
Abstract
Invasive fungal infections are responsible for millions of deaths worldwide each year. Therefore, focusing on innovative approaches to developing therapeutics that target fungal pathogens is critical. Here, we discuss targeting the fungal trehalose biosynthesis pathway with antifungal therapeutics, which may lead to the improvement of human health globally, especially as fungal pathogens continue to emerge due to fluctuations in the climate.
Similar content being viewed by others

Introduction
Pathogenic fungal microorganisms can cause both superficial and invasive diseases in human hosts, contributing to approximately 6.5 million infections and 2.5 million directly attributable deaths worldwide each year1. Invasive fungal infections often occur in immunocompromised hosts, most notably those patients undergoing immunosuppressive therapies for cancer treatment, solid-organ transplants or those who are immunocompromised due to diseases like HIV infection or autoimmune disorders2,3. However, there are also cases where apparently immunocompetent individuals have suffered from invasive fungal diseases4,5. For example, the Pacific Northwest epidemic of infections due to Cryptococcus gattii resulted in multiple cases of pulmonary cryptococcosis in otherwise healthy individuals6,7,8,9. Therefore, invasive fungal infections cause significant morbidity and mortality across the globe in a wide variety of individuals, making invasive fungal infections as much of a public health threat as tuberculosis and malaria3.
According to the World Health Organization, the most critical class of human fungal pathogens include those from the genera Cryptococcus and Candida10. Among these, Cryptococcus neoformans is a basidiomycete yeast that is responsible for approximately 19% of AIDS-related deaths worldwide, despite the widespread use of antiretrovirals11. C. neoformans can survive as a saprophyte in the environment and is often found in soil and associated with pigeon guano12. Initial infection typically occurs by the inhalation of C. neoformans spores or desiccated yeast cells that may remain dormant in the lungs as an asymptomatic infection13. Alternatively, the fungus may be cleared at this stage of the infection. In fact, given that C. neoformans is ubiquitously found in the environment, 70% of individuals greater than 5 years old have serological evidence of prior C. neoformans exposure, many with no clinical evidence of disease14. However, in the absence of a sufficient immune protection, C. neoformans can disseminate from the lungs to other organs15,16,17,18,19, including the central nervous system to cause meningoencephalitis20. In the absence of treatment, these infections of the brain and surrounding structures are uniformly fatal21.
In contrast, Candida albicans is one of the most common fungi in the host microbiota and commonly colonizes epithelial surfaces such as the gastrointestinal tract, genitourinary tract, mouth and skin in more than 70% of the world population22,23. C. albicans and host interactions often lead to asymptomatic commensalism. However, in immunocompromised hosts, C. albicans can cause life-threatening invasive candidiasis. In fact, C. albicans is the fourth most frequent causative agent of healthcare-associated bloodstream infections, which result in a mortality rate of approximately 40%24,25.
While C. neoformans and C. albicans are currently among the most critical fungal pathogens, new fungal pathogens are emerging due to rapid changes in the global climate. Increases in global temperatures will likely preferentially select for environmental fungi that can survive at higher temperatures, resulting in fungal species acquiring the ability to survive at human body temperatures26,27,28. Rising global temperatures and the coincident extreme weather events can also result in the disruption of environments where fungi live, resulting in an increased dissemination of fungi to new environments and hosts29. As a result, many environmentally derived and emerging fungi, such as Candida auris, are now causing deadly diseases in humans30,31. Newly emerged isolates of C. auris are naturally resistant to the antifungal therapeutics that are currently utilized32.
The three major classes of antifungal drugs are the polyenes, azoles and echinocandins. The polyenes are derived from natural products and target and deplete the essential membrane lipid, ergosterol, from the plasma membrane by functioning as a sponge33. Polyenes have broad-spectrum activity but suffer from significant host toxicities and poor oral bioavailability34,35,36. The azoles block ergosterol biosynthesis directly by inhibiting the function of lanosterol-14α-demethylase. While azoles have good oral bioavailability, they result in only a fungistatic inhibition of fungal growth. This lack of fungicidal activity promotes the possible development of antifungal drug resistance37. The echinocandin class of antifungal drugs disrupts the integrity of the cell surface by blocking the production of a key component of the cell wall, (1,3)-β-D-glucan38,39. Echinocandins function as the first line of treatment for Candida species but are clinically ineffective against many other fungi including Cryptococcus40. Therefore, it is critical to develop new antifungal drugs, preferably those that are broad-spectrum, have fungicidal effects and are less prone to development of antifungal drug resistance41. Given that fungi and the human hosts are both eukaryotes, and therefore share similar cellular mechanisms, it can prove challenging to identify fungal drug targets that would not also result in off-target effects in the host. Therefore, it is of the utmost importance to develop antifungal drugs that are safe, especially for the immunocompromised patients that are most susceptible to invasive fungal infections.
In searching for new targets for antifungal drugs several stress response pathways in fungal pathogens have been investigated, including the calcineurin, Hsp90 and trehalose biosynthesis pathways42. The trehalose biosynthesis pathway has been implicated in aiding many organisms in the ability to tolerate a wide range of both biotic and abiotic stresses43. Here, we explore the potential of developing an antifungal drug that targets the trehalose biosynthesis pathway in fungal pathogens. Given that the trehalose biosynthesis pathway in fungal pathogens is not found in humans, targeting trehalose biosynthesis may result in an antifungal therapeutic with low toxicity44. Additionally, studies revealing trehalose biosynthesis protein structures have primed the enzymatic targets for structure-based drug design studies45,46,47,48.
The trehalose biosynthesis pathway contributes to the virulence of fungal pathogens
Trehalose (α-D-glucopyranosyl-(1 → 1)-α-D-glucopyranoside) is a non-reducing disaccharide composed of two glucose molecules linked by an α,α-1,1-glycosidic linkage43. Fungal pathogens employ a two-step trehalose biosynthesis pathway to generate trehalose (Fig. 1a). The first step is initiated when uridine-diphosphate glucose (UDPG) and glucose-6-phosphate (G6P) are converted to trehalose-6-phosphate (T6P) by trehalose-6-phosphate synthase (Tps1). Next, trehalose-6-phosphate phosphatase (Tps2) removes the phosphate group from T6P to generate trehalose. Several fungi contain additional regulatory proteins that lack known enzymatic activity and are hypothesized to contribute to complex formation in cells48,49,50,51,52,53,54.

a The canonical, two-step trehalose biosynthesis pathway in fungal pathogens. Tps1 (shown as the C. neoformans homo-tetramer) converts UDPG and G6P into T6P, releasing the byproduct UDP45. Tps2 is a phosphatase that removes the phosphate group from T6P to generate trehalose. Trehalases can breakdown trehalose into two glucose molecules. Shown here are representations of trehalases from C. neoformans (Nth1 and Nth2)66. b There are multiple fungal virulence phenotypes attributed to Tps1 in several fungi including cryptococcal species and C. albicans. c Known fungal virulence phenotypes attributed to Tps2 amongst fungal pathogens are fewer than Tps1. This figure was created in Biorender.
Trehalose plays key roles in cells as a stress response molecule and an energy source. These two biological functions may be closely connected, as the protective nature of trehalose is utilized the most while fungi are exposed to stressful environments. In several biological systems, trehalose interacts with proteins and phospholipids resulting in the protection of membranes. Interactions between trehalose and proteins prevent the proteolysis of intracellular proteins43,55,56. As a result, trehalose and the trehalose biosynthesis pathway are critical for the survival of fungal pathogens under multiple stresses, including temperature stress, osmotic stress and dehydration57,58,59. Fungi can utilize trehalose as an energy source through trehalase-dependent degradation of trehalose (Fig. 1a). As a result of this degradation, trehalose is converted into two glucose molecules which can be shuttled into energy-generating processes for the cell43.
For the purposes of this review, we will focus mainly on the contribution of trehalose biosynthesis proteins to virulence. In both mice and humans infected with C. neoformans, a significant amount of trehalose was detected that was tightly associated with the brain cryptococcomas60,61. The genes required for trehalose biosynthesis in Cryptococcus have also been shown to be upregulated during infections62. Genetic approaches have been utilized to determine in a detailed manner, the requirement of trehalose biosynthesis for virulence in Cryptococcus and Candida species.
Several phenotypes associated with loss of Tps1 have been described in multiple species of Cryptococcus, as well as C. albicans (Fig. 1b). Tps1 is required for hyphal growth transitions in C. albicans, Cryptococcus deneoformans and C. gattii63,64,65. The ability of C. albicans, C. neoformans and C. gattii to grow at elevated temperatures is dependent on Tps163,65,66. The thermotolerance displayed by these fungal pathogens contributes to their virulence in multiple hosts63,65,66,67. However, Tps1 is also required for C. neoformans virulence in both the zebrafish infection model and C. elegans infection models, indicating that the lack of thermotolerance is not solely responsible for Tps1-mediated virulence phenotypes66,68. C. gattii tps1Δ mutations are fungistatic in vitro65. In C. neoformans deletion of the TPS1 gene results in fungicidal effects, meaning that the infected immunosuppressed rabbits rapidly clear the fungi66. The lethal phenotype of C. neoformans tps1Δ in vivo is a critical indication of the promise of targeting Tps1 with an antifungal drug. Cryptococcal tps1Δ mutants have defects in the production of virulence factors such as melanin and capsule65,67. Additionally, C. deneoformans and C. gattii tps1Δ mutants are unable to sporulate64,65. Although the function of Tps1 is well characterized in the laboratory fungal strains, very little is known about the role of Tps1 in diverse species and strains of Cryptococcus, including environmental strains and clinical strains. Given the various niches filled by different strains of Cryptococcus and Candida, studying various environmental and clinical isolates may yield interesting insights into trehalose roles and potential effectiveness of trehalose-targeting drugs. Similarly, the role of Tps1 in Candida species has been limited to the wild-type C. albicans SC5314 strain47,63. In Saccharomyces cerevisiae, changes in Tps1-associated phenotypes were dependent on the yeast strain, highlighting the importance of investigating the trehalose biosynthesis pathway across multiple fungal species and strains69.
Tps2 also contributes to virulence in fungal pathogens (Fig. 1c). Like Tps1, Tps2 contributes to stress tolerance. In C. neoformans Tps2 is required for high temperature growth66. In C. albicans, disruption of the TPS2 gene causes multiple defects including increased sensitivity to both high temperatures and oxidative stress70,71. C. neoformans Tps2 is required for tolerance of oxidative and osmotic stress66. Deletion of TPS2 from C. albicans results in pH-dependent defects in cell wall integrity, which could not be fully differentiated by investigating the cell wall ultrastructure63,70. C. albicans tps2Δ can grow at higher temperatures in vitro, albeit with a reduced rate of growth compared to wild-type C. albicans cells72. Interestingly, Tps2 is dispensable for many phenotypes that are associated with Tps1, such as dimorphic transition, chlamydospore formation and the formation of C. neoformans virulence factors melanin and capsule66,70,71. Additionally, in C. gatti, tps2Δ mutants have no defects in filamentation or sporulation65. These discrepancies may be explained by a signaling role of the trehalose biosynthesis intermediate, T6P (Fig. 1a). However, deletion of TPS2 also leads to a cytotoxic hyperaccumulation of T6P, likely as a result of phosphate sequestration and deregulation of metabolic processes66,73. Perhaps most importantly for the development of Tps2 as an antifungal drug target is that disruption of TPS2 in both Cryptococcus and Candida species results in critical and severe virulence attenuation. C. albicans tps2Δ mutants have significantly reduced virulence in a mouse infection model71,72. The cryptococcal tps2Δ mutant cannot grow in vitro at 37 °C and is therefore unable to survive in the mammalian host66. C. neoformans tps2Δ has no virulence defects in the C. elegans model of infection66. This result suggests that the lack of thermotolerance alone may be sufficient to explain the lack of C. neoformans tps2Δ virulence in the mouse infection model.
Structures of the trehalose biosynthesis enzymes
The multiple structures of the trehalose biosynthesis proteins that have been determined will enable structure-based drug design approaches to design an antifungal therapeutic that targets the trehalose biosynthesis pathway. Therefore, the details of these structures require discussion.
The Tps1 proteins in fungal pathogens adopt the highly conserved GT-B fold of the retaining glycosyltransferase family (Fig. 2a and Table 1)45,47,74,75. Glycosyltransferases in the GT-B family contain two modified Rossmann fold domains76. The N-terminal lobe binds the acceptor molecule, which for Tps1 is glucose-6-phoshate (G6P). The C-terminal lobe binds the donor, for example uridine diphosphate glucose (UDPG). The majority of the movement in the Tps1 proteins occurs in the N-terminal domain, which rotates upon substrate binding and is less structurally conserved to enable the binding of diverse acceptor molecules45,47,74,75. The two lobes are connected by a C-terminal α-helix that extends to the N-terminal lobe, thereby connecting the two subdomains. Interestingly, the kink in the C-terminal α-helix is conserved and is also characteristic of GT-B-type glycosyltransferases75. Difficulties with crystallization may have hampered the determination of GT-B glycosyltransferase structures using x-ray crystallography45,77. However, the use of cryo-electron microscopy (cryo-EM) has enabled the structural determination of the C. neoformans Tps1 enzyme and even revealed a homo-tetrameric complex formation (Fig. 2a, b)45. There is a deep substrate-binding cleft between the two subdomains of Tps1 (Fig. 2c). Although GT-B glycosyltransferases do not share any one specific, conserved residue for catalysis, there is excellent conservation of the substrate-binding residues amongst fungal and bacterial Tps1 enzymes (Fig. 2d).

a Tps1 enzymes are GT-B family glycosyltransferases that consist of a N-terminal lobe and a C-terminal lobe. The two lobes are connected by a kinked C-terminal α-helix. Shown in light brown is E. coli OtsA-UDP-2-fluoroglucose (PDB: 1UQU). Shown in blue is M. oryzae MoTps1-UDP structure (PDB: 6JBW). In purple is the C. albicans Tps1 bound to UDP and G6P (PDB: 5HUU). Shown in pink is M. thermoresistible MtrOtsA in complex with GDPG (PDB: 5K42). In green is the A. fumigatus AfTps1A-UDP-validoxylamine A structure (PDB: 5HVM). In dark beige is a protomer from the C. neoformans cryo-EM homo-tetramer structure (PDB: 8FO1). For clarity, substrates are not shown in the binding pocket of these structures. b The cryo-EM structure of the C. neoformans Tps1 homo-tetramer (PDB: 8FO1)45. c The substrate-binding pocket of M. thermoresistible MtrOtsA reveals the nearly buried binding site of GDP-glucose. d Conservation of selected substrate-binding residues with the numbers listed from the CaTps1-UDP-G6P (PDB: 5HUU) structure. Residues highlighted with the red stars have been implicated in substrate-binding45,47. This figure was created in Biorender with structures generated in ChimeraX108.
Tps2 is a phosphatase in the haloacid dehalogenase superfamily (HADSF) of phosphatases (Fig. 3a and Table 2)78. Generally, phosphatases, including those in the HADSF, are known to be promiscuous and therefore not considered to be great antifungal drug targets78. However, it has been determined that the C. neoformans Tps2PD phosphatase is highly specific and unable to catabolize other sugar donor substrates48. Therefore, Tps2 from fungal pathogens may be able to be targeted with an antifungal compound without off-target toxicity. The Tps2 structure consists of a cap domain and a core domain (Fig. 3a)48,79,80,81,82. The core domain is a hydrolase consisting of a conserved Rossmann fold domain. The substrate-binding site for T6P is a deep, charged and highly conserved pocket in a cleft between the cap and core domain (Fig. 3b). The substrate-binding domain of Tps2 enzymes contains four conserved motifs that are characteristic of HADSF phosphatases83. These active site motifs coordinate the binding of the requisite magnesium cofactor as well as the binding of the substrate phosphoryl group for catalysis. There is a nucleophilic aspartate residue that is conserved amongst Tps2 enzymes from fungal pathogens (Fig. 3c). The conserved nature of these substrate-binding pockets indicates that it might be possible to generate a broad-spectrum antifungal drug that inhibits the activity of Tps2.

a Tps2 enzymes are HAD superfamily phosphatases that consist of a cap and core domain. The two domains are connected by a flexible linker to enable movement during catalysis. Shown in magenta is C. neoformans Tps2PD (D24N)-T6P (PDB: 5DX9) and in teal is the A. fumigatus Tps2PD (PDB: 5DXL). Shown in yellow is the S. typhimurium StT6PP-T6-sulfate (PDB: 6UPB). In light gray, is the Brugia malayi Tps2 structure (PDB: 4OFZ). b Conservation of the residues in C. neoformans Tps2PD (D24N)-T6P (PDB: 5DX9) demonstrates that the most conserved residues are near the substrate-binding pocket, while the variable regions are surface-exposed. The heat map is shown in the legend. c The details of the substrate-binding pocket of C. neoformans Tps2PD (D24N)-T6P (PDB: 5DX9), demonstrating the interaction between the Mg2+ molecule, T6P and the conserved DXD motif of HADSF enzymes. This figure created in Biorender with structures generated in ChimeraX108. Conservation analysis was completed with the ConSurf server109.
The cap and the core domains are connected by a flexible hinge region that enables the movement between the two domains, which is required for the dynamic catalytic process. For example, binding of T6P leads to the closure of the cap domain (Fig. 3a)48,79,80. Interestingly, several Tps2 enzymes contain N-terminal extensions that go beyond the conserved HADSF protein domains. For example, CaTps2 and CnTps2 N-terminal extensions contain a nonfunctional Tps1-like domain that may be important for complex formation amongst fungal Tps1 and Tps2 enzymes48,80,81. The Mycobacterium thermoresistible Tps2 (MtbTPP) contains an approximately 120 amino acid N-terminal extension that is hypothesized to contain autophosphorylation activity81. Further understanding of this N-terminal extension may provide more information of the functions of Tps2 proteins and, therefore, potential additional methods for targeting Tps2 with antifungal drugs.
Exploring novel antifungal strategies to develop the trehalose biosynthesis pathway as an antifungal drug target
The trehalose biosynthesis pathway, due to the multiple critical roles of trehalose in fungal cells as well as its lack of presence in humans, is an attractive antifungal drug target. In fact, the trehalose biosynthesis pathway has been ranked amongst the top antifungal drug target candidates by computer-aided target selection84.
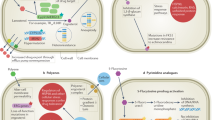
Current efforts to identify inhibitors of the trehalose biosynthesis pathway include a combination of high-throughput screens and “rational design” of inhibitors (Fig. 4)85. The ability to generate large amounts of highly-pure, recombinant and enzymatically active Tps1 and Tps2 protein also helps make the high-throughput screen approach feasible85. There are also numerous libraries of either bioactive molecules or lower molecular weight lead compounds that can be evaluated in high-throughput screens for either binding or reduction of activity of the target enzyme. The medium to high-resolution structures of Tps1 and Tps2 enzymes, discussed above, can be used for the structure-based drug design approach (Tables 1 and 2). Importantly, the structures of Tps1 and Tps2 have not just been solved in the apo or unbound form. There are now structures of fungal Tps1 and Tps2 enzymes that have been solved bound to either inhibitors, substrate analogs or native substrates, revealing details of conformational changes and key interactions between substrates and the target45,47,48,74. Data from multiple Tps1 and Tps2 structures have demonstrated to exquisite detail the amount of specificity in the active sites of these enzymes. Additionally, subsequent biochemical analyses reveal the crucial residues, usually via single point mutations, that are sufficient to render the enzyme inactive. This information is key and can be exploited to design an antifungal drug that targets the trehalose biosynthesis pathway. The iterative approach of using structures generated either via x-ray crystallography or cryo-EM and medicinal chemistry to generate derivative compounds may generate a specific or broad-spectrum antifungal drug that targets trehalose biosynthesis in fungal pathogens.

There are multiple methods to utilize to develop an inhibitor of trehalose biosynthesis, some of which are outlined in this figure. Structure-based approaches combined with high-throughput inhibitor screening can lead to the development of compounds that inhibit Tps1 and Tps2. This approach is enhanced with the use of in silico screening. There are currently multiple compounds that can act as scaffolds to begin the process of designing Tps1 inhibitors47,85,86. Additionally, if trehalose biosynthesis enzymes form complexes that contribute to their function, inhibiting complex formation if another method to inhibit trehalose biosynthesis. Lastly, there may be synergy between current antifungal drugs, particularly those that target the plasma membrane, and an inhibitor of trehalose biosynthesis. This figure was generated in Biorender.
There are already several compounds that have been demonstrated to bind trehalose biosynthesis proteins and inhibit their activity (Table 3)47,85,86,87,88,89. Several of the compounds were identified in Drosophila melanogaster, in screens of biologicals for activity against Tps1 and for use in structural biology studies. These compounds may be ideal scaffolds for the development of more effective trehalose biosynthesis-targeting antifungal drugs (Fig. 4).
High-throughput screens and structure-based approaches generally yield compounds that targets the substrate-binding or catalytic pockets of enzymes. However, since it has been hypothesized that trehalose biosynthesis proteins form a complex, the complex formation of trehalose biosynthesis proteins can be targeted (Fig. 4)48,49,50,51,52,53,54. In both Cryptococcus and Candida, the Tps2 proteins contain an enzymatically inactive Tps1-like domain. Therefore, Tps2 may be a Rosetta Stone protein, indicative of interactions between Tps1 and Tps290,91,92,93. In S. cerevisiae, there are four proteins in the trehalose biosynthesis pathway. Tps1 and Tps2 are the synthesis enzymes, while Tps3 and Tsl1 are the regulatory proteins. Tps1, Tps2 and Tsl1 have been found in the same fractions from a size exclusion chromatography fractionation94. Furthermore, in a bacterial two-hybrid experiment, there have been interactions between trehalose biosynthesis proteins identified (E.J.W., unpublished data). It remains to be seen whether complex formation is required for function. However, if the complex formation is required for the most efficient trehalose biosynthesis pathway, an allosteric inhibitor that disrupts the complex formation and, therefore, the substrate-binding pocket could be an effective antifungal drug. Additionally, targeting both the catalytic pocket directly and allosterically could work as a variation on the theme of Knudson’s two-hit hypothesis95. Several therapeutics have been identified that inhibit complex formation where complex formation is required for the function of the target96. By combining medicinal chemistry, structural biology and structure-based drug design, new potent yet safe antifungal drugs may be designed.
There is also the possibility to target trehalose biosynthesis in combination with current antifungal drugs97,98. It has been hypothesized that trehalose may contribute to the stabilization of fungal membranes. If so, weakening fungal membranes with fluconazole, for example, may predispose fungal cells to be more susceptible to compounds targeting trehalose biosynthesis. Such synergism has been demonstrated for other combinations of antifungal drugs99. In addition, such a dual mode-of-action formulation would decrease the likelihood of drug resistance developing. Similarly, on the theme of duality, a dual-target drug that inhibits both Tps1 and Tps2 might be incredibly effective, given that there is only partial overlap in their functions in fungal cells (Fig. 1b, c). By combining these multiple approaches and taking advantage of advances in medicinal chemistry and structural biology, the potential of developing a novel potent yet safe antifungal drug targeting the trehalose biosynthesis pathway may be realized.
What remains to be learned about trehalose biosynthesis in pathogenic fungi?
In Aspergillus fumigatus, it was determined, surprisingly, that deletion of two of the Tps1-like proteins, TpsA and TpsB, results in a moderately hypervirulent strain100. This genetic approach was instrumental in determining that Tps1 may not be an ideal antifungal drug target in A. fumigatus. Consequently, further understanding of the function of Tps1 in A. fumigatus was determined using multiple approaches including immunoprecipitation assays coupled with liquid chromatography-tandem mass spectrometry43,101. The example of A. fumigatus tpsA/tpsBΔ null mutant phenotypes highlights the importance of understanding the complete function of the trehalose biosynthesis proteins in the context of the fungal cell. The identification of trehalose biosynthesis pathway interacting partners is key. It is important to note that these might be species-specific. These proteins might be a novel set of proteins in fungi that may also be novel targets for antifungal drug development.
Several other pathways of note intersect with the trehalose biosynthesis pathway proteins. As mentioned earlier, there is a tight association and regulation of glycolysis and trehalose biosynthesis in fungal cells. The neutral trehalases in C. neoformans have been demonstrated to breakdown trehalose into two glucose molecules (Fig. 1a)66. Trehalase enzymes appear to only play minor roles in fungal virulence66. However, the glucose molecules are then phosphorylated by hexokinases. C. neoformans has two hexokinases, Hxk1 and Hxk2. Work by Michael Price has demonstrated the importance of hexokinases to fungal virulence in C. neoformans102. In S. cerevisiae the intermediate of the trehalose biosynthesis pathway, T6P, inhibits the activity of Hxk1 and Hxk2 and therefore has been proposed by the John Perfect laboratory as a potential molecular mechanism of T6P in Cryptococcus species66,85,103,104.
It is also likely that trehalose biosynthesis proteins have moonlighting functions105. Identification of these moonlighting functions using protein-protein interaction studies will reveal the interesting pathways that intersect with the trehalose biosynthesis pathway proteins in Cryptococcus and Candida, as has been demonstrated in A. fumigatus.
Conclusions
Here we conclude that the trehalose biosynthesis pathway is an important pathway to be investigated as a potential antifungal drug target. Trehalose enables organisms to tolerate multiple stresses. Many of the stresses are evolving as environmental fungi are exposed to changing conditions due to the climate. Understanding and modifying trehalose biosynthesis may be important in preventing the emergence of new pathogens that are developing thermotolerance and are being selected for crossing the thermal restriction zone31. In Cryptococcus and Candida species trehalose biosynthesis is required for virulence, resulting in fungicidal phenotypes when Tps1 and Tps2 are deleted. Importantly, the machinery for generating trehalose is not found in mammals106. With the tools and structures that have been developed, including the use of cryo-EM, there are multiple approaches that can be used to develop an inhibitor of trehalose biosynthesis in fungal pathogens. However, there is more work to be done, including the study the trehalose biosynthesis pathway in multiple different species and also a selection of clinical and environmental isolates107. The intersection of the trehalose biosynthesis pathway with other cellular pathways and processes is critical to understand and may well reveal novel antifungal drug targets.
Data availability
No datasets were generated or analyzed during the current study.
References
Denning, D. W. Global incidence and mortality of severe fungal disease. Lancet Infect Dis. 24, e428–e438 (2024).
Wang, L. R., Barber, C. E., Johnson, A. S. & Barnabe, C. Invasive fungal disease in systemic lupus erythematosus: a systematic review of disease characteristics, risk factors, and prognosis. Semin. Arthritis. Rheum. 44, 325–330 (2014).
Brown, G. D. Hidden killers: human fungal infections. Sci. Transl. Med. 4, 165rv13 (2012).
Speed, B. & Dunt, D. Clinical and host differences between infections with the two varieties of Cryptococcus neoformans. Clin. Infect. Dis. 21, 28–34 (1995).
Montoya, M. C., Magwene, P. M. & Perfect, J. R. Associations between Cryptococcus Genotypes, Phenotypes, and Clinical Parameters of Human Disease: A Review. J. Fungi 7, 260 (2021).
Kidd, S. E. et al. A rare genotype of Cryptococcus gattii caused the cryptococcosis outbreak on Vancouver Island (British Columbia, Canada). Proc. Natl. Acad. Sci. USA 101, 17258–17263 (2004).
Byrnes, E. J. 3rd, Bildfell, R. J., Dearing, P. L., Valentine, B. A. & Heitman, J. Cryptococcus gattii with bimorphic colony types in a dog in western Oregon: additional evidence for expansion of the Vancouver Island outbreak. J. Vet. Diagn. Invest 21, 133–136 (2009).
Datta, K. et al. Spread of Cryptococcus gattii into Pacific Northwest region of the United States. Emerg. Infect. Dis. 15, 1185–1191 (2009).
Hoang, L. M. N., Maguire, J. A., Doyle, P., Fyfe, M. & Roscoe, D. L. Cryptococcus neoformans infections at Vancouver Hospital and Health Sciences Centre (1997-2002): epidemiology, microbiology and histopathology. J. Med. Microbiol. 53, 935–940 (2004).
WHO Fungal Priority Pathogens List to Guide Research, Development and Public Health Action (World Health Organization, 2022).
Rajasingham, R. The global burden of HIV-associated cryptococcal infection in adults in 2020: a modelling analysis. Lancet Infect. Dis. 22, 1748–1755 (2022).
Ecology of Cryptococcus neoformans. In: Cryptococcus neoformans 41–70 (John Wiley & Sons, Ltd). https://doi.org/10.1128/9781555818241.ch3 (1998).
Lin, X. & Heitman, J. The biology of the Cryptococcus neoformans species complex. Annu. Rev. Microbiol. 60, 69–105 (2006).
Goldman, D. L. et al. Serologic evidence for Cryptococcus neoformans infection in early childhood. Pediatrics 107, E66 (2001).
Bailly, M. P. et al. Persistence of Cryptococcus neoformans in the prostate: failure of fluconazole despite high doses. J. Infect. Dis. 164, 435–436 (1991).
Barber, B. A., Crotty, J. M., Washburn, R. G. & Pegram, P. S. Cryptococcus neoformans myositis in a patient with AIDS. Clin. Infect. Dis. 21, 1510–1511 (1995).
Ghigliotti, G. & De Marchi, R. Cutaneous involvement with Cryptococcus neoformans in AIDS. J. Am. Acad. Dermatol 32, 820–821 (1995).
Seaton, R. A., Verma, N., Naraqi, S., Wembri, J. P. & Warrell, D. A. Visual loss in immunocompetent patients with Cryptococcus neoformans var. gattii meningitis. Trans. R. Soc. Trop. Med. Hyg. 91, 44–49 (1997).
Sobel, J. D. & Vazquez, J. A. Fungal infections of the urinary tract. World J. Urol. 17, 410–414 (1999).
Chang, Y. C. et al. Cryptococcal yeast cells invade the central nervous system via transcellular penetration of the blood-brain barrier. Infect. Immun. 72, 4985–4995 (2004).
Stott, K. E. et al. Cryptococcal meningoencephalitis: time for action. Lancet Infect. Dis. 21, e259–e271 (2021).
Romo, J. A. & Kumamoto, C. A. On Commensalism of Candida. J. Fungi 6, 16 (2020).
Kumamoto, C. A., Gresnigt, M. S. & Hube, B. The gut, the bad and the harmless: Candida albicans as a commensal and opportunistic pathogen in the intestine. Curr. Opin. Microbiol. 56, 7–15 (2020).
Pfaller, M. A., Diekema, D. J., Turnidge, J. D., Castanheira, M. & Jones, R. N. Twenty years of the SENTRY antifungal surveillance program: results for Candida species from 1997–2016. Open. Forum. Infect. Dis. 6, S79–S94 (2019).
Wisplinghoff, H. et al. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis. 39, 309–317 (2004).
Casadevall, A. Fungi and the rise of mammals. PLoS Pathog. 8, e1002808 (2012).
Casadevall, A. Global warming could drive the emergence of new fungal pathogens. Nat. Microbiol. 8, 2217–2219 (2023).
Nnadi, N. E. & Carter, D. A. Climate change and the emergence of fungal pathogens. PLoS Pathog. 17, e1009503 (2021).
Seidel, D. et al. Impact of climate change and natural disasters on fungal infections. Lancet Microbe S2666-S5247, https://doi.org/10.1016/S2666-5247(24)00039-9 (2024).
Casadevall, A., Kontoyiannis, D. P. & Robert, V. Environmental Candida auris and the Global Warming Emergence Hypothesis. mBio 12, e00360–21 (2021).
Casadevall, A., Kontoyiannis, D. P. & Robert, V. On the Emergence of Candida auris: Climate Change, Azoles, Swamps, and Birds. mBio 10, e01397–19 (2019).
Lockhart, S. R. et al. Simultaneous Emergence of Multidrug-Resistant Candida auris on 3 Continents Confirmed by Whole-Genome Sequencing and Epidemiological Analyses. Clin. Infect. Dis. 64, 134–140 (2017).
Anderson, T. M. Amphotericin forms an extramembranous and fungicidal sterol sponge. Nat. Chem. Biol. 10, 400–406 (2014).
Fanos, V. & Cataldi, L. Amphotericin B-induced nephrotoxicity: a review. J. Chemother. 12, 463–470 (2000).
Ostrosky-Zeichner, L., Casadevall, A., Galgiani, J. N., Odds, F. C. & Rex, J. H. An insight into the antifungal pipeline: selected new molecules and beyond. Nat. Rev. Drug Discov. 9, 719–727 (2010).
Ostrosky-Zeichner, L., Marr, K. A., Rex, J. H. & Cohen, S. H. Amphotericin B: time for a new ‘gold standard’. Clin. Infect. Dis. 37, 415–425 (2003).
Zavrel, M., Esquivel, B. D. & White, T. C. The Ins and Outs of Azole Antifungal Drug Resistance: Molecular Mechanisms of Transport. In: Handbook of Antimicrobial Resistance (eds. Berghuis, A., Matlashewski, G., Wainberg, M. A., Sheppard, D. & Gotte, M.) 423–452 (Springer New York, 2017). https://doi.org/10.1007/978-1-4939-0694-9_29.
Taft, C. S., Stark, T. & Selitrennikoff, C. P. Cilofungin (LY121019) inhibits Candida albicans (1-3)-beta-D-glucan synthase activity. Antimicrob. Agents Chemother. 32, 1901–1903 (1988).
Kurtz, M. B. et al. Morphological effects of lipopeptides against Aspergillus fumigatus correlate with activities against (1,3)-beta-D-glucan synthase. Antimicrob. Agents Chemother. 38, 1480–1489 (1994).
Pappas, P. G. et al. Executive Summary: Clinical Practice Guideline for the Management of Candidiasis: 2016 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 62, 409–417 (2016).
Perfect, J. R., et al. Editorial: Antifungal Pipeline: Build It Strong; Build It Better!. Front. Cell Infect. Microbiol. 12, 881272 (2022).
LeBlanc, E. V., Polvi, E. J., Veri, A. O., Privé, G. G. & Cowen, L. E. Structure-guided approaches to targeting stress responses in human fungal pathogens. J. Biol. Chem. 295, 14458–14472 (2020).
Thammahong, A., Puttikamonkul, S., Perfect, J. R., Brennan, R. G. & Cramer, R. A. Central Role of the Trehalose Biosynthesis Pathway in the Pathogenesis of Human Fungal Infections: Opportunities and Challenges for Therapeutic Development. Microbiol. Mol. Biol. Rev. 81, e00053–16 (2017).
Gancedo, C. & Flores, C. -L. The importance of a functional trehalose biosynthetic pathway for the life of yeasts and fungi. FEMS Yeast Res. 4, 351–359 (2004).
Washington, E. J. et al. Structures of trehalose-6-phosphate synthase, Tps1, from the fungal pathogen Cryptococcus neoformans: A target for antifungals. Proc. Natl. Acad. Sci. USA 121, e2314087121 (2024).
Chen, X. et al. Trehalose Phosphate Synthase Complex-Mediated Regulation of Trehalose 6-Phosphate Homeostasis Is Critical for Development and Pathogenesis in Magnaporthe oryzae. mSystems 6, e0046221 (2021).
Miao, Y. et al. Structural and In Vivo Studies on Trehalose-6-Phosphate Synthase from Pathogenic Fungi Provide Insights into Its Catalytic Mechanism, Biological Necessity, and Potential for Novel Antifungal Drug Design. mBio 8, e00643–17 (2017).
Miao, Y. et al. Structures of trehalose-6-phosphate phosphatase from pathogenic fungi reveal the mechanisms of substrate recognition and catalysis. Proc. Natl. Acad. Sci. USA 113, 7148–7153 (2016).
Vuorio, O. E., Kalkkinen, N. & Londesborough, J. Cloning of two related genes encoding the 56-kDa and 123-kDa subunits of trehalose synthase from the yeast Saccharomyces cerevisiae. Eur. J. Biochem. 216, 849–861 (1993).
Ferreira, J. C., Silva, J. T. & Panek, A. D. A regulatory role for TSL1 on trehalose synthase activity. Biochem. Mol. Biol. Int. 38, 259–265 (1996).
Reinders, A. et al. Structural analysis of the subunits of the trehalose-6-phosphate synthase/phosphatase complex in Saccharomyces cerevisiae and their function during heat shock. Mol. Microbiol. 24, 687–695 (1997).
Trevisol, E. T. V., Panek, A. D., De Mesquita, J. F. & Eleutherio, E. C. A. Regulation of the yeast trehalose-synthase complex by cyclic AMP-dependent phosphorylation. Biochim. Biophys. Acta 1840, 1646–1650 (2014).
Cao, Y. et al. Trehalose is an important mediator of Cap1p oxidative stress response in Candida albicans. Biol. Pharm. Bull. 31, 421–425 (2008).
Svanström, Å., van Leeuwen, M. R., Dijksterhuis, J. & Melin, P. Trehalose synthesis in Aspergillus niger: characterization of six homologous genes, all with conserved orthologs in related species. BMC Microbiol. 14, 90 (2014).
Meikle, A. J., Chudek, J. A., Reed, R. H. & Gadd, G. M. Natural abundance 13C-nuclear magnetic resonance spectroscopic analysis of acyclic polyol and trehalose accumulation by several yeast species in response to salt stress. FEMS Microbiol. Lett. 66, 163–167 (1991).
Crowe, J. H., Crowe, L. M. & Chapman, D. Preservation of membranes in anhydrobiotic organisms: the role of trehalose. Science 223, 701–703 (1984).
Elliott, B., Haltiwanger, R. S. & Futcher, B. Synergy between trehalose and Hsp104 for thermotolerance in Saccharomyces cerevisiae. Genetics 144, 923–933 (1996).
Singer, M. A. & Lindquist, S. Thermotolerance in Saccharomyces cerevisiae: the Yin and Yang of trehalose. Trends Biotechnol. 16, 460–468 (1998).
Tapia, H., Young, L., Fox, D., Bertozzi, C. R. & Koshland, D. Increasing intracellular trehalose is sufficient to confer desiccation tolerance to Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 112, 6122–6127 (2015).
Vanherp, L. et al. Trehalose as quantitative biomarker for in vivo diagnosis and treatment follow-up in cryptococcomas. Transl. Res. 230, 111–122 (2021).
Himmelreich, U. et al. Cryptococcomas distinguished from gliomas with MR spectroscopy: an experimental rat and cell culture study. Radiology 220, 122–128 (2001).
Steen, B. R. et al. Cryptococcus neoformans gene expression during experimental cryptococcal meningitis. Eukaryot. Cell 2, 1336–1349 (2003).
Zaragoza, O., Blazquez, M. A. & Gancedo, C. Disruption of the Candida albicans TPS1 gene encoding trehalose-6-phosphate synthase impairs formation of hyphae and decreases infectivity. J. Bacteriol. 180, 3809–3815 (1998).
Lin, X. & Heitman, J. Chlamydospore formation during hyphal growth in Cryptococcus neoformans. Eukaryot. Cell 4, 1746–1754 (2005).
Ngamskulrungroj, P. et al. The trehalose synthesis pathway is an integral part of the virulence composite for Cryptococcus gattii. Infect. Immun. 77, 4584–4596 (2009).
Petzold, E. W. et al. Characterization and regulation of the trehalose synthesis pathway and its importance in the pathogenicity of Cryptococcus neoformans. Infect. Immun. 74, 5877–5887 (2006).
Goughenour, K. et al. Cryptococcus neoformans trehalose-6-phosphate synthase (tps1) promotes organ-specific virulence and fungal protection against multiple lines of host defenses. Front. Cell Infect. Microbiol. 14, 1392015 (2024).
Tenor, J. L., Oehlers, S. H., Yang, J. L., Tobin, D. M. & Perfect, J. R. Live Imaging of Host-Parasite Interactions in a Zebrafish Infection Model Reveals Cryptococcal Determinants of Virulence and Central Nervous System Invasion. mBio 6, e01425–01415 (2015).
Chen, A., Vargas-Smith, J., Tapia, H. & Gibney, P. A. Characterizing phenotypic diversity of trehalose biosynthesis mutants in multiple wild strains of Saccharomyces cerevisiae. G3 Bethesda 12, jkac196 (2022).
Martínez-Esparza, M. et al. Role of trehalose-6P phosphatase (TPS2) in stress tolerance and resistance to macrophage killing in Candida albicans. Int. J. Med. Microbiol. 299, 453–464 (2009).
Van Dijck, P., De Rop, L., Szlufcik, K., Van Ael, E. & Thevelein, J. M. Disruption of the Candida albicans TPS2 gene encoding trehalose-6-phosphate phosphatase decreases infectivity without affecting hypha formation. Infect. Immun. 70, 1772–1782 (2002).
Zaragoza, O., de Virgilio, C., Pontón, J. & Gancedo, C. Disruption in Candida albicans of the TPS2 gene encoding trehalose-6-phosphate phosphatase affects cell integrity and decreases infectivity. Microbiology 148, 1281–1290 (2002).
De Virgilio, C. et al. Disruption of TPS2, the gene encoding the 100-kDa subunit of the trehalose-6-phosphate synthase/phosphatase complex in Saccharomyces cerevisiae, causes accumulation of trehalose-6-phosphate and loss of trehalose-6-phosphate phosphatase activity. Eur. J. Biochem. 212, 315–323 (1993).
Wang, S. et al. Crystal structures of Magnaporthe oryzae trehalose-6-phosphate synthase (MoTps1) suggest a model for catalytic process of Tps1. Biochem. J. 476, 3227–3240 (2019).
Mendes, V. et al. Mycobacterial OtsA Structures Unveil Substrate Preference Mechanism and Allosteric Regulation by 2-Oxoglutarate and 2-Phosphoglycerate. mBio 10, e02272–19 (2019).
Klutts, J. S., Yoneda, A., Reilly, M. C., Bose, I. & Doering, T. L. Glycosyltransferases and their products: cryptococcal variations on fungal themes. FEMS Yeast Res. 6, 499–512 (2006).
Breton, C., Snajdrová, L., Jeanneau, C., Koca, J. & Imberty, A. Structures and mechanisms of glycosyltransferases. Glycobiology 16, 29R–37R (2006).
Kuznetsova, E. et al. Functional Diversity of Haloacid Dehalogenase Superfamily Phosphatases from Saccharomyces cerevisiae: Biochemical, Structural, and Evolutionary Insights. J. Biol. Chem. 290, 18678–18698 (2015).
Suthisawat, S., Gourlay, L. J., Bolognesi, M., Boonyuen, U. & Vanaporn, M. Functional and structural analysis of trehalose-6-phosphate phosphatase from Burkholderia pseudomallei: Insights into the catalytic mechanism. Biochem. Biophys. Res. Commun. 523, 979–984 (2020).
Farelli, J. D. et al. Structure of the trehalose-6-phosphate phosphatase from Brugia malayi reveals key design principles for anthelmintic drugs. PLoS Pathog. 10, e1004245 (2014).
Shan, S., Min, H., Liu, T., Jiang, D. & Rao, Z. Structural insight into dephosphorylation by trehalose 6-phosphate phosphatase (OtsB2) from Mycobacterium tuberculosis. FASEB J. 30, 3989–3996 (2016).
Harvey, C. M. et al. Structural Analysis of Binding Determinants of Salmonella typhimurium Trehalose-6-phosphate Phosphatase Using Ground-State Complexes. Biochemistry 59, 3247–3257 (2020).
Burroughs, A. M., Allen, K. N., Dunaway-Mariano, D. & Aravind, L. Evolutionary genomics of the HAD superfamily: understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J. Mol. Biol. 361, 1003–1034 (2006).
Spaltmann, F., Blunck, M. & Ziegelbauer, K. Computer-aided target selection-prioritizing targets for antifungal drug discovery. Drug Discov. Today 4, 17–26 (1999).
Perfect, J. R., Tenor, J. L., Miao, Y. & Brennan, R. G. Trehalose pathway as an antifungal target. Virulence 8, 143–149 (2017).
Kern, C. et al. Trehalose-6-phosphate synthase from the cat flea Ctenocephalides felis and Drosophila melanogaster: gene identification, cloning, heterologous functional expression and identification of inhibitors by high throughput screening. Insect Mol. Biol. 21, 456–471 (2012).
Pan, Y. T. & Elbein, A. D. Inhibition of the trehalose-P synthase of mycobacteria by various antibiotics. Arch. Biochem. Biophys. 335, 258–266 (1996).
Cross, M. et al. A suicide inhibitor of nematode trehalose-6-phosphate phosphatases. Sci. Rep. 9, 16165 (2019).
Chen, Y. et al. Dual-Specificity Inhibitor Targets Enzymes of the Trehalose Biosynthesis Pathway. J. Agric. Food Chem. 72, 209–218 (2024).
Marcotte, E. M. et al. Detecting protein function and protein-protein interactions from genome sequences. Science 285, 751–753 (1999).
Enright, A. J., Iliopoulos, I., Kyrpides, N. C. & Ouzounis, C. A. Protein interaction maps for complete genomes based on gene fusion events. Nature 402, 86–90 (1999).
Marcotte, C. J. V. & Marcotte, E. M. Predicting functional linkages from gene fusions with confidence. Appl. Bioinforma. 1, 93–100 (2002).
Yanai, I., Derti, A. & DeLisi, C. Genes linked by fusion events are generally of the same functional category: a systematic analysis of 30 microbial genomes. Proc. Natl. Acad. Sci. USA 98, 7940–7945 (2001).
Bell, W. et al. Composition and functional analysis of the Saccharomyces cerevisiae trehalose synthase complex. J. Biol. Chem. 273, 33311–33319 (1998).
Hino, O. & Kobayashi, T. M. o. u. r. n. i. n. g. D. r. Alfred G. Knudson: the two-hit hypothesis, tumor suppressor genes, and the tuberous sclerosis complex. Cancer Sci. 108, 5–11 (2017).
Neckers, L. et al. Methods to validate Hsp90 inhibitor specificity, to identify off-target effects, and to rethink approaches for further clinical development. Cell Stress Chaperones 23, 467–482 (2018).
Iyer, K. R. An oxindole efflux inhibitor potentiates azoles and impairs virulence in the fungal pathogen Candida auris. Nat. Commun. 11, 6429 (2020).
Robbins, N. & Cowen, L. E. Antifungal discovery. Curr. Opin. Microbiol. 69, 102198 (2022).
Cui, J., Ren, B., Tong, Y., Dai, H. & Zhang, L. Synergistic combinations of antifungals and anti-virulence agents to fight against Candida albicans. Virulence 6, 362–371 (2015).
Al-Bader, N. et al. Role of trehalose biosynthesis in Aspergillus fumigatus development, stress response, and virulence. Infect. Immun. 78, 3007–3018 (2010).
Thammahong, A., Dhingra, S., Bultman, K. M., Kerkaert, J. D. & Cramer, R. A. An Ssd1 Homolog Impacts Trehalose and Chitin Biosynthesis and Contributes to Virulence in Aspergillus fumigatus. mSphere 4, e00244–19 (2019).
Price, M. S. et al. Cryptococcus neoformans requires a functional glycolytic pathway for disease but not persistence in the host. mBio 2, e00103–e00111 (2011).
Blázquez, M. A., Lagunas, R., Gancedo, C. & Gancedo, J. M. Trehalose-6-phosphate, a new regulator of yeast glycolysis that inhibits hexokinases. FEBS Lett. 329, 51–54 (1993).
Magalhães, R. S. S. et al. Hexokinase 2: The preferential target of trehalose-6-phosphate over hexokinase 1. J. Cell Biochem. 123, 1808–1816 (2022).
Thammahong, A., Caffrey-Card, A. K., Dhingra, S., Obar, J. J. & Cramer, R. A. Aspergillus fumigatus Trehalose-Regulatory Subunit Homolog Moonlights To Mediate Cell Wall Homeostasis through Modulation of Chitin Synthase Activity. mBio 8, e00056–17 (2017).
Argüelles, J. -C. Why can’t vertebrates synthesize trehalose?. J. Mol. Evol. 79, 111–116 (2014).
Desjardins, C. A. et al. Population genomics and the evolution of virulence in the fungal pathogen Cryptococcus neoformans. Genome Res. 27, 1207–1219 (2017).
Meng, E. C. et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Armon, A., Graur, D. & Ben-Tal, N. ConSurf: an algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J. Mol. Biol. 307, 447–463 (2001).
Gibson, R. P., Turkenburg, J. P., Charnock, S. J., Lloyd, R. & Davies, G. J. Insights into trehalose synthesis provided by the structure of the retaining glucosyltransferase OtsA. Chem. Biol. 9, 1337–1346 (2002).
Gibson, R. P., Tarling, C. A., Roberts, S., Withers, S. G. & Davies, G. J. The donor subsite of trehalose-6-phosphate synthase: binary complexes with UDP-glucose and UDP-2-deoxy-2-fluoro-glucose at 2 A resolution. J. Biol. Chem. 279, 1950–1955 (2004).
Acknowledgements
The author thanks all members and friends of the Washington laboratory for helpful discussions. Thanks to Dr. Andrew Alspaugh and Dr. David A. Hubert for reviewing the manuscript before submission. E.J.W. is supported by the Duke Next Generation Fellowship funded by the Duke Science and Technology Initiative in the Duke University School of Medicine. The funder played no role in study design, data collection, analysis and interpretation of data, or the writing of this manuscript.
Author information
Authors and Affiliations
Contributions
E.J.W. researched data for the article and wrote the article. E.J.W. reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Washington, E.J. Developing the trehalose biosynthesis pathway as an antifungal drug target. npj Antimicrob Resist 3, 30 (2025). https://doi.org/10.1038/s44259-025-00095-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44259-025-00095-2
