyond, while remaining zero for all other tokens. While the concept based on GPT-3 is one-dimensional, pathological images are two-dimensional; hence we analogize the distance (K = k, 2k, 3k, . . .) in 1D context to the radius (Rad = 0 ~ r, r ~ 2r, 2r ~ 3r, . . . , (m − 1)r ~ mr) in 2D context. The illustration of the RDSA mechanism is depicted in Supplementary Fig. 11. The key idea is to reduce the dense attention matrix to a sparse representation by restricting attention computation to a limited subset of query-key (QK) pairs.
In this paper, we first obtain the attention matrix based on patches from the model, as the [CLS] token does not contain WSI coordinate-related information. Subsequently, we compute the relative distances D between patches within WSIs, as illustrated below:
where (xi, yi) represents the coordinate of the ith patch in the WSI, and max(x) denotes the maximum value of the coordinates x among all patches. We first mark the patches within each radius range by:
To implement the patch selection within the radius range, we utilize a randomly initialized unit matrix R within the (0, 1) range to assign a selection probability to the patches within the different radius ranges, as illustrated by S.
where ⊗ denotes element-wise operation. Let \({t}_{i}^{(n)}\) denote the kth largest values of range n in the ith row, and k is the hyper-parameter. To select the top-k values of S in the different radius ranges Rad, we use the mask operation M(.), which is illustrated as:
We concatenate the {M(n); n = 1, 2, …, m} to form M = [Mij]:
After concatenation, the RDSA is defined as follows:
Data availability
The data supporting the findings of this study are publicly available online through the TCGA’s Genomic Data Commons (https://portal.gdc.cancer.gov/) with the project IDs TCGA-BRCA, TCGA-OV, and TCGA-PAAD. The ChosenMed-BRCA dataset and the dataset used for DINO8K self-supervised pretraining during the current study may be obtained from the corresponding author upon reasonable request.
Code availability
The code required to reproduce these findings is available for download from https://github.com/Luan-zb/SuRe-Transformer.
References
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. J. CA Cancer J. Clin. 74, 12–49 (2024).
Schaeffer, E. et al. Nccn guidelines insights: prostate cancer, version 1.2021: Featured updates to the nccn guidelines. J. Natl. Compr. Cancer Netw. 19, 134–143 (2021).
Lips, E. H. et al. Quantitative copy number analysis by multiplex ligation-dependent probe amplification (mlpa) of brca1-associated breast cancer regions identifies brcaness. BREAST Cancer Res. 13, 1–9 (2011).
Davies, H. et al. Hrdetect is a predictor of brca1 and brca2 deficiency based on mutational signatures. Nat. Med. 23, 517–525 (2017).
Tumiati, M. et al. A functional homologous recombination assay predicts primary chemotherapy response and long-term survival in ovarian cancer patients. Clin. Cancer Res. 24, 4482–4493 (2018).
Bergstrom, E. N. et al. Deep learning predicts hrd and platinum response from histology slides in breast and ovarian cancer. medRxiv, https://www.medrxiv.org/content/10.1101/2023.02.23.23285869v1 (2023).
Lazard, T. et al. Deep learning identifies morphological patterns of homologous recombination deficiency in luminal breast cancers from whole slide images. Cell Rep. Med. 3, 100872 (2022).
Li, B., Li, Y. & Eliceiri, K. W. Dual-stream multiple instance learning network for whole slide image classification with self-supervised contrastive learning. In Proceedings of the IEEE/CVF conference on computer vision and pattern recognition, 14318–14328 (IEEE, 2021).
Lu, M. Y. et al. Ai-based pathology predicts origins for cancers of unknown primary. Nature 594, 106–110 (2021).
Li, R., Yao, J., Zhu, X., Li, Y. & Huang, J. Graph cnn for survival analysis on whole slide pathological images. In: International conference on medical image computing and computer-assisted intervention, 174–182 (Springer, 2018).
Raju, A., Yao, J., Haq, M. M., Jonnagaddala, J. & Huang, J. Graph attention multi-instance learning for accurate colorectal cancer staging. In: Medical image computing and computer assisted intervention-MICCAI, 529–539 (Springer, 2020).
Adnan, M., Kalra, S. & Tizhoosh, H. R. Representation learning of histopathology images using graph neural networks. In Proceedings of the IEEE/CVF conference on computer vision and pattern recognition workshops, 988–989 (IEEE, 2020).
Wang, X. et al. Weakly supervised deep learning for whole slide lung cancer image analysis. IEEE Trans. Cybern. 50, 3950–3962 (2019).
Chen, X., Li, T.-H., Zhao, Y., Wang, C.-C. & Zhu, C.-C. Deep-belief network for predicting potential mirna-disease associations. Brief. Bioinforma. 22, bbaa186 (2021).
Ha, J., Park, C., Park, C. & Park, S. Imipmf: inferring mirna-disease interactions using probabilistic matrix factorization. J. Biomed. Inform. 102, 103358 (2020).
Ha, J. & Park, S. Ncmd: Node2vec-based neural collaborative filtering for predicting mirna-disease association. IEEE/ACM Trans. Computational Biol. Bioinforma. 20, 1257–1268 (2022).
Ha, J. Mdmf: Predicting mirna–disease association based on matrix factorization with disease similarity constraint. J. Personalized Med. 12, 885 (2022).
Ha, J. Smap: Similarity-based matrix factorization framework for inferring mirna-disease association. Knowl. Based Syst. 263, 110295 (2023).
Ha, J. & Park, C. Mlmd: metric learning for predicting mirna-disease associations. IEEE Access 9, 78847–78858 (2021).
Ha, J. Graph convolutional network with neural collaborative filtering for predicting mirna-disease association. Biomedicines 13, 136 (2025).
Ha, J. Deepwalk-based graph embeddings for mirna–disease association prediction using deep neural network. Biomedicines 13, 536 (2025).
Unger, M. & Kather, J. N. Deep learning in cancer genomics and histopathology. Genome Med. 16, 44 (2024).
Schirris, Y., Gavves, E., Nederlof, I., Horlings, H. M. & Teuwen, J. Deepsmile: contrastive self-supervised pre-training benefits MSI and HRD classification directly from h&e whole-slide images in colorectal and breast cancer. Med. Image Anal. 79, 102464 (2022).
Bergstrom, E. N. et al. Deep learning artificial intelligence predicts homologous recombination deficiency and platinum response from histologic slides. J. Clin. Oncol. 42, 3550–3560 (2024).
Vaswani, A. et al. Attention is all you need. In: Advances in neural information processing systems, 30 (NIPS, 2017).
Han, K. et al. A survey on vision transformer. IEEE Trans. Pattern Anal. Mach. Intell. 45, 87–110 (2022).
Dosovitskiy, A. et al. An image is worth 16x16 words: Transformers for image recognition at scale. In: International conference on learning representations, 1–22 (OpenReview.net, 2020).
Xiong, Y. et al. Nyströmformer: A nyström-based algorithm for approximating self-attention. Proc. AAAI Conf. Artif. Intell. 35, 14138–14148 (2021).
Shao, Z. et al. Transmil: Transformer based correlated multiple instance learning for whole slide image classification. Adv. Neural Inf. Process. Syst. 34, 2136–2147 (2021).
Luan, H. et al. Multi-class cancer classification of whole slide images through transformer and multiple instance learning. In: International symposium on bioinformatics research and applications, 150–164 (Springer, 2023).
Choromanski, K. et al. Rethinking attention with performers. In: International conference on learning representations, 1–38 (OpenReview.net, 2021).
Liu, Z. et al. Swin transformer: hierarchical vision transformer using shifted windows. In: Proceedings of the IEEE/CVF international conference on computer vision, 10012–10022 (IEEE, 2021).
Xia, Z., Pan, X., Song, S., Li, L. E. & Huang, G. Vision transformer with deformable attention. In: Proceedings of the IEEE/CVF conference on computer vision and pattern recognition, 4794–4803 (IEEE, 2022).
Knijnenburg, T. A. et al. Genomic and molecular landscape of dna damage repair deficiency across the cancer genome atlas. Cell Rep. 23, 239–254 (2018).
Birkbak, N. J. et al. Telomeric allelic imbalance indicates defective dna repair and sensitivity to dna-damaging agents. Cancer Discov. 2, 366–375 (2012).
Popova, T. et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with brca1/2 inactivation. Cancer Res. 72, 5454–5462 (2012).
Abkevich, V. et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 107, 1776–1782 (2012).
Wagner, S. J. et al. Transformer-based biomarker prediction from colorectal cancer histology: A large-scale multicentric study. Cancer Cell 41, 1650–1661 (2023).
Ilse, M., Tomczak, J. & Welling, M. Attention-based deep multiple instance learning. In: International conference on machine learning, 2127–2136 (PMLR, 2018).
Yan, R. et al. Sparse and hierarchical transformer for survival analysis on whole slide images. IEEE J. Biomed. Health Inf. 28, 7–18 (2023).
Yan, R. et al. Histopathological bladder cancer gene mutation prediction with hierarchical deep multiple-instance learning. Med. Image Anal. 87, 102824 (2023).
Loshchilov, I. & Hutter, F. Decoupled weight decay regularization. In: International conference on learning representations, 1–19 (OpenReview.net, 2019).
Zhao, E. Y. et al. Homologous recombination deficiency and platinum-based therapy outcomes in advanced breast cancer. Clin. Cancer Res. 23, 7521–7530 (2017).
Sztupinszki, Z. et al. Migrating the snp array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. NPJ Breast Cancer 4, 16 (2018).
Børresen-Dale, A.-L. Tp53 and breast cancer. Hum. Mutat. 21, 292–300 (2003).
Blackwell, K. L. et al. Randomized study of lapatinib alone or in combination with trastuzumab in women with erbb2-positive, trastuzumab-refractory metastatic breast cancer. J. Clin. Oncol. 28, 1124–1130 (2010).
Miller, T. W., Rexer, B. N., Garrett, J. T. & Arteaga, C. L. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 13, 1–12 (2011).
Fong, P. C. et al. Poly (adp)-ribose polymerase inhibition: Frequent durable responses in brca carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 28, 2512–2519 (2010).
Asch-Kendrick, R. & Cimino-Mathews, A. The role of gata3 in breast carcinomas: a review. Hum. Pathol. 48, 37–47 (2016).
Widmann, C., Gibson, S., Jarpe, M. B. & Johnson, G. L. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiological Rev. 79, 143–180 (1999).
Jiang, Y.-Z. et al. Integrated multiomic profiling of breast cancer in the chinese population reveals patient stratification and therapeutic vulnerabilities. Nat. Cancer 5, 673–690 (2024).
Qu, H. et al. Genetic mutation and biological pathway prediction based on whole slide images in breast carcinoma using deep learning. NPJ Precis. Oncol. 5, 87 (2021).
Wölflein, G. et al. A good feature extractor is all you need for weakly supervised learning in histopathology. Preprint at https://arxiv.org/abs/2311.11772 (2023).
Wang, X. et al. Transformer-based unsupervised contrastive learning for histopathological image classification. Med. Image Anal. 81, 102559 (2022).
Chen, X., Xie, S. & He, K. An empirical study of training self-supervised vision transformers. In Proceedings of the IEEE/CVF international conference on computer vision, 9640–9649 (IEEE, 2021).
Chen, R. J. et al. Towards a general-purpose foundation model for computational pathology. Nat. Med. 30, 850–862 (2024).
Oquab, M. et al. Dinov2: Learning robust visual features without supervision. Trans. Mach. Learn. Res J 1–31 (2024).
Valieris, R. et al. Deep learning predicts underlying features on pathology images with therapeutic relevance for breast and gastric cancer. Cancers 12, 3687 (2020).
Caron, M. et al. Emerging properties in self-supervised vision transformers. In Proceedings of the IEEE/CVF international conference on computer vision, 9650–9660 (IEEE, 2021).
Brown, T. et al. Language models are few-shot learners. Adv. Neural Inf. Process. Syst. 33, 1877–1901 (2020).
Gu, A. & Dao, T. Mamba: Linear-time sequence modeling with selective state spaces. In: International conference on learning representations, 1–27 (OpenReview.net, 2023).
Kather, J. N. et al. Pan-cancer image-based detection of clinically actionable genetic alterations. Nat. Cancer 1, 789–799 (2020).
El Nahhas, O. S. et al. Regression-based deep-learning predicts molecular biomarkers from pathology slides. Nat. Commun. 15, 1253 (2024).
Loeffler, C. M. L. et al. Direct prediction of homologous recombination deficiency from routine histology in ten different tumor types with attention-based multiple instance learning: a development and validation study. medRxiv, https://www.medrxiv.org/content/10.1101/2023.03.08.23286975v1.full.pdf (2023).
Acknowledgements
This study was funded by the National Natural Science Foundation of China (92259101), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB38040100), the National Natural Science Foundation of China (62402473, 62271465), and the Suzhou Basic Research Program (SYG202338). The funder played no role in study design, data collection, analysis and interpretation of data, or the writing of this manuscript.
Author information
Authors and Affiliations
Contributions
H.-J.L. conceived the idea of this work and designed the methodology for this work. H.-J.L. and T.-Y.H. prepared and wrote the manuscript. H.-J.L. and J.-F.H. prepared the visualization of the manuscript. H.-J.L., W.-E.L., K.-X.Y., J.-Y.H., and R.-L.L. reviewed the manuscript. Y.-J.G., D.-W.S. X.-H.D. participated in the curation of the dataset. R.Y., S.-K.Z., and B.-F.N. supervised this work. B.-F.N. acquired funding for this work. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Luan, H., Hu, T., Hu, J. et al. Breast cancer homologous recombination deficiency prediction from pathological images with a sufficient and representative Transformer. npj Precis. Onc. 9, 160 (2025). https://doi.org/10.1038/s41698-025-00950-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-025-00950-5
