
Abstract
In the present study, a new type of Caspase-1 homolog is identified from Crassostrea gigas (defined as CgCas1-2D). It is composed of 2×DSRM-CASc domain and has closer evolutionary relationship with mammalian Caspase-1s. The mRNA expressions of CgCas1-2D increase significantly after Vibrio splendidus or LPS stimulation. Recombinant CgCas1-2D and its 2×DSRM and CASc domains all bind various PAMPs and bacteria. rCgCas1-2D shows the highest binding activity to human Caspase-1 substrate. Upon recognizing bacteria, CgCas1-2D co-localizes and interacts with CgGSDME, while it has no cleavage activity to CgGSDME. CgCas1-2D inhibits the histone methylation and acetylation levels and CgNF-κB/Rel nuclear translocation mediated by CgGSDME. In addition, CgCas1-2D suppresses the mRNA expression levels of cytokines mediated by GSDME-NF-κB/Rel axis. The results demonstrate that a new type of anti-inflammatory Caspase-1 identified from oyster upon recognizing various bacteria interacts with GSDME to inhibit the histone modification and NF-κB signaling to suppress the inflammation.
Similar content being viewed by others

Introduction
Caspase-1 is one of the main mediators of inflammatory Caspases and has become a correspondent with inflammation and pyroptosis1,2. Caspase-1, a cysteinyl aspartate specific proteinase, is composed of the Caspase activation and recruitment domain (CARD) and Cysteine aspartase catalytic (CASc) domain3. Caspase-1 is cleaved after the inflammasome is activated to recruit the adaptor apoptosis-associated speck-like protein containing a CARD (ASC) protein. The activated Caspase-1 promoted the maturation and secretion of inflammatory cytokines such as interleukin (IL)-1β and IL-18 and induced GSDM-mediated pyroptosis to support inflammation, immunity, and tissue repair4,5,6,7.
Caspase-1 is the first member of the Caspase family to be discovered and is a key protein for transmitting inflammatory signals and triggering pyroptosis8,9. Caspase-1 is composed of a classical CARD and a CASc domain. In mammals, inflammasome sensors recruited adaptor protein ASC to lead to the activation of Caspase-110. The activated Caspase-1 cleaved GSDMD to lead to the release of IL-1β and IL-182. In bony fish, Caspase-1 was found to directly cleave GSDME to induce pyroptosis in Cynoglossus semilaevis11. In invertebrates, the expansion of Caspase family members has been observed in various species and multiple Caspase-1 genes have been found in some species. However, only GSDME and PJVK were found in invertebrate species12,13, and GSDME-mediated pyroptosis was also observed in hydra14 and coral15. There are few studies on the relationship between Caspase-1 and GSDME in invertebrates. Recently, NLRC4 from Crassostrea hongkongensis can mediate pyroptosis in HEK293T cells through the Caspase-1-GSDME pathway16.
Some Caspases were found to function as pattern recognition receptors (PRRs) to sense the conserved bacterial molecules termed pathogen-associated molecular patterns (PAMPs). For example, human Caspase-4/Caspase-5 and mouse Caspase-11 were identified as intracellular PRRs to directly sense intracellular LPS during macrophage-mediated inflammatory responses. Direct recognition of intracellular LPS facilitated the rapid oligomerization of Caspase-11/4/5, which resulted in the secretion of IL-1β and IL-18 and pyroptosis17. In bony fish, Caspy2 in zebrafish directly bound LPS through the N-terminal pyrin domain (PYD) to lead to Caspy2 oligomerization18. In Hydra, CaspA could recognize LPS through its LPS-binding domain (LBD)14. In C. gigas, Caspase-3 bound LPS through its N-terminal domain, thereby inhibiting its proteolytic activity toward substrates19. These findings suggested that there may exist more complex functions of Caspases in invertebrates in the regulation of pyroptosis.
Histone modification and NF-κB signaling pathway play crucial roles in inflammation through inducing the expression of inflammatory cytokines20,21. In RAW264.7 cells (mouse mononuclear macrophage cell line), the histone acetylation and NF-κB signaling pathway inhibited the virus-induced inflammation22. In zebrafish, the histone modification (H3K9ac and H3K4me3) and NF-κB activation could protect the larvae from microbial infection by inducing IL-1β expression23. In invertebrates, the inflammation response and inflammatory cytokines have also been reported in several species, and they play an important role in anti-bacterial infection24,25,26. For example, in oysters, the H3K4me3 modification and NF-κB signaling pathway also regulated the mRNA expressions of IL17s in hemocytes27. These results indicated that histone modification and NF-κB signaling pathway are conserved in regulating inflammatory response.
Invertebrates mainly rely on innate immunity to resist pathogen invasion28. Caspase plays an important role in the innate immune response3. There is an expansion phenomenon of Caspase family genes annotated in genomes of species in invertebrates, especially in molluscs29. Thirty-five potential Caspase homologous genes were annotated in C. gigas genome30. In the present study, a new type of Caspase-1 with two tandem DSRM domains was screened and identified from C. gigas (defined as CgCas1-2D) with the objectives of studying the recognition function of CgCas1-2D to various PAMPs and bacteria, revealing the association and function of CgCas1-2D to CgGSDME, and clarifying the mechanism of Cas1-2D-GSDME signals for inflammation, which would provide new insights for understanding the roles of Caspase for GSDME-dependent pyroptosis and inflammation.
Results
CgCas1-2D with two DSRM domains was identified from C. gigas
A Caspase-1 homolog with two N-terminal tandem DSRM domains and a C-terminal CASc domain was identified from C. gigas, defined as CgCas1-2D. The full-length cDNA sequence of CgCas1-2D was of 1653 bp. The open reading frame (ORF) was of 1278 bp encoding a polypeptide of 425 amino acid residues, with a predicted molecular weight of 48.3 kDa. In the resultant phylogenetic tree, CgCas1-2D from C. gigas was grouped within the same clade as the Caspase-1 orthologs of Homo sapiens, Mus musculus, and Xenopus tropicalis. While it exhibited distant evolutionary relationship with the other predicted Caspase-1s from C. gigas. This clustering pattern strongly suggested a close evolutionary affinity between CgCas1-2D and vertebrate Caspase-1s (Fig. 1A). All Caspase-1s contained a conserved CASc domain, while only CgCas1-2D contained two tandem DSRM domains (Fig. 1B). Multiple sequence comparison between CgCas1-2D and Caspase-1s from other species showed that the active sites of the pentapeptide in the CASc domains were relatively conserved, and its active sites were “QSCRG” (Supplementary Fig. S1).

A The evolutionary analysis of CgCas1-2D and Caspase-1s from other species. The clustered genes were labeled with the same color, and CgCas1-2D was labeled in red font. The numbers at the forks represented the bootstrap. B The structural domains of CgCas1-2D and Caspase-1s from other species. CgCas1-2D was labeled in red font.
The mRNA and protein expressions of CgCas1-2D were induced after immune stimuli
The distribution of CgCas1-2D mRNA in different tissues was investigated by Reverse transcription quantitative PCR (RT-qPCR). CgCas1-2D mRNA was constitutively expressed in all the tested tissues. The transcripts of CgCas1-2D mRNA were relatively higher expressed in hepatopancreas, haemolymph and mantle, which were 19.16-fold (p = 0.0029), 9.63-fold (p = 0.043) and 9.36-fold (p = 0.0018) of that in adductor muscle (Fig. 2A). The temporal expressions of CgCas1-2D mRNA in hemocytes after LPS or V. splendidus stimulation were investigated by RT-qPCR. After LPS stimulation, the mRNA expressions of CgCas1-2D increased significantly at 3 h and 48 h, which were 315.49-fold (p = 0.011) and 36.94-fold (p = 0.0115) of that in the seawater group (Fig. 2B). The mRNA expressions of CgCas1-2D in hemocytes increased significantly at 3, 6 and 48 h after V. splendidus stimulation, which were 272.72-fold (p = 1.35e-03), 3.98-fold (p = 0.012) and 70.44-fold (p = 0.045) of that in the seawater group (Fig. 2C). The expression of CgCas1-2D protein was analyzed by Western blotting at different time points after V. splendidus stimulation. The protein expressions of CgCas1-2D were increased at 3 h after V. splendidus stimulation (Fig. 2D). After V. splendidus stimulation, the expression levels of CgCas1-2D protein exhibited a dynamic change trend of first increasing, then decreasing, and finally increasing again (Fig. 2E). The positive signals of CgCas1-2D were mainly located in hemocytes cytoplasm (Fig. 2F).

A The mRNA transcripts of CgCas1-2D in different tissues. B The mRNA expressions of CgCas1-2D in hemocytes after LPS stimulation. C The mRNA expressions of CgCas1-2D in hemocytes after V. splendidus stimulation. CgEF was used as the internal control. D The protein expression levels of CgCas1-2D at different time points after V. splendidus stimulation. E Statistical analysis of CgCas1-2D protein expression levels at different time points after V. splendidus stimulation. Vertical bars show the mean ± S.D. (n = 3). Asterisks indicate significant differences (*p < 0.05). The different letters indicated significant differences among different groups (p < 0.05, Tukey). F The subcellular localization of CgCas1-2D in hemocytes. The haemocyte nuclei stained with DAPI were in blue. Anti-CgCas1-2D conjugated to Alexa-fluor 647 was in red. The scale bar was 5 μm.
The intracellular CgCas1-2D had the ability to bind various PAMPs and bacteria
Recombinant CgCas1-2D (rCgCas1-2D), the 2×DSRM of CgCas1-2D (rCgCas1-2D-p16) and the CASc domain (rCgCas1-2D-p28) were expressed and purified using the Ni-NTA affinity chromatography and identified by SDS-poly-acrylamide gel electrophoresis (SDS-PAGE). There were evident bands observed, which was consistent with the predicted molecular weights of rCgCas1-2D-p16, rCgCas1-2D-p28 and rCgCas1-2D, respectively (Fig. 3A–C). rCgCas1-2D was verified by Native PAGE Gel and there were three bands, which were identical to the prediction of molecular weights of rCgCas1-2D, rCgCas1-2D-p28 and rCgCas1-2D-p16, respectively (Fig. 3D). The specificity of anti-CgCas1-2D was examined with the haemocyte lysates by Western blotting. There were two obvious bands revealed, which were identical to the prediction of molecular weights of CgCas1-2D-p28 and CgCas1-2D, respectively (Fig. 3E). The protein expression levels of CgCas1-2D and its activated fragments increased significantly after 3 h of V. splendidus stimulation (Fig. 3F, G). After incubation of rCgCas1-2D with purchased human Caspase-1 substrate (Ac-YVAD-pNA), Caspase-3 substrate (Ac-DEVD-pNA), Caspase-6 substrate (Ac-VEID-pNA), Caspase-8 substrate (Ac-IETD-pNA), Caspase-9 substrate (Ac-LEHD-pNA), the bind activity of rCgCas1-2D to human Caspase-1 substrate (Ac-YVAD-pNA) was the highest, which was 1.2-fold of that in Caspase-8 substrate (Ac-IETD-pNA) group (Fig. 3J).

A–C SDS-PAGE analysis of rCgCas1-2D and its mutant. Lane M: standard protein molecular weight marker; Lane 1: the whole cell lysates of positive transformants before induction with IPTG; Lane 2: the expressed protein after induction with IPTG; Lane 3: purified rCgCas1-2D-p16 (A), rCgCas1-2D-p28 (B), rCgCas1-2D (C). D rCgCas1-2D verified by Native PAGE Gel. Band 1: the expressed rCgCas1-2D; Band 2: the activated fragment rCgCas1-2D-p28; Band 3: the activated fragment rCgCas1-2D-p16. E The specificity of anti-CgCas1-2D in hemocytes by Western blotting. Band 1: native CgCas1-2D in hemocytes; Band 2: the cleavage fragment of CgCas1-2D in hemocytes. F The protein expression of CgCas1-2D and its cleavage fragments after V. splendidus stimulation for 3 h. G Statistical analysis of CgCas1-2D and its cleavage fragment protein expression levels. Vertical bars show the mean ± S.D. (n = 3). The PAMP binding activity of rCgCas1-2D (H), rCgCas1-2D-p28 (I) and rCgCas1-2D-p16 (J) to LPS, GLU and MAN, respectively. “P/N” means that P (sample) − B (blank) / N (negative) − B (blank). Vertical bars show the mean ± S.D. (n = 3). K The proteolytic specificity of rCgCas1-2D to Caspase substrates. Vertical bars show the mean ± S.D. (n = 3). The different letters indicated significant differences among different groups (p < 0.05, Tukey).
The binding activities of rCgCas1-2D, rCgCas1-2D-p28 and rCgCas1-2D-p16 to LPS, Mannose (MAN), and Glucose (GLU) were investigated by Enzyme-linked immunosorbent assay (ELISA) (Fig. 3G–I). When the concentrations of rCgCas1-2D, rCgCas1-2D-p28 and rCgCas1-2D-p16 were 12.9 μmol/L, 3.1 μmol/L and 2.3 μmol/L, respectively, the P/N values in LPS, GLU and MAN groups were all more than 2.1, indicating that there were binding activities of rCgCas1-2D and its mutants to PAMPs. The P/N values of rCgCas1-2D-p16 group for LPS, GLU and MAN at the lowest concentration were higher than those of rCgCas1-2D and rCgCas1-2D-p28 groups, indicating that rCgCas1-2D-p16 had better binding activity with PAMPs. The binding activities of rCgCas1-2D-p16, rCgCas1-2D-p28 and rCgCas1-2D to various bacteria were analyzed by Western blotting. After rCgCas1-2D, rCgCas1-2D-p28 and rCgCas1-2D-p16 were incubated with bacteria, respectively, the specific bands were observed in Escherichia coli, V. splendidus, Vibrio alginolyticus, Aeromonas hydrophila, Staphylococcus aureus, and Micrococcus luteus groups. Compared with rCgCas1-2D group, the rCgCas1-2D-p28 had no specific band in S. aureus and V. splendidus (Fig. 4A–C). There was no band detected in the rTrx group. Meanwhile, there was no band observed in the bacterial protein group (Fig. 4D). The co-localizations of CgCas1-2D and fluorescein isothiocyanate (FITC)-labelled bacteria (V. splendidus and V. alginolyticus) in hemocytes were observed by immunocytochemistry assay. The positive red signals of CgCas1-2D were co-localized with the green signals of FITC-labelled V. splendidus and V. alginolyticus. In the negative serum group, there were no positive co-localization signals of CgCas1-2D with V. splendidus and V. alginolyticus, while only FITC-labelled bacteria were observed in hemocytes. Statistical analysis of co-localization fluorescence intensity of CgCas1-2D with V. splendidus and V. alginolyticus were shown, respectively (Fig. 4D–G).

Bacterial binding activity of rCgCas1-2D (A), rCgCas1-2D-p28 (B), and rCgCas1-2D-p16 (C). The binding spectrum of rCgCas1-2D and its mutants to bacteria were detected by Western blotting and rTrx was employed as control. D Bacterium without any treatment was used as loading control. E The co-localization of CgCas1-2D and V. splendidus in hemocytes. The scale bar was 2 μm. F Statistical analysis of co-localization fluorescence intensity of CgCas1-2D and V. splendidus. G The co-localization of CgCas1-2D and V. alginolyticus in hemocytes. The scale bar was 2 μm. H Statistical analysis of co-localization fluorescence intensity of CgCas1-2D and V. alginolyticus. The negative serum was used as control.
CgCas1-2D interacted with CgGSDME while had no cleavage activity to CgGSDME
CgCas1-2D was represented as a pink cartoon model, CgGSDME was shown as a purple cartoon model, and their binding sites were shown as the corresponding-colored stick structure. The binding site was visualized as a representation of the associated protein in Pymol. The predicted binding free energy of CgCas1-2D to CgGSDME was −32.749 kcal/mol. When the binding energy of protein docking is less than −15 kcal/mol, it is considered that there is a certain binding tendency. There were multiple residues between CgCas1-2D and CgGSDME forming hydrogen bonds or salt bridges, such as the hydrogen bond formed by ARG 350 of CgCas1-2D and GLU 334 of CgGSDME (Fig. 5A). Immunocytochemistry was used to observe the co-localization of CgCas1-2D and CgGSDME in hemocytes at 3 h after V. splendidus stimulation. The anti-CgCas1-2D conjugated to Alexa Fluor 647 was observed in red and anti-CgGSDME conjugated to Alexa-fluor 488 was observed in green. The positive red signals of CgCas1-2D were co-localized with the green signals of CgGSDME after V. splendidus stimulation (Fig. 5B, C). One hundred cells were used for the statistical analyses and there were three replications in each group. The haemocyte number containing co-localization signals increased significantly after V. splendidus stimulation, which was 1.70-fold of that in the seawater group (p = 0.039) (Fig. 5D). However, in the negative serum and CgGSDME co-localization group, there was no positive signal of negative serum in the seawater or V. splendidus group, while only the positive signals of CgGSDME were observed (Supplementary Fig. S2). Co-IP assay was used to confirm the interaction between CgCas1-2D and CgGSDME. There was a band of CgCas1-2D and CgGSDME after the hemocyte lysates were coimmunoprecipitated by CgCas1-2D and CgGSDME antibodies, respectively. Compared with the seawater group, the interaction between CgCas1-2D and CgGSDME was enhanced after V. splendidus stimulation (Fig. 5E).

A The molecular docking of CgCas1-2D and CgGSDME. B The co-localization of CgCas1-2D and CgGSDME in hemocytes by using immunocytochemistry. Seawater was used as the control. The scale bar was 5 μm. C Statistical analysis of co-localization fluorescence intensity of CgCas1-2D and CgGSDME. D The statistical analysis of hemocyte number containing co-localization signals of CgCas1-2D and CgGSDME. One hundred hemocytes were used for the statistical analyses and there were three replications in each group. Vertical bars show the mean ± S.D. (n = 3). The different letters indicated significant differences among different groups (p < 0.05, Tukey). E The Co-IP assay of CgCas1-2D and CgGSDME in hemocytes.
The protein expression of the active fragment of rCgGSDME (rCgGSDME-N) was detected after incubation of rCgGSDME and rCgCas1-2D in vitro, the his-tagged antibody was used as primary antibody. The control groups were rCgCas1-2D and rCgGSDME incubated with TBS, respectively. The specific bands of rCgGSDME and rCgGSDME-N were all observed in rCgGSDME incubated with rCgCas1-2D and TBS group, respectively (Fig. 6A). The Image J analysis and statistical results showed that there was no significant change in the rCgGSDME-N band after the co-incubation of rCgCas1-2D and rCgGSDME (Fig. 6B). Transfection assay was performed in HEK293T cell lines to detect whether CgCas1-2D-CASc cleaved CgGSDME. The cell lysates after transfection analyzed by Western blotting showed that there was no significant change in the band intensity of rCgGSDME-N in the mCherry-tagged CgCas1-2D-CASc and EGFP-tagged CgGSDME co-transfection group and the EGFP-tagged CgGSDME transfection group (Fig. 6C). The transfected plasmids of mCherry-tagged CgCas1-2D-CASc were observed in red and that of EGFP-tagged CgGSDME was observed in green (Fig. 6D).

A The cleavage of rCgGSDME after incubation with rCgCas1-2D by Wstern blotting with Anti-his antibody. B The statistical analysis of protein expression of rCgGSDME-N/rCgGSDME using Image J. Vertical bars show the mean ± S.D. (n = 3). C The cleavage of CgGSDME after co-transfection with mCherry-tagged CgCas1-2D-CASc and EGFP-tagged CgGSDME in HEK293T cells by Western blotting with anti-EGFP-tag. D The transfection of mCherry-tagged CgCas1-2D-CASc and EGFP-tagged CgGSDME in HEK293T cells. The scale bar was 50 μm.
CgCas1-2D reduced the histone H3 modification and CgRel nuclear translocation after V. splendidus stimulation
The mRNA expression of CgCas1-2D was investigated in CgCas1-2D-RNAi oysters to confirm the efficiency of RNA interference (RNAi). The mRNA expression of CgCas1-2D decreased significantly in CgCas1-2D-RNAi oysters, which was 0.20-fold (p = 0.017) of that in NC-RNAi oysters (Fig. 7A). H3 antibody was used for quantitative analysis of the sample to ensure that each group contained the equivalent amounts of histones. The methylation and acetylation of histone H3 in CgCas1-2D-RNAi oysters were analyzed by Western blotting. The band intensities of the methylation of H3K4 and the acetylation of H3K9 and H3K27 were all induced in hemocytes of CgCas1-2D-RNAi oysters after V. splendidus stimulation (Fig. 7B).

A The mRNA expression of CgCas1-2D in hemocytes of CgCas1-2D-RNAi oysters after V. splendidus stimulation. The NC-RNAi oysters were used as control. Vertical bars show the mean ± S.D. (n = 3). The different letters indicated significant differences among different groups (p < 0.05, Tukey). B The levels of CgH3, CgH3K4me1, CgH3K4me2, CgH3K4me3, CgH3K9ac and CgH3K27ac in hemocytes of CgCas1-2D-RNAi oysters after V. splendidus stimulation. C Subcellular localization of CgRel in hemocytes of CgCas1-2D-RNAi oysters after V. splendidus stimulation. Anti-CgRel conjugated to Alexa-fluor 488 was in green. The hemocyte nuclei stained with DAPI were in blue. The scale bar was 10 μm. D The hemocyte number containing co-localization signals of CgRel and nucleus. One hundred hemocytes were used for the statistical analyses and there were three replications in each group. Vertical bars show the mean ± S.D. (n = 3). The different letters indicated significant differences among different groups (p < 0.05, Tukey). Statistical analysis of co-localization fluorescence intensity of CgRel and nucleus in blank (E), EGFP-RNAi (F) and CgGSDME-RNAi (G) oysters, respectively. The NC-RNAi oysters were used as control.
The immunofluorescence assay was conducted to determine the subcellular localization of CgRel in hemocytes of CgCas1-2D-RNAi oysters after V. splendidus stimulation. Anti-CgRel conjugated to Alexa Fluor 488 was in green and the nuclei stained by 4’,6-diamidino-2-phenylindole (DAPI) were in blue. In blank oysters, the green signals of CgRel were distributed in the haemocyte cytoplasm. In CgCas1-2D-RNAi oysters, the positive signals of CgRel in the nucleus became stronger, compared with that of NC-RNAi oysters at 3 h after V. splendidus stimulation (Fig. 7C). The haemocyte number containing co-localization signals of CgRel and nucleus increased significantly in CgCas1-2D-RNAi oysters, which was 1.6-fold (p = 0.06) of that in NC-RNAi oysters (Fig. 7D). Statistical analysis of co-localization fluorescence intensity of CgRel and nucleus in each group were shown, respectively (Fig. 7E–G).
CgGSDME promoted the histone H3 modification and CgRel nuclear translocation after V. splendidus stimulation
The mRNA expressions of CgGSDME decreased significantly in hemocytes of CgGSDME-RNAi oysters, which was 0.34-fold (p = 3.7e−04) of that in EGFP-RNAi oysters (Fig. 8A). The methylation and acetylation levels of histone H3 in CgGSDME-RNAi oysters were analyzed by Western blotting. The band intensities of the methylation of H3K4 and the acetylation of H3K9 and H3K27 were all reduced in hemocytes of CgGSDME-RNAi oysters (Fig. 8B). The immunofluorescence assay was conducted to determine the subcellular localization of CgRel in hemocytes of CgGSDME-RNAi oysters after V. splendidus stimulation. Anti-CgRel conjugated to Alexa Fluor 488 was in green signals and the nuclei stained by DAPI were in blue. In blank group, the green signals of CgRel were distributed in the haemocyte cytoplasm. In CgGSDME-RNAi oysters, the positive signals of CgRel in the nucleus became weaker, compared with that of EGFP-RNAi oysters at 3 h after V. splendidus stimulation (Fig. 8C). The haemocyte number containing co-localization signals of CgRel and nucleus decreased significantly in CgGSDME-RNAi oysters, which was 0.42-fold (p = 3.07e−04) of that in EGFP-RNAi oysters (Fig. 8D). Statistical analysis of co-localization fluorescence intensity of CgRel and nucleus in each group were shown, respectively (Fig. 8E–G).

A The mRNA expressions of CgGSDME in hemocytes of CgGSDME-RNAi oysters after V. splendidus stimulation. The EGFP-RNAi oysters were used as control. Vertical bars show the mean ± S.D. (n = 3). The different letters indicated significant differences among different groups (p < 0.05, Tukey). B The levels of CgH3, CgH3K4me1, CgH3K4me2, CgH3K4me3, CgH3K9ac and CgH3K27ac in hemocytes of CgGSDME-RNAi oysters after V. splendidus stimulation. The EGFP-RNAi oysters were used as control. C Subcellular localization of CgRel in hemocytes of CgGSDME-RNAi oysters after V. splendidus stimulation. The scale bar was 10 μm. D The hemocyte number containing co-localization signals of CgRel and nucleus. One hundred hemocytes were used for the statistical analyses and there were three replications in each group. Vertical bars show the mean ± S.D. (n = 3). The different letters indicated significant differences among different groups (p < 0.05, Tukey). Statistical analysis of co-localization fluorescence intensity of CgRel and nucleus in blank (E), EGFP-RNAi (F) and CgGSDME-RNAi (G) oysters, respectively.
CgCas1-2D reduced the mRNA expression levels of inflammatory cytokines mediated by GSDME-NF-κB/Rel axis and GSDME-histone H3 modification
The mRNA expression levels of CgGSDME, high mobility group box 1 protein (CgHMGB1) and tumor necrosis factor-2 (CgTNF-2) in CgCas1-2D-RNAi oysters significantly increased, which were 1.57-fold (p = 0.017), 2.63-fold (p = 0.002) and 2.0-fold (p = 0.033) of that in NC-RNAi oysters, respectively (Fig. 9A–C). The mRNA expressions of CgHMGB1 and CgTNF-2 in CgGSDME-RNAi oysters were significantly decreased, which were 0.42-fold (p = 8.37e−05) and 0.44-fold (p = 0.0039) of that in EGFP-RNAi oysters, respectively (Fig. 9D, E). The mRNA expression of CgRel in CgRel-RNAi oysters was 0.07-fold (p = 0.0016) of that in NC-RNAi oysters, and the interference efficiency was 93% (Fig. 9F). The mRNA expressions of CgHMGB1 and CgTNF-2 in CgRel-RNAi oysters were significantly decreased, which were 0.45-fold (p = 0.0017) and 0.71-fold (p = 0.006) of that in NC-RNAi oysters, respectively (Fig. 9G, H). Compared with that in DMSO group, the H3 methylation level in MTA group was significantly reduced. The expression levels of H3K4me1, H3K4me2 and H3K4me3 in MTA group were 0.78-fold (p = 6.19e−04), 0.81-fold (p = 0.01) and 0.54-fold (p = 3.97e−04) of that in DMSO group, respectively (Fig. 10A–D). The mRNA expression levels of CgHMGB1 and CgTNF-2 in MTA group were significantly reduced, which were 0.67-fold (p = 0.011) and 0.66-fold (p = 0.005) of that in DMSO group, respectively (Fig. 10E, F). Compared with that of DMSO group, the histone H3 acetylation level in the HY-100734 group was significantly reduced. The expression levels of H3K9ac and H3K27ac in HY-100734 group were 0.70-fold (p = 1.57e−04) and 0.80-fold (p = 0.008) of that in DMSO group, respectively (Fig. 10G–I). The mRNA expression levels of CgHMGB1 and CgTNF-2 in HY-100734 group were significantly reduced, which were 0.61-fold (p = 0.0057) and 0.65-fold (p = 0.0053) of that in DMSO group, respectively (Fig. 10J, K).

The mRNA expressions of CgGSDME (A), CgHMGB1 (B) and CgTNF-2 (C) in hemocytes of CgCas1-2D-RNAi oysters after V. splendidus stimulation. The NC-RNAi oysters were used as control. D, E The mRNA expressions of CgHMGB1 (D) and CgTNF-2 (E) in hemocytes of CgGSDME-RNAi oysters after V. splendidus stimulation. The EGFP-RNAi oysters were used as control. The mRNA expressions of CgRel (F), CgHMGB1 (G) and CgTNF-2 (H) in hemocytes of CgRel-RNAi oysters after V. splendidus stimulation. The NC-RNAi oysters were used as control. Vertical bars show the mean ± S.D. (n = 3). The different letters indicated significant differences among different groups (p < 0.05, Tukey).

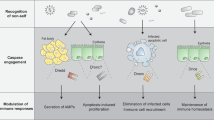
A The levels of CgH3K4me1, CgH3K4me2 and CgH3K4me3 in hemocytes of MTA-treated oysters after V. splendidus stimulation. Statistical analysis of the levels of CgH3K4me1 (B), CgH3K4me2 (C) and CgH3K4me3 (D) in MTA-treated group after V. splendidus stimulation, respectively. The mRNA expressions of CgHMGB1 (E) and CgTNF-2 (F) in hemocytes of MTA-treated oysters after V. splendidus stimulation. G The levels of CgH3K9ac and CgH3K27ac in hemocytes of HY-100734-treated oysters after V. splendidus stimulation. Statistical analysis of CgH3K9ac (H) and CgH3K27ac (I) levels in HY-100734-treated group after V. splendidus stimulation, respectively. The mRNA expressions level of CgHMGB1 (J) and CgTNF-2 (K) in hemocytes of HY-100734-treated oysters after V. splendidus stimulation. DMSO-treated oysters were used as control. Vertical bars show the mean ± S.D. (n = 3). Asterisks indicate significant differences (*p < 0.05, **p < 0.01, ***p < 0.001). L CgCas1-2D upon recognizing bacteria inhibited GSDME-mediated H3 modification and GSDME-NF-κB axis to suppress inflammatory responses. Figdraw was used to assist in drawing patterns.
Discussion
The mammalian inflammatory Caspases play important roles in regulating programmed cell death and inflammatory response31,32. Caspase-1/4/5/11 can cleave GSDMD to produce GSDMD-N to induce pyroptosis and inflammatory response33. The cleavage of GSDME mediated by Caspase-3 to induce pyroptosis was found in corals, which was firstly proposed the existence of pyroptosis in invertebrates15. Subsequently, the GSDME-dependent pyroptosis was reported in other invertebrates, such as oyster, abalone, and hydra12,14,34. In the present study, a novel of Caspase-1 homolog with two tandem DSRM domains (defined as CgCas1-2D) was identified from C. gigas. Upon binding PAMPs and bacteria, CgCas1-2D interacted with CgGSDME to inhibit its cleavage, thereby suppressing the histone modification- and NF-κB-mediated the mRNA expressions of inflammatory cytokines.
All inflammatory Caspases in vertebrates, including Caspase-1/4/5/11/12, contain CARD and CASc domains35. In contrast, only in cyprinid fish, Caspase-1 homolog was composed of PYD-CASc domains, instead of CARD-CASc domains36. Caspase-1 homologs have been identified in various invertebrates37,38,39. Caspase-1 homolog only containing CASc domain was identified in C. gigas40. In the present study, Caspase-1s from vertebrates and invertebrates were selected to construct the evolutionary tree. CgCas1-2D had a closer relationship with Caspase-1s from vertebrates than those from invertebrates. Caspase-1 homologs from invertebrates only had one or two CASc domains. However, CgCas1-2D was composed of 2×DSRM-CASc domains. DSRM was usually used as a sensor for innate immunity by recognizing intracellular dsRNA41,42. Caspase-1 homolog containing 2×DSRM only existed in molluscs, and it might function as intracellular recognition molecules.
Inflammatory Caspases have been found to be mainly involved in pyroptosis and inflammatory response3,43,44. Caspase-1 was activated after being recruited to various inflammasomes by the adaptor ASC protein through its CARD domain45,46. There is increasing evidence that Caspases can act as intracellular PRRs to recognize LPS through their CARD domains, including human Caspase-4/5 and mouse homologous Caspase-1147,48. Caspase-1 in humans and mice relied on a central scaffold protein (AIM2, NAIPs, Pyrin or NLRP1B) to sense bacteria49. CgCas1-2D and its 2×DSRM and CASc domain could all bind to various PAMPs (MAN, GLU, and LPS) and bacteria (E. coli, A. hydrophila, V. splendidus, V. alginolyticus, S. aureus, and M. luteus) in vitro. Among which, 2×DSRM domain had relatively higher binding activity to PAMPs. In mammals, Caspase-4/5/11 were also identified as the intracellular noncanonical inflammasomes for their binding activity to LPS50. In the present study, CgCas1-2D was co-localized with V. splendidus and V. alginolyticus in hemocyte cytoplasm. These results demonstrated that CgCas1-2D could function as an intracellular recognition molecule to sense various PAMPs and bacteria in hemocytes. The mRNA transcripts of CgCas1-2D were detected in all the tested tissues with relatively higher expression in haemolymph, mantle, and hepatopancreas. Among them, hemocytes are the main immune component of most aquatic invertebrates and play a key role in mediating innate immunity51. The mRNA expressions of CgCas1-2D in hemocytes increased significantly after LPS or V. splendidus stimulation, indicating that CgCas1-2D played critical roles in oyster anti-bacterial immunity. The mRNA and protein expression levels of CgCas1-2D increased significantly at 3 h after V. splendidus stimulation, indicating that CgCas1-2D might mainly function at 3 h of V. splendidus stimulation. When V. splendidus stimulated for 12 and 24 h, there was a robust inflammatory response. Although the mRNA expression level of CgCas1-2D increased, its protein expression level decreased, suggesting that it may play a negative regulatory role. There was no significant change about the activated fragments of CgCas1-2D after V. splendidus stimulation, which suggested that CgCas1-2D might mainly function as a full-length protein in hemocytes.
In 2015, GSDMD was identified as the substrate of all inflammatory Caspases and the true executor of pyroptosis33,52. Since then, the members of GSDM family have been identified, and the relationship between the inflammatory Caspases and GSDMs has been further studied. Activated Caspase-1/4/5/11 can directly cleave GSDMD to produce GSDMD-N53. Caspase-3 directly targeted GSDME, leading to its cleavage54. In bony fish, Caspase-1/3/7 could cleave GSDME to induce pyroptosis11. In corals, OfCaspase-3 cleaved OfGSDME to induce pyroptosis. In hydra, HyCaspA cleaved apoptotic caspase HyCARD2 to activate HyGSDME. CgCaspase-3 promoted the translocation of CgGSDME to the haemocyte membrane in oyster12,14,15. In the present study, the full length CgCas1-2D could interact and co-localize with CgGSDME, but it could not lead to the cleavage of CgGSDME, suggested that the interaction of CgCas1-2D and CgGSDME might competitively inhibit the interaction of CgGSDME with other Caspases.
The cleavage of GSDMs can induce the inflammatory response, and histone modification and NF-κB signaling pathway play an indispensable role in mediating inflammatory responses55,56. Histone modification mainly regulated chromatin state and gene transcription57. Moreover, inflammatory response can also be regulated by histone methylation and acetylation in macrophages58. Histone modification cooperated with NF-κB signaling pathway to promote the transcription of target genes59,60,61. The NF-κB signaling pathway was demonstrated to regulate the Caspase-1-GSDMD-mediated or Caspase-3-GSDME-mediated pyroptosis and inflammatory response62,63,64,65,66. Also, in the study of nonalcoholic steatohepatitis, the active fragment GSDMD-N could exacerbate NF-κB activation, thereby promoting the inflammatory response67. In our previous study, histone modification and NF-κB signaling pathway could both regulate the expressions of various inflammatory cytokines27,68,69. In the present study, CgCas1-2D could inhibit the H3 methylation and the acetylation levels as well as NF-κB/Rel nuclear translocation, while CgGSDME could induce their levels in hemocytes after V. splendidus stimulation. These results suggested that CgCas1-2D upon binding to various PAMPs and bacteria, interacted with CgGSDME to inhibit H3 modification and NF-κB signaling pathway. CgCas1-2D might compete for the binding sites of CgGSDME with other Caspases, thereby inhibiting GSDME-histone modification and GSDME-NF-κB axis to lead to immune suppression. In mammals, there was evidence that Caspase-3 could directly cleave the p65 subunit of NF-κB to regulate apoptosis70,71, and whether CgCas1-2D could directly interact with NF-κB need to be further confirmed in future.
The production of various inflammatory cytokines was induced by H3 modification and NF-κB signaling pathway exerts final inflammatory effects21,22,72,73. In oysters, CgHMGB1 and CgTNF-2 were identified from C. gigas and CgHMGB174 and CgGSDME (data unpublished) had been demonstrated to induce the inflammatory response and display obvious antibacterial activity. In the present study, the mRNA expressions of CgGSDME, CgHMGB1 and CgTNF-2 were significantly increased in CgCas1-2D-RNAi oysters. The mRNA expressions of CgHMGB1 and CgTNF-2 were significantly decreased in CgGSDME-RNAi oysters and CgRel-RNAi oysters, respectively. The methylation of H3K4 can regulate the expression of a variety of cytokines and TLR4 expression to induce inflammatory response75,76. The acetyltransferase p300 can mediate the acetylation of H3K9 and H3K2777, thereby mediating the expression of inflammatory cytokines78,79. In the present study, the mRNA expression levels of CgHMGB1 and CgTNF-2 were significantly decreased after inhibiting the methylation and acetylation levels of histone H3, respectively. These results proved that CgCas1-2D upon binding to various PAMPs and bacteria interacted with CgGSDME to inhibit histone H3 modification and NF-κB signaling pathway, which eventually suppressed the mRNA expressions of CgGSDME, CgHMGB1 and CgTNF-2 to reduce inflammatory response.
In conclusion, CgCas1-2D could act as an intracellular PRR to recognize various intracellular PAMPs and bacteria. Then, the activated CgCas1-2D interacted with CgGSDME to inhibit CgGSDME cleavage, which eventually inhibited the histone H3 modification and NF-κB signaling pathway to suppress the mRNA expressions of inflammatory cytokines (Fig. 10L). These results indicated that there was a Caspase-mediated anti-inflammatory mechanism in molluscs and the anti-inflammatory Cas1-2D directly interacted with GSDME to inhibit the GSDME-dependent pyroptosis and suppressed the histone H3 modification and NF-κB signaling pathway to inhibit inflammatory responses. The findings provided new insights for studying about mammalian Caspases and also offered a new strategy for the treatment of diseases dependent on pyroptosis and inflammation.
Materials and methods
Animals and strain
The Pacific oyster C. gigas (two-year-old, average shell length of 13.0 ± 2 cm) used in this study were all healthy and collected from an aquaculture farm in Dalian, Liaoning province, China. The oysters were cultured in the filtered and aerated seawater at 18–22 °C for a week prior to the experiments. The experiments were randomly grouped. Four to six weeks old specific pathogen free (SPF) female KM mice were obtained from Dalian Medical University for the polyclonal antibody preparation. The mice were placed in standard experimental animal cages. All the strains used in the experiment were pure strains preserved in the laboratory. All the experiments were performed following the animal ethics guidelines approved by the Ethics Committee of Dalian Ocean University.
Sequence and structure analysis of CgCas1-2D
The nucleotide sequence of CgCas1-2D was obtained from the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/). The structural domain of CgCas1-2D was predicted by the Simple Modular Architecture Research Tool (http://smart.embl-heidelberg.de/index2.cgi). The schematic diagram of each protein domain was drawn using IBS1.0.3 software. The amino acid sequences of Caspase-1 homologs from species were obtained from NCBI database, which were then used for constructing the evolutionary tree using MEGA6 software. The evolutionary tree was beautified using Interactive tree of life (https://itol.embl.de/).
The temporal expression and tissue distribution of CgCas1-2D
The 180 oysters were divided into three groups for receiving an injection with 100 μL sterilized seawater, 100 μL V. splendidus (1 × 107 CFU mL−1 in seawater) and 100 μL LPS (0.5 mg mL−1 in seawater) from E. coli (O222:B44, Sigma Aldrich, USA), respectively. The hemocytes were collected from each group at 0, 3, 6, 12, 24, 48 and 72 h after immune stimulation. The hemocytes from three oysters were pooled together as one sample and there were three replicates for each time point. The tissues including hemocytes, gills, mantle, hepatopancreas, gonads, labial palps and adductor muscle were from untreated oysters. All the samples were harvested by centrifugation at 800 × g, 4 °C for 10 min, and then stored at -80 °C for subsequent RNA extraction to examine the mRNA expression of CgCas1-2D. Twenty-four oysters received an injection with 100 μL V. splendidus (1 × 107 CFU mL−1 in seawater), and hemocytes were collected at 0, 3, 6, 12, 24, 48 and 72 h after stimulation. The protein expressions of CgCas1-2D in hemocytes were analyzed by Western blotting.
RNA extraction and cDNA synthesis
Total RNAs were extracted from oyster tissues with Trizol reagent (Thermo Fisher Scientific, USA)69. The concentration of extracted RNAs was examined by Nanodrop 2000. The cDNA synthesis reaction was performed according to the protocol of manufacturer (TransGen Biotech, China). The cDNA mix liquor was diluted to 1:20 and stored at −80 °C for subsequent experiments.
Recombinant expression, purification and antiserum preparation of CgCas1-2D
The sequences of CgCas1-2D, CgCas1-2D-p16 and CgCas1-2D-p28 were amplified from haemocyte samples using the primers (CgCas1-2D-Ex-F/R, CgCas1-2D-p16-Ex-F/R, and CgCas1-2D-p28-Ex-F/R) (Supplementary Data 1). The PCR products of CgCas1-2D and its mutants were inserted into the pET-30a vector, which were transformed into E. coli transetta (DE3) (TransGen, China). Then the rCgCas1-2D and its mutants were purified by affinity chromatography using His-Bind resin (Beyotime, China) following the manufacturer’s instructions. The purified rCgCas1-2D and its mutants were identified by 12.5% SDS-PAGE.
Antiserum preparation of CgCas1-2D was referred to previous report by using rCgCas1-2D80. Blood samples from treated mouse were collected after the fourth booster and then placed overnight at 4 °C to obtain the antiserum. The haemocyte samples were then used to confirm the specificity of CgCas1-2D antibody by using Western blotting. The antisera of CgGSDME and CgRel were prepared in our lab12,68.
The PAMP binding activity of rCgCas1-2D and its mutants
ELISA was used to detect the PAMP binding activity of rCgCas1-2D and its mutants. PAMPs including LPS (Sigma-Aldrich, USA), MAN (Sigma-Aldrich, USA), GLU (Sangon Biotech, China) were dissolved in Na2CO3-NaHCO3 buffer solution (35 mM NaHCO3,15 mM Na2CO3, PH 9.6) and dissolved to coat 96-well plate (Corning, USA). One hundred microliters of rCgCas1-2D and its mutants with gradient dilution, were added to the wells, respectively. The TBS and tag protein (rTrx) were used as blank and negative control, respectively. The reaction was incubated with 100 μL of TMB Single-Component Substrate solution (Solarbio, China) at room temperature for 10 min and then stopped by adding 100 μL of Stop Solution for TMB Substrate (450 nm, Sulfuric acid free) (Beyotime, China) per well. Three biological replicates and three technical replicates were performed for all measurements. The dates were presented as mean ± SD (n = 3). Samples with P (sample)-B (blank)/N (negative)-B (blank) >2.1 were considered as positive, that is P/N > 2.1, it is considered to have binding activity to PAMP.
rCgCas1-2D specificity
rCgCas1-2D was incubated with purchased human Caspase-1 substrate (Ac-YVAD-ρNA), Caspase-3 substrate (Ac-DEVD-ρNA), Caspase-6 substrate (Ac-VEID-ρNA), Caspase-8 substrate (Ac-IETD-ρNA), Caspase-9 substrate (Ac-LEHD-ρNA) (Beyotime, Shanghai, China) at 37 °C for 2 h. The release of ρNA was monitored at 405 nm to determine the cleavage specificity of rCgCas1-2D.
The bacterial binding assay of rCgCas1-2D and its mutants
The bacteria such as E. coli, V. splendidus, V. alginolyticus, A. hydrophila, S. aureus, B. subtilis and M. luteus, were cultured and collected according to previous description81. The bacteria were suspended with 200 μL TBS and then incubated with 200 μL rCgCas1-2D and its mutants, respectively. The rTrx was used as negative control. The bacterial protein was used as loading control. The mixture was then under slight rotation at room temperature for 3 h and then centrifuged at 6000 × g for 5 min. After washed five times with TBS, the bacteria were resuspended with 80 μL TBS and 20 μL 5× loading buffer (Takara, China) for SDS-PAGE and then analyzed by Western blotting. Anti-His antibody (Sangon Biotech, China) and HRP-labeled Goat Anti-mouse IgG (Beyotime, China) were used as the primary antibody and secondary antibody, respectively.
Co-localization assay of CgCas1-2D and bacteria
The bacteria (V. splendidus and V. alginolyticus) were collected at the mid-log phase. After washed three times with sterile seawater, the bacteria were incubated with FITC (1 mg mL−1, Sigma) dissolved in NaHCO3 (0.1 M, pH 9.0) at 4 °C overnight. After washed four times with TBS, the FITC-labeled V. splendidus and V. alginolyticus were resuspended in TBS at a concentration of 1 × 107 CFU mL−1. One hundred microliters of FITC-labeled V. splendidus and V. alginolyticus were incubated with 1 mL of hemocytes resuspended in anticoagulant from six oysters, respectively, which were then placed at room temperature with slight rotation in dark for 0.5 h. The hemocytes were washed three times with anticoagulants, and then deposited onto glass slides. Anti-CgCas1-2D and Alexa Fluor 647-labeled Goat Anti-mouse IgG (Beyotime, China) were used as primary antibody and secondary antibody, respectively. Fluorescence was observed under the laser scanning confocal microscopy (Axio Imager A2, ZEISS).
The cleavage activity assay of rCgCas1-2D to rCgGSDME
rCgGSDME with His-tag was offered by Zihan Wang in our lab. One hundred microliter rCgCas1-2D was incubated with 100 µL rCgGSDME at 37 °C for 1 h. The control groups were rCgCas1-2D and rCgGSDME incubated with TBS, respectively. The mixture was analyzed by Western blotting with Anti-His antibody as the primary antibody and HRP-labeled Goat Anti-mouse IgG (Beyotime, China) as the secondary antibody.
HEK293T cell lines and co-transfection assay
HEK293T cell lines (Mcellbank, Shanghai) were grown in DMEM medium (Hyclone, USA) supplemented with 10% fetal bovine serum (Hyclone, USA) at 37 °C in a 5% CO2 incubator. The transfected plasmids were CgCas1-2D-CASc ligated to the mCherry vector and CgGSDME ligated to the EGFP vector. Control groups included CgCas1-2D-CASc transfection group, CgGSDME transfection group, EGFP vector transfection group, mCherry vector transfection group, EGFP vector and mCherry vector co-transfection group. HEK293T cells without any transfection were used as the loading control. The process of transfection assay was referred to EntransterTM-H4000 instructions (Engreen, China). Fluorescence was observed under the laser scanning confocal microscopy (LSCM) (Axio Imager A2, ZEISS). After co-transfection of CgCas1-2D-CASc and CgGSDME, the cell protein samples were analyzed by Western blotting. The EGFP tag antibody (Beyotime, China) was used as the primary antibody, and HRP-labeled Goat anti-Mouse IgG as the secondary antibody.
Molecular docking simulation
The structures of CgCas1-2D and CgGSDME were predicted by I-TASSER. The ZDOCK online website was used for rigid docking, and based on this result, RosettaDock was used for flexible docking. The selection of the results depends on the score module built in Rosetta. Ligplot software was used to analyze the interaction between the two proteins, and Rosetta’s Interface analyzer module was used to analyze the overall interaction binding energy of the two proteins. Pymol software was used to display the conformation of the protein after docking.
Real-time quantitative PCR (RT-qPCR) analysis of related genes
The RT-qPCR with primers CgCas1-2D-RT-F/R (Supplementary Data 1) were performed to examine the tissue distribution and temporal expressions of CgCas1-2D mRNA. The fragments of elongation factor (CgEF, NP_001292242.2) amplified with primers CgEF-RT-F/R (Supplementary Data 1) were used as the control. The RT-qPCR with corresponding primers (Supplementary Data 1) was performed to analyze the mRNA expressions of CgGSDME, CgTNF-2, CgHMGB1 and CgRel by using the 2−ΔΔCt method82.
Western blotting assay of target genes
The total haemocyte lysates were separated by 12.5% SDS-PAGE and transferred onto PVDF membranes. The membranes were blocked with TBST (50 mmol L−1 Tris-HCl, 150 mmol L−1 NaCl and 0.1% Tween-20) in 3% bovine serum albumin (BSA) at room temperature for 3 h. The membrane was incubated with anti-CgCas-1-2D, anti-CgGSDME, anti-CgH3, anti-CgH3K4me1, anti-CgH3K4me2, anti-CgH3K4me3, anti-CgH3K9ac, anti-CgH3K27ac and anti-Tubulin (Beyotime, China) (1/1000 diluted into 3% BSA in TBST) as the primary antibodies, respectively, and HRP-labeled Goat Anti-mouse IgG as the secondary antibody. Tubulin and H3 antibody were used for quantitative analysis of samples, respectively. After washed with TBST for three times, the membrane was immersed into the reaction system (1:1 mixture of BeyoECL Moon A and B) in dark for 1 min. The signal bands were imaged by Amersham Imager 600 (GE Healthcare).
Co-immunoprecipitation (Co-IP) assay of CgCas1-2D and CgGSDME
The hemocytes from thirty-six oysters were divided into two groups and collected after stimulation with sterilized seawater and V. splendidus, respectively. Input was used as the control and sample quantification. Subsequently, two groups of protein samples were incubated with protein (A + G) agar and negative serum to remove non-specific binding proteins. They were then incubated with CgCas1-2D and CgGSDME antibodies at 4 °C overnight, respectively. The mixture was incubated with protein (A + G) agar at 4 °C for 3 h, and the resulting pellet (bound protein, antibody and CgCas1-2D or CgGSDME protein) was analyzed by Western blotting after washing five times with TBS. Anti-CgCas1-2D and anti-CgGSDME was used as primary antibody, respectively, and IPKine™ HRP Goat Anti-mouse IgG LCS and IPKine™ HRP Mouse Anti-rabbit IgG LCS (Abbkine, China) was as secondary antibody.
Immunocytochemical assay of CgCas1-2D and CgRel
Immunocytochemistry was used to observe the subcellular localization of CgCas1-2D and CgRel, respectively. The specific experimental steps were referred to the previous report83. Anti-CgCas1-2D and anti-CgRel were used as primary antibody, respectively. Alexa Fluor 647-labeled Goat Anti-mouse IgG (Beyotime, China) and Alexa Fluor 488-labeled Goat Anti-mouse IgG (Beyotime, China) were as secondary antibody, respectively. After washed with PBST (140 mM NaCl, 10 mM sodium phosphate, 0.1% Tween-20, pH 7.4) for five times, the hemocytes were incubated with DAPI (Beyotime, China) for 20 min. Fluorescence was observed under LSCM (Axio Imager A2, ZEISS).
Co-localization assay of CgCas1-2D and CgGSDME
The hemocytes from three oysters were extracted and mixed with anticoagulants after V. splendidus stimulation for 3 h. Three oysters were stimulated with sterilized seawater as control. Negative serum co-localized with CgGSDME was used as control. For the co-localization of CgCas1-2D and CgGSDME, the anti-CgCas1-2D (1:500 in 3% BSA) and anti-CgGSDME (1:500 in 3% BSA) were used as primary antibody. Alexa Fluor 647 Anti-mouse IgG (1:1000 in 3% BSA) and Alexa Fluor 488-labeled Goat Anti-rabbit IgG (1:1000 in 3% BSA) (Beyotime, China) were used as secondary antibody. Fluorescence was observed under LSCM (Axio Imager A2, ZEISS).
RNAi of CgCas1-2D, CgRel, and CgGSDME
The specific small interfering RNAs (siRNAs) of CgCas1-2D (CgCas1-2D-RNAi-F/R) and CgRel (CgRel-RNAi-F/R) were designed and synthesized (GenePharma, China) to inhibit the expressions of CgCas1-2D and CgRel, respectively. The untreated oysters were employed as blank group. The oysters received the injection of siRNAs of negative control were employed as the control (NC-RNAi group). Each oyster in CgCas1-2D-RNAi, CgRel-RNAi and NC-RNAi group received 100 μL siRNAs (33 μg siRNA dissolved in 125 µL DEPC-treated water) of CgCas1-2D, CgRel and NC, respectively. In 12 h of siRNAs injection, the hemocytes were collected at 3 h after V. splendidus stimulation. The hemocytes of nine oysters were collected in blank, CgCas1-2D-RNAi, CgRel-RNAi and NC-RNAi group, respectively. The interference efficiencies of CgCas1-2D-RNAi and CgRel-RNAi oysters were evaluated by RT-qPCR, respectively.
In our previous study, double-strand RNAs (dsRNAs) of CgGSDME injection had been successfully used to inhibit the expression of CgGSDME. CgGSDME fragments were amplified with primers CgGSDME-Fi/Ri (Supplementary Data 1) and then used as a template to synthesize dsRNAs of CgGSDME using T7 polymerase (Takara, China) according to the instruction. The primers EGFP-Fi/Ri (Supplementary Data 1) were used to synthesize dsRNAs of EGFP from pEGFP vector as control. The steps of oysters receiving the injection with dsRNAs were referred as previous reported12. The untreated oysters were employed as blank group. The hemocytes of nine oysters were collected in blank, EGFP-RNAi and CgGSDME-RNAi oysters, respectively. The interference efficiencies of CgGSDME-RNAi oysters were evaluated by RT-qPCR.
Treatment with histone acetylation and methylation inhibitor
The twenty-four oysters were employed for the treatment with HY-100734 (histone acetyltransferase inhibitor II, p300 inhibitors, MedChemExpress) and MTA (histone methylation inhibitor, Sigma), respectively. The oysters in the HY-100734 and MTA group received an injection of HY-100734 and MTA at a dose of 96 µmol kg−1 body weight, respectively27. The oysters treated with 100 µL of dimethyl sulfoxide (DMSO, 5% in PBS) were used as a control. After the treatments with HY-100734, MTA and DMSO 24 h, 100 µL of V. splendidus were used to treat each oyster. Hemolymph samples were collected from treated oysters to examine the methylation and acetylation levels of histones, as well as the mRNA expression levels of CgHMGB1 and CgTNF-2.
Statistics and reproducibility
All data were shown as mean ± standard deviation (SD). Multiple group comparisons analyzed by one-way ANOVA and multiple comparisons using Statistical Package for Social Sciences (SPSS) 20. Statistically significant difference was considered significant at p < 0.05 (N = 3). Student’s t test was used for the comparison of the data between the two groups. The criteria for determining statistical significance were as follows: *p < 0.05 (significant), **p < 0.01 (highly significant), ***p < 0.001 (extremely highly significant).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All study data are included in the article. The source data behind the graphs in the paper can be found in Supplementary information and Supplementary Data 2.
References
Martinon, F., Burns, K. & Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426 (2002).
Franchi, L., Eigenbrod, T., Muñoz-Planillo, R. & Nuñez, G. The inflammasome: a Caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247 (2009).
Kesavardhana, S., Malireddi, R. K. S. & Kanneganti, T. D. Caspases in cell death, inflammation, and pyroptosis. Annu. Rev. Immunol. 38, 567–595 (2020).
Huang, Y., Xu, W. & Zhou, R. NLRP3 inflammasome activation and cell death. Cell Mol. Immunol. 18, 2114–2127 (2021).
Fang, Y. et al. Pyroptosis: a new frontier in cancer. Biomed. Pharmacother. 121, 109595 (2020).
Song, Z., Gong, Q. & Guo, J. Pyroptosis: mechanisms and links with fibrosis. Cells 10, 3509 (2021).
Burdette, B. E., Esparza, A. N., Zhu, H. & Wang, S. Gasdermin D in pyroptosis. Acta Pharm. Sin. B 11, 2768–2782 (2021).
Kanneganti, T. D. Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 10, 688–698 (2010).
Wei, S., Feng, M. & Zhang, S. Molecular characteristics of cell pyroptosis and its inhibitors: a review of activation, regulation, and inhibitors. Int. J. Mol. Sci. 23, 16115 (2022).
Xue, Y., Enosi Tuipulotu, D., Tan, W. H., Kay, C. & Man, S. M. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol. 40, 1035–1052 (2019).
Jiang, S., Gu, H., Zhao, Y. & Sun, L. Teleost gasdermin E is cleaved by Caspase 1, 3, and 7 and induces pyroptosis. J. Immunol. 203, 1369–1382 (2019).
Li, X. et al. CgCaspase-3 activates the translocation of CgGSDME in hemocytes of Pacific oyster Crassostrea gigas. Fish. Shellfish Immunol. 131, 757–765 (2022).
Wang, X. et al. Characterization of GSDME in amphioxus provides insights into the functional evolution of GSDM-mediated pyroptosis. PLoS Biol. 21, e3002062 (2023).
Chen, S. et al. Caspase-mediated LPS sensing and pyroptosis signaling in Hydra. Sci. Adv. 9, eadh4054 (2023).
Jiang S., Zhou Z., Sun Y., Zhang T. & Sun L. Coral gasdermin triggers pyroptosis. Sci. Immunol. 5, eabd2591 (2020).
Lin, T. et al. ChNLRC4, a cytoplasmic pattern recognition receptor, activates the pyroptosis signaling pathway in Mollusca. Int. J. Biol. Macromolecules 296, 139632 (2025).
Yi, Y. S. Caspase-11 non-canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology 152, 207–217 (2017).
Yang, D. et al. Sensing of cytosolic LPS through caspy2 pyrin domain mediates noncanonical inflammasome activation in zebrafish. Nat. Commun. 9, 3052 (2018).
Xu, J. et al. Caspase-3 serves as an intracellular immune receptor specific for lipopolysaccharide in oyster Crassostrea gigas. Dev. Comp. Immunol. 61, 1–12 (2016).
Chen, X. et al. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 109, E2865–E2874 (2012).
Chung, S. et al. NF-κB inducing kinase, NIK mediates cigarette smoke/TNFα-induced histone acetylation and inflammation through differential activation of IKKs. PloS One 6, e23488 (2011).
Yang, J., Cao, M. X., Hu, W. Y., Wei, Y. Y. & Hu, T. J. Sophorasubprosrate polysaccharide suppress the inflammatory reaction of RAW264.7 cells infected with PCV2 via regulation NF-κB/MAPKs/c-Jun signal pathway and histone acetylation modification. Int. J. Biol. Macromol. 159, 957–965 (2020).
Galindo-Villegas, J., García-Moreno, D., de Oliveira, S., Meseguer, J. & Mulero, V. Regulation of immunity and disease resistance by commensal microbes and chromatin modifications during zebrafish development. Proc. Natl. Acad. Sci. USA 109, E2605–E2614 (2012).
Xin, L. et al. The characterization of an interleukin-12 p35 homolog involved in the immune modulation of oyster Crassostrea gigas. Dev. Comp. Immunol. 123, 104145 (2021).
Wu, S. Z., Huang, X. D., Li, Q. & He, M. X. Interleukin-17 in pearl oyster (Pinctada fucata): molecular cloning and functional characterization. Fish. Shellfish Immunol. 34, 1050–1056 (2013).
Rosani, U., Varotto, L., Gerdol, M., Pallavicini, A. & Venier, P. IL-17 signaling components in bivalves: Comparative sequence analysis and involvement in the immune responses. Dev. Comp. Immunol. 52, 255–268 (2015).
Lian, X. et al. The modification of H3K4me3 enhanced the expression of CgTLR3 in hemocytes to increase CgIL17-1 production in the immune priming of Crassostrea gigas. Int. J. Mol. Sci. 25, 1036 (2024).
Wang, L., Song, X. & Song, L. The oyster immunity. Dev. Comp. Immunol. 80, 99–118 (2018).
Gao, L. et al. Species-specific CgCaspase-Cg-5 in the pacific oyster induces haemocyte apoptosis by regulating the mRNA expression of apoptosis-related genes in the early stage of immune response. Fish. Shellfish Immunol. 138, 108856 (2023).
Vogeler, S., Carboni, S., Li, X. & Joyce, A. Phylogenetic analysis of the caspase family in bivalves: implications for programmed cell death, immune response and development. BMC Genomics 22, 80 (2021).
Cohen, G. M. Caspases: the executioners of apoptosis. Biochem J. 326, 1–16 (1997).
McIlwain, D. R., Berger, T. & Mak, T. W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 5, a008656 (2013).
Shi, J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015).
Qin, K., Jiang, S., Xu, H., Yuan, Z. & Sun, L. Pyroptotic gasdermin exists in Mollusca and is vital to eliminating bacterial infection. Cell Rep. 42, 112414 (2023).
Shalini, S., Dorstyn, L., Dawar, S. & Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 22, 526–539 (2015).
Morimoto N., Kono T., Sakai M., Hikima J. I. Inflammasomes in Teleosts: Structures and Mechanisms That Induce Pyroptosis during Bacterial Infection. Int. J. Mol. Sci. 22, 4389 (2021).
Ying, Z. et al. The Spodoptera frugiperda effector caspase Sf-caspase-1 becomes unstable following its activation. Arch. Insect Biochem. Physiol. 83, 195–210 (2013).
Jearaphunt, M. et al. Caspase-1-like regulation of the proPO-system and role of ppA and Caspase-1-like cleaved peptides from proPO in innate immunity. PLoS Pathog. 10, e1004059 (2014).
Wang, Q., Ju, X., Chen, L. & Chen, K. Caspase-1 from the silkworm, Bombyx mori, is involved in Bombyx mori nucleopolyhedrovirus infection. Z. Naturforsch. C. J. Biosci. 72, 147–153 (2017).
Lu, G. et al. The self-activation and LPS binding activity of executioner Caspase-1 in oyster Crassostrea gigas. Dev. Comp. Immunol. 77, 330–339 (2017).
Hur, S. Double-stranded RNA sensors and modulators in innate immunity. Annu. Rev. Immunol. 37, 349–375 (2019).
Lv, X. et al. The Dicer from oyster Crassostrea gigas functions as an intracellular recognition molecule and effector in anti-viral immunity. Fish. Shellfish Immunol. 95, 584–594 (2019).
Van Opdenbosch, N. & Lamkanfi, M. Caspases in cell death, inflammation, and disease. Immunity 50, 1352–1364 (2019).
Martinon, F. & Tschopp, J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell 117, 561–574 (2004).
Makoni, N. J. & Nichols, M. R. The intricate biophysical puzzle of Caspase-1 activation. Arch. Biochem. Biophys. 699, 108753 (2021).
de Vasconcelos, N. M., Van Opdenbosch, N. & Lamkanfi, M. Inflammasomes as polyvalent cell death platforms. Cell Mol. Life Sci. 73, 2335–2347 (2016).
Shi, J. et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 (2014).
Matikainen, S., Nyman, T. A. & Cypryk, W. Function and regulation of noncanonical caspase-4/5/11 inflammasome. J. Immunol. 204, 3063–3069 (2020).
Zhao, Y. & Shao, F. Diverse mechanisms for inflammasome sensing of cytosolic bacteria and bacterial virulence. Curr. Opin. Microbiol. 29, 37–42 (2016).
Yang, J., Zhao, Y. & Shao, F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr. Opin. Immunol. 32, 78–83 (2015).
Ben Cheikh, Y., Travers, M. A. & Le Foll, F. Infection dynamics of a V. splendidus strain pathogenic to Mytilus edulis: In vivo and in vitro interactions with hemocytes. Fish. Shellfish Immunol. 70, 515–523 (2017).
Kayagaki, N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015).
Lu, F. et al. Emerging insights into molecular mechanisms underlying pyroptosis and functions of inflammasomes in diseases. J. Cell Physiol. 235, 3207–3221 (2020).
Wu, J. et al. TNF-α contributes to sarcopenia through Caspase-8/Caspase-3/GSDME-mediated pyroptosis. Cell Death Discov. 9, 76 (2023).
Zhang, Y. et al. Histone demethylase KDM5B licenses macrophage-mediated inflammatory responses by repressing Nfkbia transcription. Cell Death Differ. 30, 1279–1292 (2023).
Leus, N. G., Zwinderman, M. R. & Dekker, F. J. Histone deacetylase 3 (HDAC 3) as emerging drug target in NF-κB-mediated inflammation. Curr. Opin. Chem. Biol. 33, 160–168 (2016).
Lawrence, M., Daujat, S. & Schneider, R. Lateral thinking: how histone modifications regulate gene expression. Trends Genet 32, 42–56 (2016).
Daskalaki, M. G., Tsatsanis, C. & Kampranis, S. C. Histone methylation and acetylation in macrophages as a mechanism for regulation of inflammatory responses. J. Cell Physiol. 233, 6495–6507 (2018).
Wang, H., Cao, G., Wang, G. & Hao, H. Regulation of Mammalian UDP-Glucuronosyltransferases. Curr. Drug Metab. 19, 490–501 (2018).
Kagoshima, M., Ito, K., Cosio, B. & Adcock, I. M. Glucocorticoid suppression of nuclear factor-kappa B: a role for histone modifications. Biochem Soc. Trans. 31, 60–65 (2003).
Lu, T. & Stark, G. R. NF-κB: regulation by methylation. Cancer Res. 75, 3692–3695 (2015).
Liu, Z. et al. Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-κB/GSDMD signal in mice adipose tissue. J. Pineal. Res. 63, 12414 (2017).
Cui, X. et al. Alpha-kinase1 promotes tubular injury and interstitial inflammation in diabetic nephropathy by canonical pyroptosis pathway. Biol. Res. 56, 5 (2023).
Sha, X. et al. GSDMD mediated pyroptosis induced inflammation of Graves’ orbitopathy via the NF-κB/ AIM2/ Caspase-1 pathway. Exp. Eye Res. 240, 109812 (2024).
Mao, J. et al. Neutrophil extracellular traps induce pyroptosis of rheumatoid arthritis fibroblast-like synoviocytes via the NF-κB/Caspase 3/GSDME pathway. Inflammation 47, 921–938 (2024).
Yang, P. et al. LPS induces fibroblast-like synoviocytes RSC-364 cells to pyroptosis through NF-κB mediated dual signalling pathway. J. Mol. Histol. 52, 661–669 (2021).
Xu, B. et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 68, 773–782 (2018).
Li, Y. et al. CgRel involved in antibacterial immunity by regulating the production of CgIL17s and CgBigDef1 in the Pacific oyster Crassostrea gigas. Fish. Shellfish Immunol. 97, 474–482 (2020).
Zhang, T. et al. BCL10 regulates the production of proinflammatory cytokines by activating MAPK-NF-κB/Rel signaling pathway in oysters. Fish. Shellfish Immunol. 120, 369–376 (2022).
Kang, K. H., Lee, K. H., Kim, M. Y. & Choi, K. H. Caspase-3-mediated cleavage of the NF-kappa B subunit p65 at the NH2 terminus potentiates naphthoquinone analog-induced apoptosis. J. Biol. Chem. 276, 24638–24644 (2001).
Wier, E. M., Fu, K., Hodgson, A., Sun, X. & Wan, F. Caspase-3 cleaved p65 fragment dampens NF-κB-mediated anti-apoptotic transcription by interfering with the p65/RPS3 interaction. FEBS Lett. 589, 3581–3587 (2015).
Dou, X. et al. TLR2/4-mediated NF-κB pathway combined with the histone modification regulates β-defensins and interleukins expression by sodium phenyl butyrate in porcine intestinal epithelial cells. Food Nutr. Res. 62, 1493 (2018).
Rahman, I., Marwick, J. & Kirkham, P. Redox modulation of chromatin remodeling: impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expression. Biochem. Pharm. 68, 1255–1267 (2004).
Lv, X. et al. CgHMGB1 functions as a broad-spectrum recognition molecule to induce the expressions of CgIL17-5 and Cgdefh2 via MAPK or NF-κB signaling pathway in Crassostrea gigas. Int. J. Biol. Macromol. 211, 289–300 (2022).
Okamoto, K. et al. Regulation of cytokine and chemokine expression by histone lysine methyltransferase MLL1 in rheumatoid arthritis synovial fibroblasts. Sci. Rep. 14, 10610 (2024).
Wang, G., Xu, Y. L., Zhang, X. H., Tang, L. & Li, Y. LncRNA HOTTIP regulates TLR4 promoter methylation by recruiting H3K4 methyltransferase MLL1 to affect apoptosis and inflammatory response of fibroblast-like synoviocyte in rheumatoid arthritis. Kaohsiung J. Med. Sci. 40, 335–347 (2024).
Zhou, W. et al. Acetylation of H3K4, H3K9, and H3K27 mediated by p300 regulates the expression of GATA4 in cardiocytes. Genes Dis. 6, 318–325 (2019).
Peng, Q. M. et al. Apelin‑13 ameliorates LPS‑induced BV‑2 microglia inflammatory response through promoting autophagy and inhibiting H3K9ac enrichment of TNF‑α and IL‑6 promoter. Acta Neurobiol. Exp. 82, 65–76 (2022).
Zhang, Y. et al. RBM25 is required to restrain inflammation via ACLY RNA splicing-dependent metabolism rewiring. Cell. Mol. Immunol. 21, 1231–1250 (2024).
Sun, J., Wang, L., Yang, C. & Song, L. An ancient BCR-like signaling promotes ICP production and hemocyte phagocytosis in oyster. iScience 23, 100834 (2020).
Sun, J. et al. CgCLec-HTM-mediated signaling pathway regulates lipopolysaccharide-induced CgIL-17 and CgTNF production in oyster. J. Immunol. 203, 1845–1856 (2019).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Sun, J. J., Lan, J. F., Zhao, X. F., Vasta, G. R. & Wang, J. X. Binding of a C-type lectin’s coiled-coil domain to the Domeless receptor directly activates the JAK/STAT pathway in the shrimp immune response to bacterial infection. PLoS Pathog. 13, e1006626 (2017).
Acknowledgements
This research was supported by National Science Foundation of China (32222086, 32230110), the fund for CARS-49 and for Outstanding Talents and Innovative Team of Agricultural Scientific Research in MARA, the innovation team of Aquaculture Environment Safety from Liaoning Province (LT202009), and Liaoning Revitalization Talents Program (XLYC2203087).
Author information
Authors and Affiliations
Contributions
R.C. and J.S. designed research; R.C., J.L., Z.W., S.M., Y.L. and J.W. performed research; R.C. and J.S. analyzed data; J.S. and L.S. acquired funding; J.S. supervised the project; R.C. J.S. and L.S. wrote and edited the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Shitao Li and Mengtan Xing. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chang, R., Sun, J., Leng, J. et al. A new type of Caspase-1 upon recognizing bacteria inhibits GSDME-dependent histone modification and NF-κB signaling. Commun Biol 8, 827 (2025). https://doi.org/10.1038/s42003-025-08290-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08290-7
