 PDF(5174 KB)
PDF(5174 KB)

外胚叶发育不全基因A突变导致家族性非综合征型先天缺牙的初步研究
王慧慧, 吴情, 徐斌, 凌琪, 吴轶群
口腔疾病防治 ›› 2023, Vol. 31 ›› Issue (11) : 768-773.
PDF(5174 KB)
PDF(5174 KB)
外胚叶发育不全基因A突变导致家族性非综合征型先天缺牙的初步研究
Preliminary study of familial nonsyndromic tooth agenesis caused by ectodysplasin A mutation
目的 寻找非综合征型先天缺牙(non-syndromic tooth agenesis, NSTA)家系的致病基因,探讨其发病机制。方法 获得医院伦理审批及患者与家属知情同意,收集先证者及其家系主要成员的临床资料,采集外周静脉血,提取DNA,利用全外显子测序技术进行基因检测,运用Sanger测序验证筛查出的致病基因,运用生物信息学工具分析突变蛋白的三维结构,并与野生型进行比较分析。结果 该家系2名患者为表兄弟关系,家系中无其他先天多数牙缺失的患者,除先天缺失多颗牙外,2名患者无明显毛发异常、无指/趾异常、无出汗异常等其他外胚叶组织的异常表现。通过对此家系中的患者及主要成员进行基因测序,发现与该家系相关的突变基因是一种新的外胚叶发育不全基因A(ectodysplasin A,EDA)的错义突变c.983C>T(p. Pro328Leu),导致对应编码的氨基酸从脯氨酸(Pro)变为亮氨酸(Leu)。对该突变位点进行保守性分析发现,该位点具有高度保守性,通过三维构建模发现该位点蛋白结构发生了改变。结论 首次发现EDA基因新的错义突变位点c.983C>T(p. Pro328Leu)与非综合征型先天缺牙有关,扩大了EDA基因的突变谱。
Objective To explore the pathogenic genes in a Chinese family affected by nonsyndromic tooth agenesis so as to study the pathogenesis of oligodontia. Methods Hospital ethical approval and informed consent of the patients and family members were obtained. Clinical data of the proband and close family members were collected, peripheral venous blood was collected, and DNA was extracted. Gene sequencing was performed through whole-exome sequencing, and then the screened pathogenic genes were verified by Sanger sequencing. The three-dimensional structure of the mutant proteins was analyzed and compared with the wild-type using bioinformatics tools. Results The two patients with congenital majority tooth loss in this family were cousins, and there were no other patients with congenital majority tooth loss in the family. Besides congenital multiple tooth loss, the two patients had no obvious hair abnormalities, finger/toe abnormalities, sweating abnormalities or other abnormal manifestations of ectodermal tissue. We found a mutant gene that in this family by carrying out gene sequencing of the patients and their close family members. A novel EDA (ectodysplasin A) missense mutation c.983C>T (p. Pro328Leu) was identified, which changed the encoded amino acid from proline (Pro) to leucine (Leu). Analysis of the mutation site showed that the site was highly conserved, and three-dimensional structure modeling also found that it changed the structure of EDA. Conclusion A novel EDA missense variant (c.983C>T, p.Pro328Leu) was first identified in a Chinese family with nonsyndromic tooth agenesis, extending the mutation spectrum of the EDA gene.
外胚叶发育不全基因A / 非综合征型先天缺牙 / 综合征型先天缺牙 / 多数牙缺失 / 个别牙缺失 / 全外显子测序 / Sanger测序 / 基因组DNA / 基因突变 / 错义突变 {{custom_keyword}} /
ectodysplasin A gene / non-syndromic tooth agenesis / syndromic tooth agenesis / hypodontia / oligodontia / whole exome sequencing / Sanger sequencing / genomic DNA / gene mutation / missense mutation {{custom_keyword}} /

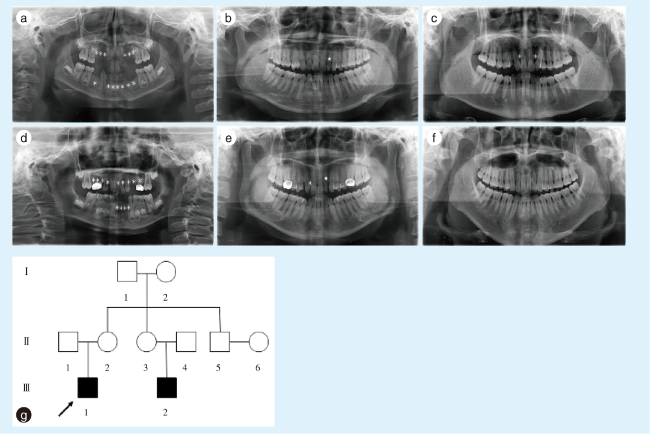
图1 非综合征型牙齿先天缺失先证者家系成员缺牙情况及家系图谱Figure 1 Information on absent teeth in the proband with nonsyndromic tooth agenesis and family pedigree * represents the location of the missing tooth; ★ represents the microdontiaa: panoramic radiograph of the proband (Ⅲ1), a 7-year-old boy, showed that permanent teeth 12~14, 22~24, 31~34, 41, 42, and 44 were missing; b: panoramic radiograph of the proband's mother (Ⅱ2) showed that the left lateral incisor in the maxilla had microdontia; c: panoramic radiograph of the proband's father (Ⅱ1) indicated that the two lateral incisors in the maxilla were missing; d: panoramic radiograph of the proband's cousin (Ⅲ2), a 6-year-old boy, showed that permanent teeth 12~15, 21~25, 31, 32, 41, and 42 were missing; e: panoramic radiograph of the cousin's mother (Ⅱ3) indicated that the right lateral incisors in the maxilla were missing and the left lateral incisor in the maxilla had microdontia; f: panoramic radiograph of the cousin's father (Ⅱ4) showed no teeth were missing; g: pedigree of the proband (arrow) showing that proband Ⅲ1 was a 7-year-old boy and proband Ⅲ2 was a 6-year-old boy who was a cousin of proband Ⅲ1 |
表1 非综合征型牙齿先天缺失的突变基因位点Table 1 Information on the mutant genes of nonsyndromic tooth agenesis |
| Chrom | Position | Reference | Call | Gene start | Gene end | Gene symbols |
|---|---|---|---|---|---|---|
| Chr1 | 150779274 | C | T | 150768683 | 150780799 | CTSK |
| Chr11 | 71148920 | G | T | 71139238 | 71163914 | DHCR7 |
| Chr3 | 57131759 | T | C | 57124009 | 57204334 | IL17RD |
| ChrX | 69255266 | C | T | 68835910 | 69259319 | EDA |
| CTSK: cathepsin K; DHCR7: 7-dehydrocholesterol reductase; IL17RD: interleukin-17 receptor D ; EDA: ectodysplasin A |
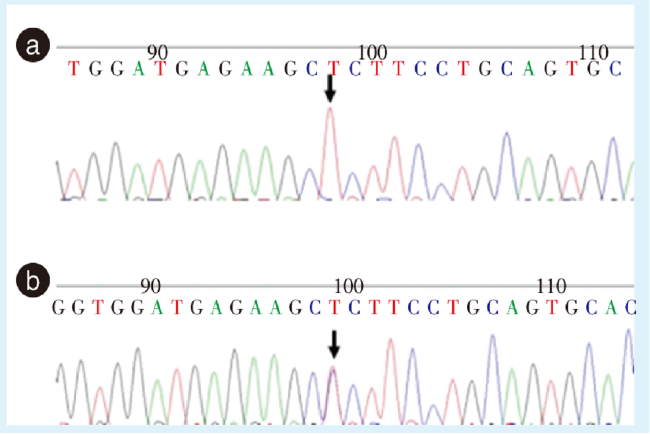
图2 Sanger测序验证非综合征型牙齿先天缺失的EDA基因突变Figure 2 Identification of the variant in the EDA gene by Sanger sequencing of nonsyndromic tooth agenesis patients a: the proband and his cousin had a missense mutation in exon 8 of the EDA gene; b: the mother of the proband and the mother of the cousin had a missense mutation in exon 8 of the EDA gene. EDA: ectodysplasin A. The arrow indicates the location of the mutation |
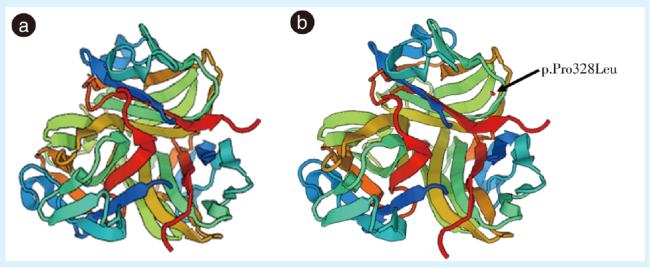
图3 非综合征型牙齿先天缺失的野生型及突变型EDA蛋白结构预测Figure 3 Structure prediction of the wild-type and mutant EDA proteins in these nonsyndromic tooth agenesis patients a: three-dimensional structure of wild-type EDA protein; b: three-dimensional structure of mutant EDA protein. EDA: ectodysplasin A. The arrow indicates the mutation (p.Pro328Leu) location |

| [1] |
Oral rare diseases are characterized by low prevalence and complex clinical features. It is not clear that which kind of diseases belong to oral rare diseases. In order to make a consensus and improve the level of diagnosis and treatment of oral rare diseases, Duan Xiaohong, the first chair of Society of Oral Genetic Diseases and Rare Diseases, Chinese Stomatological Association, progosed the first edition of oral rare diseases list, and the whole society discussed the list and finally made an agreement. The list includes 139 rare diseases with typical oral and craniofacial characteristics, and provides their Chinese names, English names, International Classification of Diseases 11th Revision (ICD-11) codes, Online Mendelian Inheritance in Man (OMIM) numbers, prevalence and simple interpretations.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [2] |
蒋彩玲, 赵彬, 吴轶群. LRP6 基因突变导致选择性先天缺牙的研究进展[J]. 口腔疾病防治, 2023, 31(3): 223-228. doi:10.12016/j.issn.2096-1456.2023.03.012.
选择性先天缺牙是由遗传或环境因素导致的牙齿数目异常,多累及恒牙列。低密度脂蛋白受体相关蛋白6(low-density lipoprotein receptor-related protein 6,LRP6)是选择性先天缺牙的常见致病基因之一,该基因突变为常染色体显性遗传,可导致非综合征型先天缺牙或综合征型先天缺牙;非综合征型先天缺牙仅表现为牙齿数目、形态异常;综合征型先天缺牙可表现为耳部发育畸形、口面裂、毛发稀少、汗腺异常等。笔者就近年来关于LRP6基因突变导致选择性先天缺牙的表型及基因突变特点的研究现况进行综述,文献收纳24个LRP6基因突变位点和38例相关先天缺牙患者,发现LRP6基因突变导致的选择性先天缺牙好发于上颌侧切牙及上下颌第二前磨牙和第一前磨牙,极少发生于第一磨牙,尤其是下颌第一磨牙,未见上颌中切牙缺失。LRP6基因在牙发育过程中主要通过WNT/β-catenin信号通路发挥重要作用,LRP6基因突变可导致蛋白表达和功能异常、信号通路破坏从而导致选择性先天缺牙。现有文献结果显示,LRP6基因突变好发于胞外段E1、E2亚结构域,影响WNT/β-catenin信号通路的传导而致病。然而目前对于选择性先天缺牙仍缺乏成熟完善的对因治疗。
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [3] |
Like all developmental processes, odontogenesis is highly complex and dynamically regulated, with hundreds of genes co-expressed in reciprocal networks. Tooth agenesis (missing one or more/all teeth) is a common human craniofacial anomaly and may be caused by genetic variations and/or environmental factors. Variants in PAX9, MSX1, AXIN2, EDA, EDAR, and WNT10A genes are associated with tooth agenesis. Currently, variants in ATF1, DUSP10, CASC8, IRF6, KDF1, GREM2, LTBP3, and components and regulators of WNT signaling WNT10B, LRP6, DKK, and KREMEN1 are at the forefront of interest. Due to the interconnectedness of the signaling pathways of carcinogenesis and odontogenesis, tooth agenesis could be a suitable marker for early detection of cancer predisposition. Variants in genes associated with tooth agenesis could serve as prognostic or therapeutic targets in cancer. This review aims to summarize existing knowledge of development and clinical genetics of teeth. Concurrently, the review proposes possible approaches for future research in this area, with particular attention to roles in monitoring, early diagnosis and therapy of tumors associated with defective tooth development.© 2020 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [4] |
The lack of treatment options for congenital (0.1%) and partial (10%) tooth anomalies highlights the need to develop innovative strategies. Over two decades of dedicated research have led to breakthroughs in the treatment of congenital and acquired tooth loss. We revealed that by inactivating, congenital tooth agenesis can be successfully ameliorated during early tooth development and that the inactivation promotes late-stage tooth morphogenesis in double knockout mice. Furthermore, Anti- antibody treatment in mice is effective in tooth regeneration and can be a breakthrough in treating tooth anomalies in humans. With approximately 0.1% of the population suffering from congenital tooth agenesis and 10% of children worldwide suffering from partial tooth loss, early diagnosis will improve outcomes and the quality of life of patients. Understanding the role of pathogenic variants, their interacting gene partners, and their protein functions will help develop critical biomarkers. Advances in next-generation sequencing, mass spectrometry, and imaging technologies will assist in developing companion and predictive biomarkers to help identify patients who will benefit from tooth regeneration.© 2023 The Japanese Society for Regenerative Medicine. Production and hosting by Elsevier B.V.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [5] |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [6] |
Oligodontia is the congenital absence of six or more teeth and comprises the more severe forms of tooth agenesis. Many genes have been implicated in the etiology of tooth agenesis, which is highly variable in its clinical presentation. The purpose of this study was to identify associations between genetic mutations and clinical features of oligodontia patients. An online systematic search of papers published from January 1992 to June 2021 identified 381 oligodontia cases meeting the eligibility criteria of causative gene mutation, phenotype description, and radiographic records. Additionally, ten families with oligodontia were recruited and their genetic etiologies were determined by whole-exome sequence analyses. We identified a novel mutation in WNT10A (c.99_105dup) and eight previously reported mutations in WNT10A (c.433 G > A; c.682 T > A; c.318 C > G; c.511.C > T; c.321 C > A), EDAR (c.581 C > T), and LRP6 (c.1003 C > T, c.2747 G > T). Collectively, 20 different causative genes were implicated among those 393 cases with oligodontia. For each causative gene, the mean number of missing teeth per case and the frequency of teeth missing at each position were calculated. Genotype-phenotype correlation analysis indicated that molars agenesis is more likely linked to PAX9 mutations, mandibular first premolar agenesis is least associated with PAX9 mutations. Mandibular incisors and maxillary lateral incisor agenesis are most closely linked to EDA mutations.© 2021. The Author(s).
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [7] |
Tooth agenesis (TA) is one of the most common developmental anomalies that affects the number of teeth. An extensive analysis of publicly accessible databases revealed 15 causative genes responsible for nonsyndromic TA, along with their signaling pathways in Wnt/β-catenin, TGF-β/BMP, and Eda/Edar/NF-κB. However, genotype-phenotype correlation analysis showed that most of the causal genes are also responsible for syndromic TA or other conditions. In a total of 198 different mutations of the 15 genes responsible for nonsyndromic TA, 182 mutations (91.9%) are derived from seven genes (AXIN2, EDA, LRP6, MSX1, PAX9, WNT10A, and WNT10B) compared with the remaining 16 mutations (8.1%) identified in the remaining eight genes (BMP4, DKK1, EDAR, EDARADD, GREM2, KREMEN1, LTBP3, and SMOC2). Furthermore, specificity analysis in terms of the ratio of nonsyndromic TA mutations versus syndromic mutations in each of the aforementioned seven genes showed a 98.2% specificity rate in PAX9, 58.9% in WNT10A, 56.6% in MSX1, 41.2% in WNT10B, 31.4% in LRP6, 23.8% in AXIN2%, and 8.4% in EDA. These findings underscore an important role of the Wnt and Wnt-associated pathways in the genetic etiology of this heterozygous disease and shed new lights on the discovery of novel molecular mechanisms associated with tooth agenesis.© 2018 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd. All rights reserved.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [8] |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [9] |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [10] |
Ectodysplasin A (EDA) is a member of the tumor necrosis factor (TNF) family of ligands that was initially reported to induce the formation of various ectodermal derivatives during normal prenatal development. EDA exerts its biological activity as two splice variants, namely, EDA-A1 and EDA-A2. The former binds to the EDA receptor (EDAR), resulting in the recruitment of the intracellular EDAR-associated death domain (EDARADD) adapter protein and the activation of the NF-κB signaling pathway, while the latter binds to a different receptor, EDA2R, also known as X-linked ectodermal dysplasia receptor (XEDAR). Inactivation mutation of the EDA gene or the genes coding for its receptors can result in hypohidrosis ectodermal dysplasia (HED), a condition that is characterized by oligotrichosis, edentulosis or oligodontia, and oligohidrosis or anhidrosis. Recently, as a new liver factor, EDA is gradually known and endowed with some new functions. EDA levels were observed to be upregulated in several metabolic diseases, such as non-alcoholic fatty liver disease (NAFLD), obesity, and insulin resistance. In addition, EDA and its receptors have been implicated in tumor pathogenesis through the regulation of tumor cell proliferation, apoptosis, differentiation, and migration. Here, we first review the role of EDA and its two-receptor system in various signaling pathways and then discuss the physiological and pathological roles of EDA and its receptors.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [11] |
Ectodermal dysplasia (ED) represents a collection of rare disorders that result from a failure of development of the tissues derived from the embryonic ectoderm. ED is often associated with hair, teeth, and skin abnormalities, which are serious conditions affecting the quality of life of the patient. To date, a large number of genes have been found to be associated with this syndrome. Here, we report a patient with hypohidrotic ED (HED) without family history. We identified that this patient's disorder arises from an X-linked HED with a mutation in the EDA gene (G299D) found by whole-exome sequencing. In addition, in this paper we summarize the disease-causing mutations based on current literature. Overall, recent clinical and genetic research involving patients with HED have uncovered a large number of pathogenic mutations in EDA, which might contribute to a full understanding of the function of EDA and the underlying mechanisms of HED caused by EDA mutations.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [12] |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [13] |
Ectodermal dysplasia (ED) is a diverse group of genetic disorders caused by congenital defects of two or more ectodermal-derived body structures, namely, hair, teeth, nails, and some glands, e.g., sweat glands. Molecular pathogenesis of ED involves mutations of genes encoding key proteins of major developmental pathways, including ectodysplasin (EDA) and wingless-type (WNT) pathways. The most common ED phenotype is hypohidrotic/anhidrotic ectodermal dysplasia (HED) featuring hypotrichosis, hypohidrosis/anhidrosis, and hypodontia. Molecular diagnosis is fundamental for disease management and emerging treatments. We used targeted next generation sequencing to study EDA, EDAR, EDARADD, and WNT10A genes in 45 Egyptian ED patients with or without hypohidrosis. We present genotype and phenotype data of 28 molecularly-characterized patients demonstrating genetic heterogeneity, variable expressivity, and intrafamilial phenotypic variability. Thirteen mutations were reported, including four novel EDA mutations, two novel EDARADD, and one novel EDAR mutations. Identified mutations congregated in exons encoding key functional domains. EDA is the most common gene contributing to 85% of the identified Egyptian ED genetic spectrum, followed by EDARADD (10%) and EDAR (5%). Our cohort represents the first and largest cohort from North Africa where more than 60% of ED patients were identified emphasizing the need for exome sequencing to explore unidentified cases.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [14] |
The Ensembl Variant Effect Predictor is a powerful toolset for the analysis, annotation, and prioritization of genomic variants in coding and non-coding regions. It provides access to an extensive collection of genomic annotation, with a variety of interfaces to suit different requirements, and simple options for configuring and extending analysis. It is open source, free to use, and supports full reproducibility of results. The Ensembl Variant Effect Predictor can simplify and accelerate variant interpretation in a wide range of study designs.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [15] |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [16] |
Genetic sequencing, or DNA sequencing, using the Sanger technique has become widely used in the veterinary diagnostic community. This technology plays a role in verification of PCR results and is used to provide the genetic sequence data needed for phylogenetic analysis, epidemiologic studies, and forensic investigations. The Laboratory Technology Committee of the American Association of Veterinary Laboratory Diagnosticians has prepared guidelines for sample preparation, submission to sequencing facilities or instrumentation, quality assessment of nucleic acid sequence data performed, and for generating basic sequencing data and phylogenetic analysis for diagnostic applications. This guidance is aimed at assisting laboratories in providing consistent, high-quality, and reliable sequence data when using Sanger-based genetic sequencing as a component of their laboratory services.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [17] |
Proline is a non-essential amino acid with key roles in protein structure/function and maintenance of cellular redox homeostasis. It is available from dietary sources, generated de novo within cells, and released from protein structures; a noteworthy source being collagen. Its catabolism within cells can generate ATP and reactive oxygen species (ROS). Recent findings suggest that proline biosynthesis and catabolism are essential processes in disease; not only due to the role in new protein synthesis as part of pathogenic processes but also due to the impact of proline metabolism on the wider metabolic network through its significant role in redox homeostasis. This is particularly clear in cancer proliferation and metastatic outgrowth. Nevertheless, the precise identity of the drivers of cellular proline catabolism and biosynthesis, and the overall cost of maintaining appropriate balance is not currently known. In this review, we explore the major drivers of proline availability and consumption at a local and systemic level with a focus on cancer. Unraveling the main factors influencing proline metabolism in normal physiology and disease will shed light on new effective treatment strategies.© 2021. The Author(s).
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [18] |
To keep pace with the rapid advancements in molecular genetics and rare diseases research, we have updated the list of ectodermal dysplasias based on the latest classification approach that was adopted in 2017 by an international panel of experts. For this purpose, we searched the databases PubMed and OMIM for the term “ectodermal dysplasia”, referring mainly to changes in the last 5 years. We also tried to obtain information about those diseases on which the last scientific report appeared more than 15 years ago by contacting the authors of the most recent publication. A group of experts, composed of researchers who attended the 8th International Conference on Ectodermal Dysplasias and additional members of the previous classification panel, reviewed the proposed amendments and agreed on a final table listing all 49 currently known ectodermal dysplasias for which the molecular genetic basis has been clarified, including 15 new entities. A newly reported ectodermal dysplasia, linked to the gene LRP6, is described here in more detail. These ectodermal dysplasias, in the strict sense, should be distinguished from syndromes with features of ectodermal dysplasia that are related to genes extraneous to the currently known pathways involved in ectodermal development. The latter group consists of 34 syndromes which had been placed on the previous list of ectodermal dysplasias, but most if not all of them could actually be classified elsewhere. This update should streamline the classification of ectodermal dysplasias, provide guidance to the correct diagnosis of rare disease entities, and facilitate the identification of individuals who could benefit from novel treatment options.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [19] |
Mutations of the Ectodysplasin-A (EDA) gene are generally associated with the syndrome hypohidrotic ectodermal dysplasia (MIM 305100), but they can also manifest as selective, non-syndromic tooth agenesis (MIM300606). We have performed an in vitro functional analysis of six selective tooth agenesis-causing EDA mutations (one novel and five known) that are located in the C-terminal tumor necrosis factor homology domain of the protein. Our study reveals that expression, receptor binding or signaling capability of the mutant EDA1 proteins is only impaired in contrast to syndrome-causing mutations, which we have previously shown to abolish EDA1 expression, receptor binding or signaling. Our results support a model in which the development of the human dentition, especially of anterior teeth, requires the highest level of EDA-receptor signaling, whereas other ectodermal appendages, including posterior teeth, have less stringent requirements and form normally in response to EDA mutations with reduced activity.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [20] |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [21] |
EDA is a tumor necrosis factor (TNF) family member, which functions together with its cognate receptor EDAR during ectodermal organ development. Mutations of EDA have long been known to cause X-linked hypohidrotic dysplasia in humans characterized by primary defects in teeth, hair and sweat glands. However, the structural information of EDA interaction with EDAR is lacking and the pathogenic mechanism of EDA variants is poorly understood. Here, we report the crystal structure of EDA C-terminal TNF homology domain bound to the N-terminal cysteine-rich domains of EDAR. Together with biochemical, cellular and mouse genetic studies, we show that different EDA mutations lead to varying degrees of ectodermal developmental defects in mice, which is consistent with the clinical observations on human patients. Our work extends the understanding of the EDA signaling mechanism, and provides important insights into the molecular pathogenesis of disease-causing EDA variants.© 2023. The Author(s).
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [22] |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [23] |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| {{custom_ref.label}} |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
PDF(5174 KB)
 粤公网安备 44010502002066号
粤ICP备18050693号
粤公网安备 44010502002066号
粤ICP备18050693号
 本作品遵循Creative Commons Attribution 4.0 License授权许可.
本作品遵循Creative Commons Attribution 4.0 License授权许可.
/
| 〈 |
|
〉 |






{kind=link}
{kind=link}
{kind=link}
{kind=link}